

Abstract
Infection by human immunodeficiency virus type 1 (HIV-1) is associated with decreases in peripheral CD4+ T cells and development of lymphadenopathy. The precise mechanisms by which HIV-1 induces these changes have not been elucidated. T-cell trafficking through lymphoid tissues is facilitated by CCL21-mediated entry and sphingosine-1-phosphate (S1P)-mediated egress. Having previously determined that HIV-1 envelop glycoprotein, gp120, directly alters T-cell migration, we investigated whether gp120 without HIV-1 infection could influence the responses of CD4+ T cells to the signals involved in T-cell trafficking through lymph tissue. Incubation of normal human T cells with gp120 for 1 h resulted in reprogramming of CD4 T-cell migratory responses by increasing sensitivity to CCL20 and CCL21 and complete inhibition of migration to S1P. Incubation of human T cells with gp120 prior to injection into NOD.CB17-Prkdcscid/J mice resulted in increases in lymph node accumulation of CD4+ T cells, with reciprocal decreases in blood and spleen compared to T cells not exposed to gp120. The effects of gp120 required CD4 signaling mediated through p56lck. These findings suggest that gp120 alone can alter CD4+ influx and efflux from lymph nodes in a fashion consistent with the development of lymphopenia and lymphadenopathy.
During primary human immunodeficiency virus (HIV) infection, there is a deletion of CD4+ T cells in the gut that is responsible for a large reduction in the total number of cells (4). After initial infection and cell loss, a homeostatic balance is achieved whereby the total number of productively infected cells is estimated to be between 1:100 and 1:400 (13). With disease progression there is a decrease in the percentage of total cells found in the peripheral blood. This lymphopenia is defined by immune cell activation coincident with tissue redistribution of CD4+ T cells (25). Lymphopenia is accompanied by and is in part due to lymphadenopathy, characterized by excessive accumulation of CD4+ T cells in secondary lymph tissue (SLT) (25, 27). The prevalence of lymphadenopathy as a clinical manifestation of infection led to the initial designation of HIV-1 as lymphadenopathy-associated virus (23). Development of lymphadenopathy is thought to facilitate enhanced infection and killing of CD4+ T cells, removal of T-helper cells from sites of infection, and dysregulation of the immune response by sequestration of peripheral T regulatory cells (11, 24). While lymphadenopathy likely plays a role in HIV-1 pathology, to date a mechanism directly responsible for this effect has yet to be reported.
Trafficking of T cells within SLT is a dynamic process that balances homing with egress. T-cell homing to the SLT requires expression of CD62L for adherence to high endothelial venules and CCR7 expression for chemotactic responsiveness to lymph node (LN)-specific chemokines, such as CCL19 and CCL21 (8). A number of studies have shown that infection of CD4+ T cells by HIV results in the upregulation of CD62L and that CD4+ T cells exposed to HIV preferentially migrate to and are sequestered in the SLT (7). Within lymph tissue high numbers of T cells accumulate for sensitization and activation, and therefore the process of recruitment is tightly regulated. Alteration of the process through either increased homing or decreased egress would potentially result in lymphadenopathy. Recent reports indicate that egress of naïve and activated T cells from the LN requires a concentration gradient of sphingosine-1-phosphate (S1P) that facilitates cell egress from the lymph tissue (15). Loss of the S1P receptor (S1P1) expression or function results in marked T-cell accumulation and developing lymphadenopathy (20). This is the mechanism for the immunosuppressive drug FTY720, an S1P1 antagonist that induces T-cell sequestration by modulating S1P1 from the cell surface (5, 31).
It is estimated that the increase in the number of CD4+ T cells sequestered within the SLT of HIV-1-infected individuals is 1 × 109 cells, or approximately 99% of the total T-cell population (13); however, it is also estimated that in an HIV-1-infected individual, 1 in every 400 CD4+ T cells is actively infected (13). As the magnitude of T cells sequestered in LNs of infected individuals exceeds the number of actively infected cells, we hypothesized that shed viral proteins and not productive infection of CD4+ T cells might be causative in altering CD4+ T-cell homing to LNs. The viral product of particular interest is the envelop glycoprotein, gp120. gp120 is shed from infected cells and has been detected in the serum of infected individuals (9, 16, 26). We along with others have previously reported that gp120 directly alters CD4+ T-cell migration, and other investigators have shown that gp120 stimulation of B lymphocytes results in altered responsiveness to the chemokines CXCL12, CCL20, and CCL21 (2, 17, 36). Therefore, we investigated the effects of gp120 on CD4+ T-cell migratory responses to LN-specific chemokines CCL19 and CCL21, to other chemokines such as CCL20 and CXCL8, and to the egress-dependent factor, S1P.
In this study we show that gp120, dependent on CD4 signaling, increases CD4+ T-cell motility to CCL20 and CCL21 while also rendering cells chemotactically unresponsive to S1P. We demonstrate further that these effects could selectively facilitate accumulation of CD4+ T cells in LNs by injecting gp120-treated human T cells into huSCID mice. gp120 stimulation induced a threefold increase in the numbers of CD4+ T cells in SLT with a reciprocal loss of circulating and splenic T cells compared to the accumulation patterns of injected untreated human T cells. These findings suggest that gp120 alone may represent a major contributing factor to HIV-1-associated lymphadenopathy.
MATERIALS AND METHODS
Cell isolation.
