

Abstract
Their extremely error-prone replication makes RNA viruses targets for lethal mutagenesis. In the case of hepatitis C virus (HCV), the standard treatment includes ribavirin, a base analog with an in vitro mutagenic effect, but the in vivo mode of action of ribavirin remains poorly understood. Here, we test the mutagenic effects of ribavirin plus interferon treatment in vivo using a new method to estimate mutation rates based on the analysis of nonsense mutations. We apply this methodology to a large HCV sequence database containing over 15,000 reverse transcription-PCR molecular clone sequences from 74 patients infected with HCV. We obtained an estimate of the spontaneous mutation rate of ca. 10−4 substitutions per site or lower, a value within the typically accepted range for RNA viruses. A roughly threefold increase in mutation rate and a significant shift in mutation spectrum were observed in samples from patients undergoing 6 months of interferon plus ribavirin treatment. This result is consistent with the known in vitro mutagenic effect of ribavirin and suggests that the antiviral effect of ribavirin plus interferon treatment is at least partly exerted through lethal mutagenesis.
RNA viruses show mutation rates that are orders of magnitude higher than those of DNA-based organisms (12, 14, 15). Although the evolutionary causes of error-prone replication remain poorly understood (4, 8), it is generally believed that high mutation rates allow RNA viruses to rapidly escape the strong selective pressure imposed by host defense mechanisms or antiviral treatments (12). On the other hand, elevations in mutation frequencies as modest as by two- or threefold can result in drastic fitness losses and frequent extinctions of large RNA virus populations in cell cultures and animal models (2). This high susceptibility to mutagens is the basis for lethal mutagenesis, an antiviral strategy which consists of overwhelming populations with an excessive mutational load. Lethal mutagenesis has been suggested as a candidate therapeutic strategy against RNA viruses (2, 13), and a formal theory of lethal mutagenesis has been recently proposed (6).
Hepatitis C virus (HCV) is a positive-stranded RNA virus of the family Flaviviridae, with a 9.6-kb genome that encodes a single polyprotein (24). HCV constitutes a global health concern, infecting an estimated 200 million people around the world. The standard treatment has evolved from interferon monotherapy to a combination of interferon and ribavirin, which has substantially increased the fraction of patients who permanently clear the infection (27, 31). However, the therapeutic mechanism of ribavirin remains unclear. Its mode of action may involve direct inhibition of the viral polymerase, inhibition of inosine monophosphate dehydrogenase, immunomodulation, or lethal mutagenesis (17). This latter possibility has attracted much recent research. Ribavirin is a guanosine analogue with an in vitro mutagenic effect and specifically increases the rate of C→U and G→A transitions and, to a lesser extent, U→C and A→G transitions (39). This effect has been confirmed in cell cultures for poliovirus (10, 30), whereas for HCV, similar but not fully conclusive results have been obtained using primer extension assays (26) and a subgenomic replicon system (9, 19, 42). In other flaviviruses, ribavirin-induced mutagenesis has been observed for GB virus B (22) but not for yellow fever virus (23).
In vivo studies have remained contradictory so far. Whereas some analyses have found increased rates of molecular evolution in NS5A and NS5B genes after initiation of the treatment (3), others have failed to detect such differences (7, 32). Similarly, comparative studies of patients receiving ribavirin treatment with patients receiving placebo or interferon monotherapy have shown either a slightly accelerated rate of molecular evolution for the NS5B gene in the former group (41) or no significant differences (25). The conclusions from analyses of mutation frequencies using reverse transcription-PCR (RT-PCR) molecular clones are similarly puzzling. Previous works have detected no changes in mutation frequencies associated with ribavirin monotherapy or interferon plus ribavirin therapy (7), very slight changes depending on the mode of treatment (19), or only exceptional increases (38).
The contradictory findings described above demonstrate the inherent difficulty of estimating mutation rates. Indeed, the use of molecular evolution rates or mutation frequencies is problematic due to the confounding effect of selection. Specifically, negative selection biases against mutant clones because these clones are generally deleterious, and consequently, the sequenced clones might not accurately reflect the mutational load induced by a viral mutagen. To avoid selection bias, one could focus on synonymous substitutions, which are often assumed to be selectively neutral. However, this approach presents two important limitations. First, neutral variation within populations depends not only on the mutation rate but also on the number of replication cycles, and this number is expected to change in response to treatment. Second, the assumption that synonymous sites are selectively neutral in single-stranded RNA viruses is not always satisfied, because RNA structure is also an important target for selection.
