

Abstract
APOBEC3G restricts Vif-deficient human immunodeficiency virus type 1 (HIV-1) by deaminating viral cDNA cytosines to uracils. This promutagenic activity is counteracted by HIV-1 Vif, which is a natural APOBEC3G antagonist. However, we previously reported that Vif-deficient HIV-1 could evolve resistance to APOBEC3G by a novel mechanism requiring an A200-to-C/T transition mutation and Vpr inactivation. A pyrimidine at nucleotide 200 in the untranslated leader region contributed to resistance by increasing virus particle production, which resulted in fewer APOBEC3G molecules per particle. Here we show that the A200-to-C/T mutation functions posttranscriptionally by inactivating an upstream start codon, which in turn enables optimal viral mRNA translation from canonical start codons.
Vif-deficient human immunodeficiency type 1 (HIV-1) is unable to replicate in cells expressing human APOBEC3G (23). Human APOBEC3G is a DNA cytosine deaminase that incorporates into Vif-deficient HIV-1 viral particles. Upon infection of a new cell, APOBEC3G deaminates cytosines to uracils within nascent viral cDNA, leading to high levels of viral G-to-A hypermutation and severely attenuated infectivity (12, 15, 23, 29). The HIV-1 Vif protein counteracts APOBEC3G by preventing its incorporation into viral particles and triggering its degradation through the proteasome (3, 13, 14, 16, 17, 19, 24, 25).
We recently reported the isolation and characterization of three variants of HIV-1IIIB (GenBank accession no. EU541617) that had evolved a Vif-independent resistance to APOBEC3G (11). All three resistant isolates had only two mutations in common: a Vpr-inactivating mutation and a noncoding A200-to-C/T transversion mutation located in the untranslated leader region of HIV-1. Importantly, both mutations are required for the resistance phenotype (Fig. 1). Our prior studies also showed that the A200-to-C/T mutation contributes to APOBEC3G resistance by increasing HIV-1 particle production. Elevated titers reduced the amount of packaged APOBEC3G to sublethal levels, which helped enable virus replication (11).
FIG. 1.

Replication kinetics of molecular clone-derived viruses with the indicated genotype. As shown previously (11), only molecular clones containing the A200-to-C/T mutation are able to grow in CEM-SS cells stably expressing APOBEC3G. Dashed lines represent virus replication on vector-control CEM-SS cells (V2), whereas solid lines represent virus replication on APOBEC3G-expressing cells (G1 and G2). Vif+ virus replicated readily on both the vector and APOBEC3G-expressing cell lines used here (11, 21; data not shown).
We had noted previously that the A200-to-C/T mutation is located immediately adjacent to a known interferon-stimulated response element (ISRE) (26). We therefore sought to determine whether this mutation created a better binding site for interferon-regulated factors or other transcription factors and thereby increased HIV-1 transcription rates. To test this hypothesis, we made a series of reporter constructs with the HIV-1 long terminal repeat (LTR) promoter region (nucleotides −455 to 335) upstream of a green fluorescent protein (GFP) reporter gene (LTR-GFP), transfected 293T cells, and monitored GFP expression by flow cytometry (11). The constructs with C200 or T200 each produced roughly eightfold more GFP fluorescence than the A200-containing plasmid (11). The converse mutation was also introduced in the LTR of an HIV-1 isolate that already has T200 (HIV-1LAI; GenBank accession no. K02013). As expected, the T200-to-A mutation in the HIV-1LAI LTR caused diminished GFP fluorescence levels (11). Together, our prior data suggested a model in which the A200-to-C/T mutation increased the level of HIV-1 particle production by increasing LTR-driven transcription (11).
To directly test whether the A200-to-C/T ISRE is bound more efficiently by cellular transcription factors, we performed a series of electrophoretic mobility shift assays by incubating T-cell nuclear lysates (as prepared in reference 27) with 6-carboxyfluorescein (FAM)-labeled HIV-1 ISRE-containing DNA duplexes (nucleotides 194 to 223 of HIV-1IIIB) with A200, C200, or T200. The samples were separated on an 8% native acrylamide gel and imaged using an FLA-5000 imaging system (Fujifilm Life Science). To our surprise, DNA duplexes containing A200 or C200 were shifted equally well by nuclear extracts, suggesting that this mutation may not enhance transcription factor binding at the HIV-1 ISRE (see Fig. S1 in the supplemental material). These results led us to consider alternative models for how A200-to-C/T increases HIV-1 particle production.
Specifically, we noted that A200 is the first base of a potential upstream translation start codon, A200UG. The leader RNA of most HIV-1 isolates lacks such upstream start codons (2). Since translation of HIV-1 mRNAs occurs in part via a ribosome scanning mechanism (6, 18, 22), this upstream A200UG could result in early translation initiation and termination before the ribosome has a chance to reach the proper viral open reading frame (ORF) start codons (Fig. 2A). Such a possibility has been documented previously for HIV-1, where the introduction of an optimal AUG at position 239 significantly decreased HIV-1 protein expression (6). Therefore, since the A200-to-C/T mutation eliminates a potential upstream translation start codon, it is possible that its effect on HIV-1 particle production is due to increased translation and not transcription. To differentiate between these two possibilities, we used quantitative reverse transcription-PCR (RT-PCR) to measure the levels of HIV-1 transcripts from 293T cells transfected with proviral plasmids containing A200, C200, or T200. While the levels of HIV-1 particle production were markedly higher with viruses containing C200 or T200, the relative HIV-1 transcript levels were constant (Fig. 2B and C; and see Fig. S2 in the supplemental material). Similarly, LTR-GFP reporter constructs with A, T, or C at position 200 showed no difference in GFP transcript levels by quantitative RT-PCR (Fig. 3A and C). In contrast, the mean levels of GFP fluorescence intensity were approximately threefold higher from constructs with a C or T at position 200 (Fig. 3A and B). These results indicated that A200-to-C/T does not contribute to APOBEC3G resistance by increasing HIV-1 transcription levels but instead does so by enhancing the efficiency of HIV-1 translation.
FIG. 2.