Human primary peripheral blood mononuclear cells were isolated from healthy volunteers as described previously (14, 15). The blood was collected in accordance with the guidelines established by the Boston University School of Medicine Institutional Review Board. Briefly, blood was collected in the presence of 100 units of heparin-sodium (American Pharmaceutical Partners) per ml of blood. Peripheral blood mononuclear cells were isolated by Ficoll-Hypaque centrifugation and suspended in M199 medium (Mediatech) supplemented with 0.4% bovine serum albumin (USB Corp.), 240 units of penicillin and 240 μg of streptomycin (Gibco), and 20 mM HEPES (Gibco); this is referred to as complete M199 medium. T cells were negatively enriched using nylon wool (Polysciences) adherence, resulting in nylon wool nonadherent T cells. Purity, as assessed by fluorescence-activated cell sorting (FACS) analysis, consistently demonstrated 96% CD3+ cells, of which 64% were CD4+ and 30% were CD8+ T cells. The cells were routinely incubated overnight in complete M199 medium before use. Further purification of T cells into CD4+ and CD8+ subsets was accomplished using a magnetic bead negative isolation technique (Dynal/Invitrogen). Resulting purity was assessed using FACS analysis and was routinely demonstrated at >98% for both CD4+ and CD8+ subsets. For in vivo analysis, 10 × 106 CD4+ T cells were stained with 5 μM CFDA SE (carboxyfluorescein diacetate-succinimidyl ester) (Invitrogen) for 15 min and washed once with phosphate-buffered saline (PBS) prior to tail vein injection in 100 μl of PBS.
Reagents.
Wild-type HIV-1IIIB gp120 was purchased from Advanced Biotechnologies, Inc., aliquoted, and stored at −80°C. HIV-1BaL gp120 was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH (catalog no. 4961), aliquoted, and stored at −80°C. Soluble CD4 (sCD4-183) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH, as generated by Pharmacia, Inc. Recombinant Tat protein was generously provided by R. Gummuluru, Boston University School of Medicine. CD4 (clone RPA-T4), P-selectin glycoprotein ligand 1 ([PSGL-1] clone KPL1), CD69 (clone FN50), CCR6 (clone 11A9), CXCR3 (clone 6C6), and CCR7 (clone 3D12) antibodies were purchased from BD Pharmingen. For labeling, 5 × 105 cells were stained with 20 μl of antibody in each test. CD3 (clone OKT3), CD4 (clone OKT4), CD8 (clone OKT8), CD62L (clone DREG 56), CXCR1 (clone eBio8F1-1-4), and mouse CD16/CD32 cocktail (clone 93) antibodies were purchased from eBiosciences. For some experiments 5.0 × 105 cells were stained with 20 μl of S1P1 rabbit anti-human polyclonal antibody (0.5 μg for 1 h) and fluorescein isothiocyanate-conjugated goat anti-rabbit antibody (1 μg for 30 min), both purchased from Caymen Chemical Company. All staining was conducted at 4°C. S1P was resuspended in 0.3 M NaOH, placed in aliquots, and stored at −80°C (Cayman Chemical). All chemokines were purchased from R&D Systems.
Cell migration.
Cell migration was measured using a 48-well Boyden Chamber (Neuro Probe) as previously described (14, 15). Briefly, human T lymphocytes were isolated as previously described using Ficoll-Hypaque density centrifugation and nylon wool adherence (14, 15) and then incubated overnight to allow for recovery at 25°C and 5% CO2 in 5% bovine serum albumin. For the migration assay, various concentrations of S1P or chemokines were added to the bottom chamber at the designated concentrations. Fifty-five microliters of a suspension of 1 × 107 cells/ml was added to the top chamber and allowed to migrate through 8-μm-pore-size cellulose nitrate filters for 1 h. Filters were removed, fixed with ethanol, and stained with hematoxylin (Sigma). Migration was calculated as the number of cells that had migrated beyond 50 μm into the filter. The cell counts from experimental conditions were compared with control cell migration with medium alone, which is expressed as 100%. In the typical migration assay for these studies 10 to 15 cells per high-powered field are counted under control conditions. Ten high-powered fields are counted for each control and experimental condition, and the data are then expressed as a percentage of control cell migration. A concentration of 50 ng/ml was used for each chemokine as this dose did not induce maximal migration but allowed for detection of augmentation and inhibition induced by gp120 treatment. For some experiments the cells were first incubated with 1 μM herbimycin A or 10 μM wortmannin for 18 to 24 h prior to extensive washing in medium, followed by pretreatment with gp120 and subsequent S1P-induced migration.
In vivo studies.
Male NOD.CB17-Prkdcscid/J mice, 6 to 8 weeks old, were obtained from Jackson Laboratories and housed under sterile conditions at Boston University School of Medicine under animal biosafety level 2 containment as approved by the Boston University School of Medicine IACUC. CFDA SE (Invitrogen)-stained T cells either untreated or treated with gp120 at a final concentration of 17 ng/ml were suspended at a final concentration of 3 × 107 cells/300 μl in PBS and injected into the tail vein. Mice were sacrificed at 4 h and spleen, LN (inguinal, axillary, and cervical), and blood were harvested. Cells were isolated from blood using Ficoll-Hypaque density centrifugation. Spleen and LNs were gently dissociated by mechanical dispersion and passed through a 40-μm-pore-size filter (BD Falcon) to obtain single-cell suspensions. Cells were analyzed for fluorescence using a BD FACScan cytometer or stained with phycoerythrin-conjugated anti-CD4 or anti-CD8 antibodies. Prior to staining, all cells were treated with a murine CD16/CD32 antibody cocktail for 15 min to block murine Fc receptors.
RESULTS
gp120 treatment does not alter the cell surface phenotype but results in reprogramming of CD4+ T-cell migration to certain chemokines.