Here, we suggest an alternative strategy based on the analysis of likely lethal mutations. In haploid populations at the mutation-selection balance, the frequency, q, of a deleterious mutation equals the ratio between the mutation rate at which it appears, μ, and its selection coefficient, s, that is,
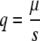 |
(1) |
For lethal mutations, s = 1, and the equilibrium frequency, q = μ, is reached instantaneously because all observed mutations have been generated in the immediately preceding generation (11). Given that the frequency of lethal mutations depends directly on the mutation rate, it is possible to estimate the mutagenic effect of ribavirin-interferon without the sampling bias introduced by selection. We used nonsense mutations as a proxy for lethal mutations. The RNA of HCV is translated as a polyprotein, and thus, nonsense mutations cause truncated translation, which ought to be fatal for the virus. We searched for these kinds of mutations in a database containing over 15,000 sequences of the E1-E2 and NS5A regions obtained by RT-PCR and molecular cloning of a cohort of 74 patients infected with HCV genotype 1 and characterized the mutation rate and spectrum of HCV in vivo. We found that both parameters are altered following ribavirin plus interferon treatment, consistent with the known in vitro mutagenic effect of ribavirin.
MATERIALS AND METHODS
Patients.
Previously published (35-37) data from 74 HCV genotype 1-infected patients were used in this study. Nucleotide sequences were obtained from a prospective study in which serum samples from 74 HCV patients, 26 infected with subtype 1a, 47 with subtype 1b, and 1 with a different, undefined subtype (5), were taken prior to (73 patients), after 6 months of (16 patients), and after 12 months of (9 patients) combined treatment with interferon plus ribavirin. These time points are referred to as T0, T6, and T12, respectively. All T6 and T12 samples came from patients who did not respond to treatment. Responders could not be included because their extremely low or null viral load precluded amplification of the viral RNA.
RNA extraction, RT-PCR, cloning, and sequencing.
Two regions were studied: a 472-nucleotide (nt) fragment encompassing the E1 and E2 genes (nt 1322 to 1793 in the reference HCV genomic sequence; GenBank accession number AF009606) and a 743-nt fragment from the NS5A gene (nt 6742 to 7484). RNA extraction, amplification, cloning, and sequencing procedures are detailed elsewhere (35). Briefly, after viral RNA extraction, reverse transcription was performed with Moloney murine leukemia virus reverse transcriptase (Promega), adding random hexadeoxynucleotides to prevent amplification bias. Pfu DNA polymerase (Promega) was used for nested PCR, with 40 cycles of amplification. This number of cycles was required to obtain a high proportion of successful PCR amplifications. Cloned products for the E1-E2 region or NS5A region were sequenced using vector-based primers KS and SK (Stratagene) to obtain two readings per clone.
The chromatograms from automatically generated sequences that contained nonsense mutations were manually revised. In total, out of 37 amplicons containing nonsense mutations, 33 were confirmed, and 4 were discarded due to poor quality of the chromatograms. All nonsense mutations were found only once in the same amplicon, except one which occurred twice (see the table in the supplemental material). The latter mutation was counted only once since it is very unlikely that different mutations appeared at the same site and in the same amplicon independently.
Sequence alignment and codon usage estimates.
Sequence alignments were performed with ClustalX (http://www.clustal.org). The first complete codon in the alignments started at the second nucleotide position for the E1-E2 region and at the third nucleotide position for the NS5A region. These initial nucleotide positions were removed from the analysis. Next, the DAMBE package (http://dambe.bio.uottawa.ca/dambe.asp) was used to calculate codon usage.
We define nonsense mutation targets (NSMTs) as sites that can generate a stop codon after a single nucleotide substitution. The total number of NSMTs was computed for each patient, viral sample, and genetic region. Since all NSMT-containing codons have only one NSMT, except UGG, which has two (Table 1), the number of NSMTs equals the sum of all codons containing an NSMT plus the number of UGG codons.
TABLE 1.