Recruitment of CD4+ T cells to the SLT requires the appropriate expression of certain chemokine receptors (CCR7) and adhesion molecules, such as CD62L, PSGL-1 (32), and CD44. To initially investigate effects of gp120 on LN-specific integrins, normal human T cells were incubated with gp120 (R5 isolate HIV-1IIIB; 17 ng/ml) for 1 h and then expression levels of CCR3, CCR6, CXCR1, CCR7, CD62L, PSGL-1, CD44, CD90, CD69, and HLA-DR were determined by FACS analysis (Fig. 1). The concentration of 17 ng/ml of gp120 was chosen because it represents the upper limit of gp120 previously detected in the plasma of infected individuals (16). Neither untreated nor gp120-treated cells were positive for the early activation markers CD69 and HLA-DR (Fig. 1). The cells were initially positive for CD4, CD44, CD90, CD62L, PSGL-1, CCR3, CCR6, CCR7, and CXCR1, and gp120 treatment had no effect on expression levels for any of these molecules (Fig. 1). Expression levels were similarly unaffected using concentrations of gp120 up to 170 ng/ml (data not shown). This suggests that based on adhesion molecule expression, both untreated and gp120-treated cells under these experimental conditions should retain the ability to bind to the high endothelial venules and enter the SLT. Interestingly, gp120 stimulation, which has previously been shown to modulate CD4 at concentrations of 500 ng/ml, had no effect on CD4 expression at 17 ng/ml (32, 33) (Fig. 1).
FIG. 1.

Effect of gp120 treatment on surface expression of activation molecules, chemokine receptors, and adhesion molecules associated with T-cell trafficking through LNs. Primary human T cells were stimulated with gp120 (17 ng/ml; HIV-1IIIB) (dashed lines) or medium control (dotted lines) for 1 h and then labeled with specific fluorescent antibody or isotype control (solid curves). For CD4 staining, isotype control and CD4-negative cells show overlapping curves. These are representative curves from four separate experiments with similar results.
It is now well established that signaling through CD4 selectively desensitizes the chemokine receptors CCR5, CXCR4, and CXCR3 following stimulation by the CD4 ligand interleukin-16 (IL-16) (30, 34). Following from those studies, we investigated whether gp120 stimulation could alter subsequent stimulation by SLT-specific chemokines CCL19 and CCL21. The effects by IL-16/CD4 are selective for certain chemokine receptors, and therefore in addition to CCL19 and CCL21, chemokines CCL20, CXCL8, and CCL11 were also investigated. CCL20 was studied because it has been reported that nonhuman primates infected with simian immunodeficiency virus demonstrated high levels of CCL20 production from lymphatic endothelial cells (28). CXCL8 was included because we have observed CD4-mediated cross-receptor desensitization of CXCR1, while CCL11/CCR3 signaling was unaffected (3). In the current study, as expected, all chemokines induced a significant migratory response of >170% of control. A concentration of 50 ng/ml was used for each chemokine as this represents an approximate 50% effective dose for induction of migration, and therefore changes through either augmentation or inhibition were likely to be detected. When the cells were treated with gp120 (17 ng/ml; HIV-1IIIB) for 1 h prior to conducting the chemotaxis assay, a significant (200%; P = 0.042) augmentation in migration to CCL20 and CCL21 was observed (Fig. 2). The same gp120 pretreatment resulted in a significant inhibition of CXCL8-induced migration, similar to an IL-16 effect (W. Cruikshank, unpublished observations), while there was no change in migration to CCL19 or to the peripheral chemokine CCL11 (Fig. 2). The differential effects on the chemokine receptors suggest that there is selective gp120-mediated chemotactic reprogramming that enhances some receptor signaling while desensitizing others. Augmentation of CCL20- and CCL21-induced migration supports the concept that exposure to gp120 may facilitate enhanced recruitment of T cells to the SLT in an HIV-1-infected individual.
FIG. 2.

Effect of gp120 on human T-cell migration to selected chemokines. Primary human T cells were treated with medium alone (filled bars) or gp120 (17 ng/ml; HIV-1IIIB) (open bars) for 1 h prior to extensive washing; the migration assay was conducted with 50 ng/ml for each chemoattractant. This experiment was conducted four separate times, and the data represent the average values (± standard deviations). *, significantly different migration from control-treated cells (P < 0.02).
gp120 stimulation inhibits S1P-induced migration.
While recruitment is an essential component of homeostatic and pathological lymphocyte accumulation in LNs, egress likely constitutes an equally important determinant. Thus, we studied the effects of gp120 on S1P-induced migration. Human T cells were again pretreated with gp120 (R5 isolate) for 1 h prior to S1P-induced migration. As shown in Fig. 3A, gp120-stimulated T cells were completely unresponsive to S1P across a wide range of concentrations. A dose-response curve for gp120 demonstrated maximal inhibition between 170 and 1.7 ng/ml, with inhibition detectable at concentrations as low as 0.017 ng/ml (approximately 0.1 pM) (Fig. 3B). No inhibitory effect was detected at concentrations of 0.0017 ng/ml or lower. A time course indicated that a 30-min exposure to gp120 was required to detect inhibition and that 60 min was sufficient for maximal inhibition (Fig. 3C).
FIG. 3.

Characterization of the inhibitory effect of gp120. (A) Human T cells were incubated with gp120 (17 ng/ml; HIV-1IIIB) (open bars) or medium control (solid bars) for 60 min prior to induced migration by the designated concentrations of S1P. (B) Dose response for the inhibitory effect of gp120 on S1P-induced migration. Human T cells were incubated with the designated concentrations of gp120 (HIV-1IIIB) for 60 min prior to induced migration by 10−8 M S1P. (C) A time course for the inhibitory effect using cells incubated with gp120 (17 ng/ml) for 30 or 60 min prior to washing and chemotaxis induced by 10−8 M S1P. For all the migration experiments, the data are expressed as a percentage of control cell migration (± standard deviations). *, significantly different migration from non-gp120-treated cells (P < 0.05). (D) gp120-induced modulation of CD4 and S1P1 in human T cells. T cells were stimulated with gp120 in a dose range from 100 ng/ml to 1.7 μg/ml for 60 min prior to assessing the surface expression of CD4 and S1P1. Loss of CD4 expression was detected at concentrations of gp120 of 500 ng/ml and higher. The representative FACS plot shows expression levels following 1.7 μg/ml gp120 stimulation. Control cells are depicted by the dashed lines while gp120-treated cells are represented by the dotted lines. This is a representative FACS plot from four separate experiments, all with similar findings.