NSMTsa
| First codon site | Second Codon site
|
|||
|---|---|---|---|---|
| U | C | A | G | |
| U | UAU | UGU | ||
| UAC | UGC | |||
| UUA | UCA | |||
| UUG | UCG | UGG | ||
| C | ||||
| CAA | CGA | |||
| CAG | ||||
| A | ||||
| AAA | AGA | |||
| AAG | ||||
| G | ||||
| GAA | GGA | |||
| GAG | ||||
There are 18 NSMT-containing codons and 19 different NSMTs (underlined; note that codon UGG contains two NSMTs). In 15/19 cases, only one of the three possible substitutions produces a stop codon, whereas in the other four cases, two of the three substitutions produce a stop codon (boldfaced).
Mutation rate estimation.
To obtain the mutation rate from mutation frequencies of NSMTs, we had to take into account that there are three possible nucleotide substitutions at each site and multiply the observed frequency for each nonsense mutation by a correction factor, C. For most NSMTs, only one of the three possible substitutions produces a stop codon, and therefore, C must equal three to obtain the mutation rate of any of the three possible nucleotides and not only of the one that produces the stop codon. However, in some cases, two of the three possible substitutions produce a stop codon (Table 1). Following the same logic as described above, the appropriate correction factor in these cases is C equals 1.5 (3/2 = 1.5). For each observed nonsense mutation, we identified the parental NSMT in order to determine the C value. We obtained, for each amplicon, the per-site mutation rate, μ:
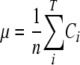 |
(2) |
where Ci is the correction factor (1.5 or 3.0) for nonsense mutation i, T is the total number of nonsense mutations in the amplicon, and n is the total number of NSMTs analyzed that are summed for all the sequences in the alignment.
Mutational pattern analysis.
To calculate the expected substitution frequencies, we first identified, for each possible substitution, the subset of NSMTs for which this substitution produces a stop codon (e.g., for U→G, stop codons can be produced from UUA or UAU), and we computed the total number of times these codons appeared in sequences derived from the same amplicon. Next, we divided this value by the total number of NSMTs in the same alignment, counting NSMTs twice where there are two possible nonsense substitutions. Finally, we averaged the results for the amplicons.
Statistical analyses.
Statistical tests were performed with SPSS version 12. Each RT-PCR amplicon was treated as an independent replicate.
RESULTS
Eighteen of the 61 sense codons of the standard genetic code contain sites that can generate a stop codon after a single nucleotide substitution (denoted here as NSMTs; see Materials and Methods), and there are, in total, 19 NSMTs in the 183 corresponding nucleotide sites (61 codons × 3 nucleotides) (Table 1). Hence, roughly 10% of all sites should a priori be NSMTs, although the actual fraction found in the sequences used for this analysis was only 8.2% due to codon usage bias. In total, 15,798 clonal sequences obtained from 74 patients infected with either HCV genotype 1a or 1b were analyzed. Samples obtained before the start of treatment (T0) were available for 73 patients, and for some patients who did not respond to treatment, additional samples were obtained after 6 or 12 months (T6 and T12 samples). Clones carrying nonsense mutations were observed in 33 of 195 total PCR amplicons. Additional information about the data set can be obtained from the table in the supplemental material.
For untreated patients, there were 22 nonsense mutations in the 560,765 NSMTs analyzed. After correcting for the three possible nucleotide substitutions per site (see Materials and Methods), the estimated spontaneous mutation rate was (1.15 ± 0.29) × 10−4 per site. However, we did not expect all types of substitutions to appear with equal probability for three reasons. First, substitutions to C and from A→G cannot produce stop codons, and hence, only 8 of the 12 possible substitutions have to be considered here. Second, codon usage bias can modify the mutational spectrum. Taking these two factors into account, it is possible to calculate the expected frequency of each substitution type (Table 2). However, transitions are biochemically more likely to occur than transversions, and consistent with this principle, the observed mutational spectrum of HCV was biased toward G→A and C→U transitions (A→G and U→C are not among the eight possible substitutions), with the latter being 1.9 times more likely to occur than expected by sheer chance (χ2 test; P = 0.049). Therefore, given that 4 of the 12 total possible substitutions are transitions, we can predict that the HCV polymerase should spontaneously produce, on average, two-thirds transitions [i.e., 1.9 × (4/12)] and one-third transversions.
TABLE 2.