Previous studies have identified that gp120 stimulation results in significant loss of CD4 surface expression with subsequent effects on the expression of associated receptors, such as TCR, CCR5, and CXCR4 (6). The time frame required for S1P inhibition of 30 to 60 min is consistent with the concept of induced S1P1 modulation. To initially investigate a mechanism for gp120-induced inhibition of S1P stimulation, effects on S1P1 expression were determined. FACS analysis indicated detectable S1P1 expression in 52.3% ± 7.72% of resting human peripheral T cells, and neither the percent positive cells nor the intensity of staining was altered following incubation with 1.7 ng/ml gp120 for 1 h (data not shown). As the concentration of gp120 present in LNs of HIV-1-infected individuals has not been well established and could conceivably be several logs higher, the concentration used in these experiments (1.7 ng/ml) may not be sufficient to reflect those in the LNs. Therefore, concentrations up to 1.7 μg/ml were assessed. Cells were treated for 1 h prior to assessment of CD4 and S1P1 surface expression by FACS analysis. Despite a significant decrease in CD4 staining, there was no change in S1P1 expression (Fig. 3D), suggesting that inhibition of S1P-induced migration is mediated through disruption of cell signaling rather than through receptor modulation.
Signaling and selectivity of gp120-mediated suppression of S1P-induced migration.
gp120 binds to cells through the formation of a complex comprised of CD4 and a chemokine coreceptor: CCR5 for macrophage-tropic (M-tropic) isolates and CXCR4 for T-cell-tropic (T-tropic) or dual-tropic isolates (19). Signaling induced by gp120 can therefore be mediated by both CD4 and CCR5 (17, 22, 36). CD4 has been shown to induce signals through both the associated kinase, p56lck, as well as through the downstream phosphatidylinositol-3 kinase with different cellular responses (21), while CCR5 has been shown to signal through activation of the GTP-binding protein Giα (18, 32). To identify which gp120-induced pathway was involved in the observed cross-receptor desensitization, T cells were incubated in the presence of herbimycin A, a selective inhibitor of p56lck enzymatic activity, prior to gp120 stimulation and then S1P-induced migration. As shown in Fig. 4A, treatment with herbimycin A completely blocked gp120-induced inhibition. The complete inhibition by herbimycin A suggests that the process is mediated through p56lck without involvement of GTPase signaling; however, further studies are required to confirm this concept.
FIG. 4.

Specificity of the gp120 effect on S1P-induced migration. (A) Effect of herbimycin A on gp120-induced inhibition of S1P-stimulated migration. Human T cells were incubated with herbimycin A (1 μM final concentration) for 18 h prior to washing and preincubation with gp120 (17 ng/ml; HIV-1IIIB) for 60 min; cells were then stimulated for migration by 10−8 M S1P. (B) Specificity of the gp120-induced inhibitory effect. Primary human T cells were incubated with medium, gp120 (17 ng/ml; HIV-1IIIB), gp120 plus 1 μg of sCD4, or Tat protein (17 ng/ml) for 1 h prior to washing and then subjected to S1P (10−8 M)-induced migration. (C) Comparison of M-tropic versus T-tropic isolates of gp120 to inhibit S1P-induced migration. Wild-type HIV-1IIIB (T tropic) and HIV-1BaL (M tropic) isolates (17 ng/ml) were used to incubate human T cells prior to induced migration by 10−8 M S1P. All migration assays were conducted at least four separate times, and the data are the averages (± standard deviations) expressed as percent migration compared with medium control-induced migration, designated as 100%. The asterisk denotes significantly different migration from untreated cells (P < 0.03).
Specificity of the gp120-mediated effect on S1P-induced migration was then addressed using two different approaches. The first was to determine whether the inhibition could be directly attributable to gp120. For these studies sCD4 (1 μg/ml) was mixed with the gp120 for 10 min before it was added to the T cells for 60 min. As expected, gp120 alone inhibited S1P-induced migration; however, this effect was completely blocked in the presence of sCD4 (Fig. 4B). sCD4 alone had no effect on S1P-induced migration (data not shown). These data confirm that the inhibitory effects are directly induced by gp120 and confirm further that a gp120-CD4 interaction is required. The second approach was to determine whether other HIV viral proteins could induce the same effect. Other viral proteins are shed from HIV-1-infected cells, and to determine whether the effect of gp120 was selective, cells were treated with HIV-1 Tat protein, shown to have direct cellular effects on T lymphocytes (1). T cells were preincubated with Tat protein in doses ranging from 10 ng/ml to 1 μg/ml for 1 h prior to S1P-induced migration. Exposure to Tat protein, however, did not demonstrate any change in responsiveness to S1P (Fig. 4B shows the results with 17 ng/ml) indicating a selective effect for gp120.
Selectivity of the gp120-mediated inhibition of S1P-induced migration was also assessed based on tropism of the protein. Studies thus far used the T-tropic isolate HIV-1IIIB. We next compared the effect of HIV-1IIIB with the M-tropic isolate HIV-1BaL. As shown in Fig. 4C, the magnitudes of inhibition for both T- and M-tropic isolates were comparable.
CD8+ T cells are unresponsive to S1P-induced migration.
A consistent observation for gp120 from both M- and T-tropic isolates was that pretreatment resulted in inhibition of all S1P-induced migration. The complete loss of S1P responsiveness was unexpected as the target cells were comprised of both CD4 (approximately 60%) and CD8 (approximately 40%) T cells, and direct effects of gp120 on unactivated CD8 T cells had not been reported. These data suggested either that gp120 was affecting both CD4+ and CD8+ T cells or that CD8+ T cells were not responsive to S1P-induced migration. While there are many studies demonstrating the migratory responsiveness of murine CD4+ and CD8+ T cells to S1P (9), this type of assessment has not been reported using human T cells. To address this, we subjected isolated CD4+ and CD8+ T cells to S1P-induced migration. Isolated populations were purified to >98% as determined by flow cytometry (data not shown). As shown in Fig. 5, CD4+ T cells demonstrated similar migratory responses to those of mixed cultures, with maximal migration seen between 10−10 M and 10−9 M; however, CD8+ T cells did not migrate to S1P at any of the concentrations used. The CD8+ T cells did respond to MIP-3α-induced migration (data not shown), suggesting an inherent difference between CD4+ and CD8+ T cells in their responsiveness to S1P-induced migration.
FIG. 5.
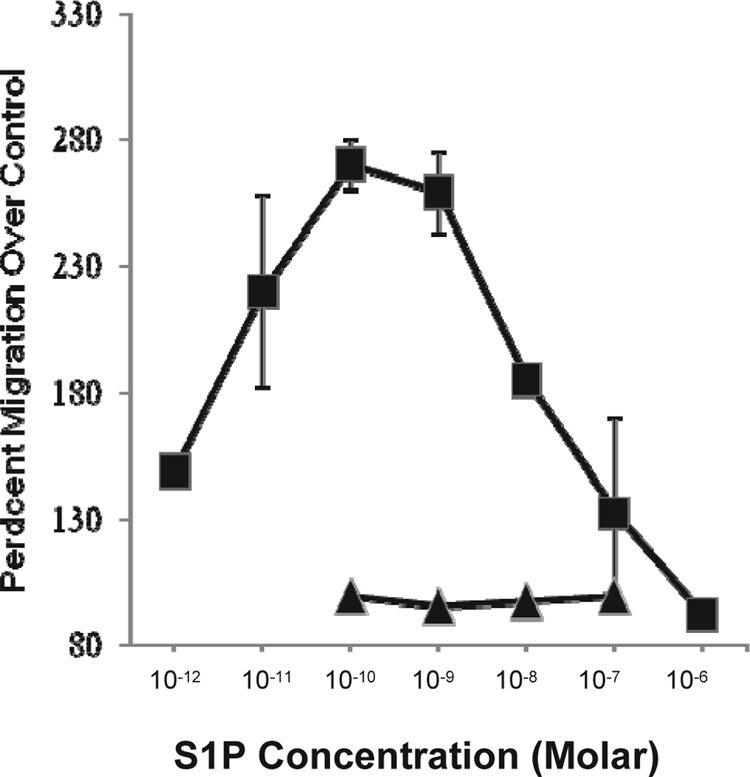
Responsiveness of CD4+ and CD8+ T cells to S1P-induced migration. Primary human T cells were isolated and fractionated into CD4+ and CD8+ T cells by negative selection, with a resultant purity of greater than 98%. The cells were then assessed for migration to various concentrations of S1P. Migration of CD4 T cells is shown by the filled squares, and migration of CD8+ T cells is depicted by the filled triangles. These data represent the average values (± standard deviations) from three separate experiments.
Treatment of human T cells with gp120 results in lymphadenopathy in a mouse model.
To address the physiologic relevance of our in vitro observations, we assessed changes in CD4+ T-cell homing to SLT in an huSCID mouse model. Fluorescently labeled human peripheral T cells were incubated without or with 17 ng/ml gp120 pretreatment for 1 h prior to washing and adoptive transfer by tail vein injection into NOD.CB17-Prkdcscid/J mice. After 4 h T cells were harvested from the spleen, peripheral blood, and SLT (inguinal, cervical, mesenteric, and axial LNs), and the number of fluorescent cells was quantified by flow cytometry. We assessed accumulation at 4 h as previous studies have shown that at that time point actively infected human T cells injected into NOD.CB17-Prkdcscid/J mice resulted in SLT sequestration (35). After 4 h, harvested tissues did not demonstrate any apparent size or weight difference between control and gp120-treated cells. Comparable tissue mass or blood volume (100 μl) was used from all mice for fluorescent cell assessment. In our studies there was an average 2.3-fold ± 1.3-fold increase in total cell numbers of gp120-treated T cells in the SLT compared to untreated control T cells (Fig. 6). In contrast, there were concomitant changes in the number of gp120-treated human T cells: (0.3 ± 0.4)-fold and (0.6 ±0.5)-fold decreases for spleen and peripheral blood, respectively. These studies confirm the concept of gp120-mediated lymphadenopathy and indicate that the response is fairly rapid as a 1-h exposure is sufficient to accomplish this effect.
FIG. 6.

Effect of gp120 on accumulation of labeled T cells in the LNs, blood, and spleen. NOD.CB17-Prkdcscid/J mice (2 mice/group/experiment) were tail vein injected with either medium-treated (filled curves) or gp120-treated (open curves) human T cells labeled with the fluorescent cell marker CFDA SE. LN, spleen, and peripheral blood T cells were harvested at 4 h and analyzed for fluorescence by flow cytometry. These are representative graphs of five separate experiments where gp120 induced an average change of a 2.3-fold ± 1.3-fold increase for LNs, 0.6-fold ± 0.5-fold decrease for the spleen, and 0.4-fold ± 0.3-fold decrease for blood.