Mutational patterns for nonsense mutations
| Substitution(s)a | Randomly expected frequencyb | Observed (expected) no. of cases forc:
|
||
|---|---|---|---|---|
| T0 | T6 | T12 | ||
| C→U | 0.124 | 4 (2.72) | 5 (0.99) | 1 (0.37) |
| G→A | 0.145 | 7 (3.18) | 2 (1.16) | 2 (0.43) |
| U→A | 0.115 | 2 (2.54) | 0 (0.92) | 0 (0.35) |
| U→G | 0.044 | 0 (0.96) | 0 (0.35) | 0 (0.13) |
| C→A | 0.172 | 4 (3.77) | 0 (1.37) | 0 (0.51) |
| C→G | 0.084 | 0 (1.84) | 0 (0.67) | 0 (0.25) |
| A→U | 0.112 | 1 (2.47) | 0 (0.90) | 0 (0.34) |
| G→U | 0.205 | 4 (4.51) | 1 (1.64) | 0 (0.61) |
| Total transitions | 0.269 | 11 (5.90) | 7 (2.15) | 3 (0.80) |
| Total transversions | 0.731 | 11 (16.10) | 1 (5.85) | 0 (2.20) |
| Total substitutions | 1 | 22 | 8 | 3 |
Mutation types that cannot produce stop codons (N→C and A→G) are not shown. The first two rows (C→U and G→A) correspond to transitions, whereas all other possible substitutions are transversions.
The expected frequencies assuming completely random mutations were calculated after accounting for codon usage bias.
The observed counts are shown for each time point, and the values in parentheses indicate the expected numbers, as calculated from the randomly expected frequencies.
We next analyzed sequences from T6 and T12 samples available from 24 patients. There were 11 nonsense mutations in samples from T6 and T12 (8 and 3 nonsense mutations, respectively) and 177,090 NSMTs in total, yielding a mutation rate of (1.9 ± 0.6) × 10−4 per site, roughly twofold higher than that for samples obtained before treatment. To test the putative mutagenic effect of the treatment on HCV mutation rate, we performed a paired test between the T0 and T6 samples from the 15 patients for whom these two time points were available, which cancels out between-patient variability and other sources of error. For this subset, mutation rate estimates were (0.6 ± 0.3) × 10−4 and (2.3 ± 0.7) × 10−4 for T0 and T6 samples, respectively, meaning that there was a nearly fourfold increase in mutation rate in T6 samples (one-tailed Wilcoxon signed-rank paired test; P = 0.025). T0 and T12 samples were available only from eight patients, and the differences in mutation rate between these two time points were not statistically significant (P = 0.715), which might be due to poor statistical power but could also indicate that the effect of treatment on mutation rate was transient. The mutational spectrum for the eight possible substitutions was strongly biased toward G→A and C→U transitions in sequences from treated patients, with the latter transition being 3.4 times more likely to occur than expected by chance, as opposed to the 1.9-fold excess observed for T0 samples. The proportion of transitions was significantly higher in T6/T12 samples than in T0 samples (one-tailed Fisher's exact test; P = 0.024) (Table 2). This shift in mutational spectrum is consistent with the effect of ribavirin in vitro.
DISCUSSION
Previous works have studied HCV genetic variability using mutation frequencies or rates of molecular evolution and addressed how treatment modifies these parameters (3, 7, 19, 25, 32, 38, 41). However, no estimates of mutation rates have been reported previously for this virus. In principle, the analysis of lethal mutations offers a direct way to estimate the mutation rate (equation 1), and the assumption that most viral genotypes carrying nonsense mutations are lethal seems realistic, especially in the case of HCV, where the genomic RNA is translated into a single polyprotein. Consistent with this view, our estimate of (1.2 ± 0.3) × 10−4 for the in vivo mutation rate is within the range generally accepted for RNA viruses (4, 12, 15, 16). In practice, however, several sources of bias could have led us to overestimate the mutation rate. First, nonsense mutations are deleterious only upon translation, and hence, several rounds of RNA copying could have been completed before nonsense mutations were selected. Second, genetic complementation could hinder the lethality of nonsense mutations. For instance, previous work has shown that a genotype of dengue virus carrying a nonsense mutation can spread in the population probably due to complementation (1). Naturally occurring HCV deletion mutants have been identified in chronic HCV patients (20, 28), although these variants could be involved in the normal replication cycle of HCV (40). Third, nonlethality might also result from stop codon suppression, although this mechanism has not been described in HCV. Finally, a fraction of the observed mutations might be RT-PCR artifacts. Taking into account the above-described sources of bias, our mutation rate estimate should be taken as an upper limit. However, it must be noted that, nevertheless, the frequency of nonsense mutations is necessarily closer to the mutation rate than that of any other kind of nucleotide substitution.
Like what has been previously established for other polymerases (18, 21, 34), the spontaneous mutation rate of HCV polymerase is biased toward transitions, and according to our data, the total mutation spectrum of HCV should be composed of two-thirds transitions and one-third transversions. Selection should alter this proportion because transitions are more often silent than transversions. Treatment-induced mutagenesis could have similar consequences, and the two factors could even have indistinguishable effects. However, here we have concentrated on nonsense mutations, and therefore, the shift in the mutation spectrum in treated patients should be attributed to mutagenesis.
Factors other than ribavirin-mediated mutagenesis could have also elevated the mutation rate of HCV, but these alternative possibilities are unlikely. First, although formally possible, a mutagenic effect of interferon is unlikely because its mode of action has not been directly or indirectly linked to RNA polymerization or editing mechanisms. Second, increased selective pressure on the virus following the onset of treatment might have favored the spread of mutators. This process has been described in bacteria (29) but not in RNA viruses, because their mutation rates are already extremely high and further elevations in mutation rates are not expected to be advantageous. Moreover, the combined interferon-ribavirin treatment specifically increased the fraction of C→U and G→A transitions, a result that would not be explained by the spread of mutators, whereas it matches the expected effect of ribavirin.
Lethal mutagenesis theory predicts that the combination of mutagenesis and viral inhibition should maximize the chances of viral clearance because the level of mutagenesis required to trigger extinction depends directly on viral yield (6). In line with this prediction, it has been shown that foot-and-mouth disease virus undergoes extinction in cell culture more frequently when the administration of mutagens is combined with viral dilution (33). In the case of HCV, ribavirin on its own has a weak effect on viral load, whereas it produces a substantial increase in the probability of sustained response to treatment when combined with interferon, a potent viral inhibitor. The mechanism for this synergistic effect between the two drugs is poorly understood (17). We suggest that this effect is indeed consistent with current lethal mutagenesis theory. Ribavirin would act as a mutagen, but its sole effect would not be sufficient to extinguish HCV in vivo. However, in the presence of interferon, the amount of mutagenesis required to clear the virus should be lower, and lethal mutagenesis would consequently have more chances of success.
Finally, it is important to notice that we did not detect an increase of the mutation rate in T12 samples and that this might indicate a transient effect of ribavirin (25). T6 and T12 samples corresponded to patients who failed to respond to treatment, and therefore, it is conceivable that the lack of sustained mutagenesis, due to the evolution of resistant viruses or to patient-specific factors, contributed to treatment failure. A detailed comparison between the change in mutation rates following the onset of treatment in responders and nonresponders might help to clarify this issue.
Supplementary Material
Acknowledgments
We thank Manuela Torres-Puente, Vicente Sentandreu, Alma Bracho, and Pilar Domingo-Calap for technical assistance and Esteban Domingo for helpful comments on the manuscript.
This work was financially supported by grant BFU2008-03978/BMC and by the Ramón y Cajal and Juan de la Cierva programs from the Spanish MICINN.