DISCUSSION
Despite the initial designation of HIV-1 as lymphadenopathy-associated virus, identification of a causative mechanism for the associated lymphadenopathy has yet to be elucidated. Previous studies using huSCID mice have determined that HIV-1-infected cells home to the LNs (35); however, in HIV-1-infected individuals the number of LN-sequestered T cells far exceeds the number of actively infected cells, suggesting that soluble factors and not direct viral infection are likely responsible for inducing the lymphadenopathy. As the HIV-1 viral envelop glycoprotein, gp120, has previously been shown to affect T-cell migration (17, 36), it became a likely candidate for this effect. With the recent understanding of T-cell trafficking through SLT, we therefore investigated the potential effects of gp120 on T-cell migratory responses to factors involved in the trafficking process. Recent evidence suggests that T-cell retention in the SLT is dependent upon a balance between SLT intrinsic recruitment signals and SLT egress signals (29). Perturbation of either would induce a change in T-cell retention within the SLT, resulting in subsequent lymphadenopathy and potentially induced immunosuppression (31).
Our studies demonstrate that gp120 alone, in association with CD4 signaling, alters T-cell trafficking through the SLT by virtue of two potentially independent effects. The first is through augmentation of CCR6 and CCR7 responsiveness. CD4 regulation of chemokine receptor signaling has been demonstrated previously and has been shown to be a selective process whereby CCR5 and CXCR4 are desensitized while other receptors such as CCR3 are unaffected (30, 34). A similar selective effect was detected following gp120 stimulation. gp120 also induced receptor cross-desensitization of CXCR1/CXCL8 and had no effect on CCR3/CCL11 signaling; however, gp120/CD4 signaling induced an enhanced migratory response to both CCR6/CCL20 and CCR7/CCL21. This enhanced effect on CCL20 and CCL21 is not detected when the cells are first stimulated by IL-16 (D. Green, unpublished observations), suggesting that gp120 ligation of CD4 results in a different signaling process than that induced by the natural ligand, IL-16. The receptor for CCL20, CCR6, has been shown to be expressed on T regulatory cells, suggesting that gp120 may preferentially augment LN recruitment of these cells. While we have not as yet investigated this, a recent study has reported that HIV-1 interaction with CD4 on T regulatory cells enhances their homing and accumulation to lymph tissue (14). It was also noteworthy that gp120 stimulation had different effects on CCL19 and CCL21 stimulation despite their use of the same receptor, CCR7. As both proteins interact differently with CCR7 due to differences in their tertiary structure, it is conceivable that different signaling pathways resulting in a migratory response are induced. Taken together, these findings suggest that the systemic presence of gp120 has the potential to reprogram CD4+ T-cell responses that affect not only LN accumulation but also cell trafficking at sites of inflammation. Additional studies, however, are required to determine the scope of this effect.
The second alteration of T-cell responsiveness induced by gp120 stimulation is through inhibition of S1P1. S1P stimulation has been found to be a major component for induction of T-cell egress from lymph tissue. This effect is thought to occur through its chemoattractant properties on T cells as well as through the induction of increased extravasation through the endothelial cell barrier within the lymph tissue (12). The in vitro studies suggest that gp120 stimulation results in loss of S1P responsiveness in close to 100% of S1P-responsive T cells. When fractionated into CD4+ and CD8+ subsets, our data indicate that CD8+ T cells appear to be unresponsive to S1P. This is in contrast to reports from previous studies using murine T cells that, while less responsive than CD4+ T cells, CD8+ T cells are capable of responding (10). Since our studies indicate that human CD8+ T cells are not responsive in the in vitro migration assay, our interpretation is that regulation of their trafficking through lymph tissue is likely mediated by other factors. The ability of gp120 to alter chemokine and S1P-induced migration may also alter peripheral CD4+ T-cell distribution within tissues. Circulating levels of gp120, similar to FTY720, could conceivably affect normal CD4+ T-cell recruitment by chemokines at sites of inflammation as well as affect the ability of S1P to facilitate cellular egress from tissues (18).
The finding that Tat protein did not induce any changes to induced migration suggests that the effect of gp120 is selective. Our data also suggest that signaling through CD4 alone is required and sufficient to induce this effect. Our findings that both R5 and X4 isolates were equally effective at blocking S1P-induced migration, experiments demonstrating that inhibition of p56lck by herbimycin A completely restored S1P responsiveness, and the observation that another CD4 ligand, IL-16, which has not been shown to bind to either CCR5 or CXCR4 inhibits S1P, support this concept. Additional studies, however, addressing the signaling pathways involved are required to firmly establish this direct relationship.
In our studies the effects of gp120 on S1P-induced migration are detectable at concentrations as low as 0.01.7 ng/ml. This is several logs lower than concentrations used in previously reported in vitro assays (16). In vivo assessment of gp120 levels has been difficult to accurately determine, with reported values of detection ranging from 2 pM to 0.8 nM (9, 26). A more accurate determination may be based on the ratio of virion-released p24 and gp120 as reported by Klasse et al. (16). Based on their calculations, circulating gp120 concentrations would be in the range of 0.07 pM. As concentrations of gp120 are predicted to be greater in LNs than in the plasma (17), a biological effect at 0.1 pM (17 ng/ml) is within a predicted LN concentration and therefore is potentially physiologically relevant.
LN accumulation was investigated and found to be significantly increased by 4 h postinjection, indicating a rapid response mechanism. It must be noted, however, that the cells were pretreated prior to injection, and therefore this time frame may not accurately reflect the kinetics of in vivo sequestration. Our data do indicate that even a short-term exposure (1 h for our studies) is sufficient to induce a significant change in T-cell homing. Our studies did not investigate the duration of the gp120-induced effect; however, based on detectable systemic levels of gp120 in HIV-1-infected individuals, one would anticipate that the T cells would be constantly exposed, and therefore even a short-term effect would be perpetuated by chronic stimulation. In addition, the magnitude of the effect of gp120 on T-cell accumulation in the SLT may not be accurately reflected in this animal model. SCID mice have LNs that are significantly reduced in size. This limitation likely reduces the capacity of the lymph tissue to sequester T cells, and therefore our observations of a 2.3-fold increase may be underrepresentative of the potential magnitude of the gp120-induced effect. Despite the limitations, the animal model clearly identifies an in vivo alteration of T-cell homing following gp120 exposure that is significant, rapid, and induced by physiologically feasible concentrations of gp120.
Overall, these findings are consistent with the concept that HIV-1 pathogenesis extends beyond the limited number of actively infected cells and for the first time identify a potential mechanism for HIV-1-associated lymphadenopathy. Our study implicates gp120 as a sole mediator of CD4+ T-cell sequestration and raises the possibility that neutralization of gp120 bioactivity could result in prolonging normal T-cell trafficking, thus potentially maintaining immunocompetence.
Acknowledgments
We thank L. Cross, K. Lee, and M. Crowley for assistance with the animal experiments, the Pulmonary Immunology Group for insightful discussions of the data, and Rahm Gummuluru for discussions and reagents. We also thank Joshua Farber for his insightful thoughts and comments.
This work was supported in part by grant AI 35680 from the NIAID/NIH.
Footnotes
Published ahead of print on 18 March 2009.
REFERENCES
- 1.Albini, A., S. Ferrini, R. Benelli, S. Sforzini, D. Giunciuglio, M. G. Aluigi, A. E. Proudfoot, S. Alouani, T. N. Wells, G. Mariani, R. L. Rabin, J. M. Farber, and D. M. Noonan. 1998. HIV-1 Tat protein mimicry of chemokines. Proc. Natl. Acad. Sci. USA 9513153-13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badr, G., G. Borhis, D. Treton, C. Moog, O. Garraud, and Y. Richard. 2005. HIV type 1 glycoprotein 120 inhibits human B cell chemotaxis to CXC chemokine ligand (CXCL) 12, CC chemokine ligand (CCL) 20, and CCL21. J. Immunol. 175302-310. [DOI] [PubMed] [Google Scholar]
- 3.Bandeira-Melo, C., K. Sugiyama, L. J. Woods, M. Phoofolo, D. M. Center, W. W. Cruikshank, and P. F. Weller. 2002. IL-16 promotes leukotriene C4 and IL-4 release from human eosinophils via CD4- and autocrine CCR3-chemokine-mediated signaling. J. Immunol. 1684756-4763. [DOI] [PubMed] [Google Scholar]
- 4.Brenchley, J. M., T. W. Schacker, L. E. Ruff, D. A. Price, J. H. Taylor, G. J. Beilman, P. L. Nguyen, A. Khoruts, M. Larson, A. T. Haase, and D. C. Douek. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200749-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinkmann, V., J. G. Cyster, and T. Hla. 2004. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 41019-1025. [DOI] [PubMed] [Google Scholar]
- 6.Cefai, D., M. Ferrer, N. Serpente, T. Idziorek, A. Dautry-Varsat, P. Debre, and G. Bismuth. 1992. Internalization of HIV glycoprotein gp120 is associated with down-modulation of membrane CD4 and p56lck together with impairment of T cell activation. J. Immunol. 149285-294. [PubMed] [Google Scholar]
- 7.Cloyd, M. W., J. J. Chen, P. Adeqboyega, and L. Wang. 2001. How does HIV cause depletion of CD4 lymphocytes? A mechanism involving virus signaling through its cellular receptors. Curr. Mol. Med. 1545-550. [DOI] [PubMed] [Google Scholar]
- 8.Cyster, J. G. 2005. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 23127-159. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert, M., J. Kirihara, and J. Mills. 1991. Enzyme-linked immunoassay for human immunodeficiency virus type 1 envelope glycoprotein 120. J. Clin. Microbiol. 29142-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graeler, M., G. Shankar, and E. J. Goetzl. 2002. Cutting edge: suppression of T cell chemotaxis by sphingosine 1-phosphate. J. Immunol. 1694084-4087. [DOI] [PubMed] [Google Scholar]
- 11.Graziosi, C., G. Pantaleo, J. F. Demarest, O. J. Cohen, M. Vaccarezza, L. Butini, M. Montroni, and A. S. Fauci. 1993. HIV-1 infection in the lymphoid organs. AIDS 7(Suppl. 2)S53-S58. [DOI] [PubMed] [Google Scholar]
- 12.Grigorova, I. L., S. R. Schwab, T. G. Phan, T. H. Pham, T. Okada, and J. G. Cyster. 2009. Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat. Immunol. 1058-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haase, A. T. 1999. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17625-656. [DOI] [PubMed] [Google Scholar]