Footnotes
Published ahead of print on 25 March 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aaskov, J., K. Buzacott, H. M. Thu, K. Lowry, and E. C. Holmes. 2006. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 311236-238. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, J. P., R. Daifuku, and L. A. Loeb. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58183-205. [DOI] [PubMed] [Google Scholar]
- 3.Asahina, Y., N. Izumi, N. Enomoto, M. Uchihara, M. Kurosaki, Y. Onuki, Y. Nishimura, K. Ueda, K. Tsuchiya, H. Nakanishi, T. Kitamura, and S. Miyake. 2005. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 43623-629. [DOI] [PubMed] [Google Scholar]
- 4.Belshaw, R., A. Gardner, A. Rambaut, and O. G. Pybus. 2008. Pacing a small cage: mutation and RNA viruses. Trends Ecol. Evol. 23188-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bracho, M. A., F. Y. Carrillo-Cruz, E. Ortega, A. Moya, and F. González-Candelas. 2006. A new subtype of hepatitis C virus genotype 1: complete genome and phylogenetic relationships of an Equatorial Guinea isolate. J. Gen. Virol. 871697-1702. [DOI] [PubMed] [Google Scholar]
- 6.Bull, J. J., R. Sanjuán, and C. O. Wilke. 2007. Theory of lethal mutagenesis for viruses. J. Virol. 812930-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chevaliez, S., R. Brillet, E. Lazaro, C. Hezode, and J. M. Pawlotsky. 2007. Analysis of ribavirin mutagenicity in human hepatitis C virus infection. J. Virol. 817732-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clune, J., D. Misevic, C. Ofria, R. E. Lenski, S. F. Elena, and R. Sanjuán. 2008. Natural selection fails to optimize mutation rates in rugged fitness landscapes. PLoS Comput. Biol. 4e1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contreras, A. M., Y. Hiasa, W. He, A. Terella, E. V. Schmidt, and R. T. Chung. 2002. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 768505-8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 986895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crow, J. F., and M. Kimura. 1970. An introduction to population genetics theory. Harper & Row, New York, NY.
- 12.Domingo, E. (ed.). 2006. Quasispecies: concept and implications for virology. Springer, Berlin, Germany.
- 13.Domingo, E. 2003. Quasispecies and the development of new antiviral strategies. Prog. Drug Res. 60133-158. [DOI] [PubMed] [Google Scholar]
- 14.Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 1481667-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 9613910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duffy, S., L. A. Shackelton, and E. C. Holmes. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9267-276. [DOI] [PubMed] [Google Scholar]
- 17.Feld, J. J., and J. H. Hoofnagle. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436967-972. [DOI] [PubMed] [Google Scholar]
- 18.Freistadt, M. S., J. A. Vaccaro, and K. E. Eberle. 2007. Biochemical characterization of the fidelity of poliovirus RNA-dependent RNA polymerase. Virol. J. 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofmann, W. P., A. Polta, E. Herrmann, U. Mihm, B. Kronenberger, T. Sonntag, V. Lohmann, B. Schonberger, S. Zeuzem, and C. Sarrazin. 2007. Mutagenic effect of ribavirin on hepatitis C nonstructural 5B quasispecies in vitro and during antiviral therapy. Gastroenterology 132921-930. [DOI] [PubMed] [Google Scholar]
- 20.Iwai, A., H. Marusawa, Y. Takada, H. Egawa, K. Ikeda, M. Nabeshima, S. Uemoto, and T. Chiba. 2006. Identification of novel defective HCV clones in liver transplant recipients with recurrent HCV infection. J. Viral Hepat. 13523-531. [DOI] [PubMed] [Google Scholar]
- 21.Lai, M. D., and K. L. Beattie. 1988. Influence of DNA sequence on the nature of mispairing during DNA synthesis. Biochemistry 271722-1728. [DOI] [PubMed] [Google Scholar]
- 22.Lanford, R. E., D. Chavez, B. Guerra, J. Y. Lau, Z. Hong, K. M. Brasky, and B. Beames. 2001. Ribavirin induces error-prone replication of GB virus B in primary tamarin hepatocytes. J. Virol. 758074-8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leyssen, P., E. De Clercq, and J. Neyts. 2006. The anti-yellow fever virus activity of ribavirin is independent of error-prone replication. Mol. Pharmacol. 691461-1467. [DOI] [PubMed] [Google Scholar]
- 24.Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436933-938. [DOI] [PubMed] [Google Scholar]