- 14.Ji, J., and M. W. Cloyd. 2009. HIV-1 binding to CD4 on CD4+ CD25+ regulatory T cells enhances their suppressive function and induces them to home to, and accumulate in, peripheral and mucosal lymphoid tissues: an additional mechanism of immunosuppression. Int. Immunol. 21283-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabashima, K., N. M. Haynes, Y. Xu, S. L. Nutt, M. L. Allende, R. L. Proia, and J. G. Cyster. 2006. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J. Exp. Med. 2032683-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klasse, P. J., and J. P. Moore. 2004. Is there enough gp120 in the body fluids of HIV-1-infected individuals to have biologically significant effects? Virology 3231-8. [DOI] [PubMed] [Google Scholar]
- 17.Kornfeld, H., W. W. Cruikshank, S. W. Pyle, J. S. Berman, and D. M. Center. 1988. Lymphocyte activation by HIV-1 envelope glycoprotein. Nature 335445-448. [DOI] [PubMed] [Google Scholar]
- 18.Ledgerwood, L. G., G. Lal, N. Zhang, A. Garin, S. J. Esses, F. Ginhoux, M. Merad, H. Peche, S. A. Lira, Y. Ding, Y. Yang, X. He, E. H. Schuchman, M. L. Allende, J. C. Ochando, and J. S. Bromberg. 2008. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat. Immunol. 942-53. [DOI] [PubMed] [Google Scholar]
- 19.Lusso, P. 2006. HIV and the chemokine system: 10 years later. EMBO J. 25447-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matloubian, M., C. G. Lo, G. Cinamon, M. J. Lesneski, Y. Xu, V. Brinkmann, M. L. Allende, R. L. Proia, and J. G. Cyster. 2004. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427355-360. [DOI] [PubMed] [Google Scholar]
- 21.Mazerolles, F., C. Barbat, C. Hivroz, and A. Fischer. 1996. Phosphatidylinositol 3-kinase participates in p56(lck)/CD4-dependent down-regulation of LFA-1-mediated T cell adhesion. J. Immunol. 1574844-4854. [PubMed] [Google Scholar]
- 22.Melar, M., D. E. Ott, and T. J. Hope. 2007. Physiological levels of virion-associated human immunodeficiency virus type 1 envelope induce coreceptor-dependent calcium flux. J. Virol. 811773-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montagnier, L., J. C. Chermann, F. Barre-Sinoussi, D. Klatzmann, S. Wain-Hobson, M. Alizon, F. Clavel, F. Brun-Vezinet, E. Vilmer, C. Rouzioux, et al. 1984. Lymphadenopathy associated virus and its etiological role in AIDS. Princess Takamatsu Symp. 15319-331. [PubMed] [Google Scholar]
- 24.Nilsson, J., A. Boasso, P. A. Velilla, R. Zhang, M. Vaccari, G. Franchini, G. M. Shearer, J. Andersson, and C. Chougnet. 2006. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 1083808-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu, M. T., D. S. Stein, and S. M. Schnittman. 1993. Primary human immunodeficiency virus type 1 infection: review of pathogenesis and early treatment intervention in humans and animal retrovirus infections. J. Infect. Dis. 1681490-1501. [DOI] [PubMed] [Google Scholar]
- 26.Oh, S. K., W. W. Cruikshank, J. Raina, G. C. Blanchard, W. H. Adler, J. Walker, and H. Kornfeld. 1992. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J. Acquir. Immune Defic. Syndr. 5251-256. [PubMed] [Google Scholar]
- 27.Pantaleo, G., C. Graziosi, J. F. Demarest, L. Butini, M. Montroni, C. H. Fox, J. M. Orenstein, D. P. Kotler, and A. S. Fauci. 1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362355-358. [DOI] [PubMed] [Google Scholar]
- 28.Pegu, A., J. L. Flynn, and T. A. Reinhart. 2007. Afferent and efferent interfaces of lymph nodes are distinguished by expression of lymphatic endothelial markers and chemokines. Lymphat. Res. Biol. 591-103. [DOI] [PubMed] [Google Scholar]
- 29.Pham, T. H., T. Okada, M. Matloubian, C. G. Lo, and J. G. Cyster. 2008. S1P1 receptor signaling overrides retention mediated by Gαi-coupled receptors to promote T-cell egress. Immunity 28122-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahangdale, S., R. Morgan, C. Heijens, T. C. Ryan, H. Yamasaki, E. Bentley, E. Sullivan, D. M. Center, and W. W. Cruikshank. 2006. Chemokine receptor CXCR3 desensitization by IL-16/CD4 signaling is dependent on CCR5 and intact membrane cholesterol. J. Immunol. 1762337-2345. [DOI] [PubMed] [Google Scholar]
- 31.Rosen, H., and E. J. Goetzl. 2005. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat. Rev. Immunol. 5560-570. [DOI] [PubMed] [Google Scholar]
- 32.Su, S. B., H. Ueda, O. M. Howard, M. C. Grimm, W. Gong, F. W. Ruscetti, J. J. Oppenheim, and J. M. Wang. 1999. Inhibition of the expression and function of chemokine receptors on human CD4+ leukocytes by HIV-1 envelope protein gp120. Chem. Immunol. 72141-160. [DOI] [PubMed] [Google Scholar]
- 33.Theodore, A. C., H. Kornfeld, R. P. Wallace, and W. W. Cruikshank. 1994. CD4 modulation of noninfected human T lymphocytes by HIV-1 envelope glycoprotein gp120: contribution to the immunosuppression seen in HIV-1 infection by induction of CD4 and CD3 unresponsiveness. J. Acquir. Immune Defic. Syndr. 7899-907. [PubMed] [Google Scholar]
- 34.Van Drenth, C., A. Jenkins, L. Ledwich, T. C. Ryan, M. V. Mashikian, W. Brazer, D. M. Center, and W. W. Cruikshank. 2000. Desensitization of CXC chemokine receptor 4, mediated by IL-16/CD4, is independent of p56lck enzymatic activity. J. Immunol. 1656356-6363. [DOI] [PubMed] [Google Scholar]
- 35.Wang, L., C. W. Robb, and M. W. Cloyd. 1997. HIV induces homing of resting T lymphocytes to lymph nodes. Virology 228141-152. [DOI] [PubMed] [Google Scholar]
- 36.Weissman, D., R. L. Rabin, J. Arthos, A. Rubbert, M. Dybul, R. Swofford, S. Venkatesan, J. M. Farber, and A. S. Fauci. 1997. Macrophage-tropic HIV and simian immunodeficiency virus envelope proteins induce a signal through the CCR5 chemokine receptor. Nature 389981-985. [DOI] [PubMed] [Google Scholar]