- 25.Lutchman, G., S. Danehower, B. C. Song, T. J. Liang, J. H. Hoofnagle, M. Thomson, and M. G. Ghany. 2007. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology 1321757-1766. [DOI] [PubMed] [Google Scholar]
- 26.Maag, D., C. Castro, Z. Hong, and C. E. Cameron. 2001. Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 27646094-46098. [DOI] [PubMed] [Google Scholar]
- 27.McHutchison, J. G., S. C. Gordon, E. R. Schiff, M. L. Shiffman, W. M. Lee, V. K. Rustgi, Z. D. Goodman, M. H. Ling, S. Cort, J. K. Albrecht, et al. 1998. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N. Engl. J. Med. 3391485-1492. [DOI] [PubMed] [Google Scholar]
- 28.Noppornpanth, S., S. L. Smits, T. X. Lien, Y. Poovorawan, A. D. Osterhaus, and B. L. Haagmans. 2007. Characterization of hepatitis C virus deletion mutants circulating in chronically infected patients. J. Virol. 8112496-12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pal, C., M. D. Macia, A. Oliver, I. Schachar, and A. Buckling. 2007. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 4501079-1081. [DOI] [PubMed] [Google Scholar]
- 30.Pfeiffer, J. K., and K. Kirkegaard. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 1007289-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poynard, T., P. Marcellin, S. S. Lee, C. Niederau, G. S. Minuk, G. Ideo, V. Bain, J. Heathcote, S. Zeuzem, C. Trepo, J. Albrecht, et al. 1998. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. Lancet 3521426-1432. [DOI] [PubMed] [Google Scholar]
- 32.Schinkel, J., M. D. de Jong, B. Bruning, B. van Hoek, W. J. Spaan, and A. C. Kroes. 2003. The potentiating effect of ribavirin on interferon in the treatment of hepatitis C: lack of evidence for ribavirin-induced viral mutagenesis. Antivir. Ther. 8535-540. [PubMed] [Google Scholar]
- 33.Sierra, S., M. Dávila, P. R. Lowenstein, and E. Domingo. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 748316-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinha, N. K., and M. D. Haimes. 1981. Molecular mechanisms of substitution mutagenesis. An experimental test of the Watson-Crick and Topal-Fresco models of base mispairings. J. Biol. Chem. 25610671-10683. [PubMed] [Google Scholar]
- 35.Torres-Puente, M., J. M. Cuevas, N. Jiménez-Hernández, M. A. Bracho, I. García-Robles, F. Carnicer, J. del Olmo, E. Ortega, A. Moya, and F. Gonzalez-Candelas. 2007. Contribution of insertions and deletions to the variability of hepatitis C virus populations. J. Gen. Virol. 882198-2203. [DOI] [PubMed] [Google Scholar]
- 36.Torres-Puente, M., J. M. Cuevas, N. Jiménez-Hernández, M. A. Bracho, I. García-Robles, F. Carnicer, J. del Olmo, E. Ortega, A. Moya, and F. Gonzalez-Candelas. 2008. Hepatitis C virus and the controversial role of the interferon sensitivity determining region in the response to interferon treatment. J. Med. Virol. 80247-253. [DOI] [PubMed] [Google Scholar]
- 37.Torres-Puente, M., J. M. Cuevas, N. Jiménez-Hernández, M. A. Bracho, I. García-Robles, B. Wróbel, F. Carnicer, J. del Olmo, E. Ortega, A. Moya, and F. González-Candelas. 2008. Genetic variability in hepatitis C virus and its role in antiviral treatment response. J. Viral Hepat. 15188-199. [DOI] [PubMed] [Google Scholar]
- 38.Vallejo, A., S. Molina-Pinelo, M. A. Abad, G. Gomez, M. Leal, A. Sánchez-Quijano, and E. Lissen. 2004. Analysis of quasispecies in the viral 5′ untranslated region of hepatitis C virus to evaluate ribavirin mutagenic effect in patients receiving ribavirin and interferon-alfa. Eur. J. Clin. Microbiol. Infect. Dis. 23923-926. [DOI] [PubMed] [Google Scholar]
- 39.Vo, N. V., K. C. Young, and M. M. Lai. 2003. Mutagenic and inhibitory effects of ribavirin on hepatitis C virus RNA polymerase. Biochemistry 4210462-10471. [DOI] [PubMed] [Google Scholar]
- 40.Yagi, S., K. Mori, E. Tanaka, A. Matsumoto, F. Sunaga, K. Kiyosawa, and K. Yamaguchi. 2005. Identification of novel HCV subgenome replicating persistently in chronic active hepatitis C patients. J. Med. Virol. 77399-413. [DOI] [PubMed] [Google Scholar]
- 41.Young, K. C., K. L. Lindsay, K. J. Lee, W. C. Liu, J. W. He, S. L. Milstein, and M. M. Lai. 2003. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 38869-878. [DOI] [PubMed] [Google Scholar]
- 42.Zhou, S., R. Liu, B. M. Baroudy, B. A. Malcolm, and G. R. Reyes. 2003. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310333-342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.