A200-to-C/T increases virus particle production by enhancing HIV-1 translation. (A) Schematic of the HIV-1 LTR and the gag ORF. For simplicity, only the gag ORF is shown. The region encompassing position 200 is blown up to illustrate the potential uORF initiated from A200TG (i.e., AUG in the viral mRNA). The HIV-1 primer-binding site (PBS) is also illustrated. It should be noted that A200UG is out of frame with gag and that read-through translation of the stop codon U323AG is unlikely. Nucleotide numbers are the same as GenBank accession no. EU541617, with +1 representing the transcription start site. (B) Relative titers of molecular clone-derived viruses with the indicated nucleotide at position 200 produced from 293T cells. Infectious titers were determined by infecting reporter cells (CEM-GFP), which express the GFP gene under the control of the HIV-1 promoter, and infectivity levels were determined by flow cytometry (e.g., see references 9 and 11). The titer of the A200 virus was normalized to 1, and each histogram bar shows the mean and standard error of two independently derived supernatants. (C) Relative transcript levels of molecular clones with the indicated nucleotide at position 200. Transcript levels were measured using a Roche Light Cycler-based quantitative RT-PCR assay. Total RNA was extracted from the corresponding virus-producing 293T cells in panel B and reverse transcribed into cDNA. HIV-1 cDNA was amplified using primers 5′-CATGAAAGCGAAAGGGAAAC and 5′-TTTGCTGGTCCTTTCCAAAC and detected using Roche Universal Probe 56 (5′-TGCTGTCC). This primer and probe set is specific to the untranslated leader and pol regions, respectively, and they flank splice junctions. To correct for variations in RNA recovery, HIV-1 transcript levels were normalized to those of the hypoxanthine phosphoribosyltransferase gene amplified with primers 5′-TGACCTTGATTTATTTTGCATACC and 5′-CGAGCAAGACGTTCAGTCCT and detected using Roche Universal Probe 73 (5′-GCTGAGGA). The RNA level of the A200 virus was normalized to 1, and each histogram bar represents the mean and standard error of two independent experiments.
FIG. 3.

A200-to-C/T enhances translation of an LTR-GFP reporter construct. (A) Schematic of the LTR-GFP reporter construct. (B) Relative GFP fluorescence measured by flow cytometry in 293T cells transfected with LTR-GFP constructs containing A200, C200, or T200. To minimize transfection variation, only the GFP-positive cell populations were used to calculate the mean fluorescence intensity. To facilitate comparison, the mean GFP fluorescence level of the A200-containing LTR-GFP reporter construct was normalized to 1. Each histogram bar represents the mean and standard error of three independent experiments. (C) Relative GFP transcript levels produced from LTR-GFP constructs containing A200, C200, or T200. Transcript levels were measured as described for Fig. 2C. GFP cDNA was amplified using primers 5′-AGAACGGCATCAAGGTGAAC and 5′-TGCTCAGGTAGTGGTTGTCG and detected using Roche Universal Probe 74 (5′-CTGCTGCC). To correct for variations in RNA recovery, HIV-1 transcript levels were normalized to those of the hypoxanthine phosphoribosyltransferase gene. GFP transcript level of the A200-containing LTR-GFP reporter construct was normalized to 1, and each histogram bar represents the mean and standard error of three independent experiments. (D) Immunoblot showing that translation can initiate at A200 and proceed through GFP, provided the uORF stop codon is removed and placed in-frame. An anti-GFP antibody (Covance) was used to detect GFP levels in 293T cells transfected with the indicated LTR-GFP construct. The membrane was stripped and reprobed with antitubulin (TUB) for a loading control (Covance). The relative GFP/TUB ratio is shown below each lane, with the ratio in lane 2 arbitrarily set to 1. The T200 or C200 reactions show approximately threefold more GFP (lanes 3 to 5). However, when the uORF stop codon is inactivated (ΔT323) and placed in-frame with GFP, all translated products were observable and the combined expression levels were similar for all reactions (lanes 6 to 10). Data from an independent experiment are shown in Fig. S3 in the supplemental material. NT, nontransfected.
If translation truly initiated at A200UG, this would produce a 41-residue peptide that terminated at the stop codon beginning with U323. To demonstrate that translation can initiate at A200, we inactivated the predicted stop codon of this upstream ORF (uORF) by deleting T323 in the LTR-GFP reporter constructs (Fig. 3A). This event also places the predicted uORF in frame with the GFP gene. Therefore, when transfected into 293T cells, the A200ΔT323 reporter constructs should produce a GFP fusion protein of 32 kDa (5-kDa uORF product plus 27-kDa GFP). As expected, anti-GFP immunoblots of transfected 293T cell lysates showed that the A200ΔT323 reporter constructs produced a GFP fusion protein of roughly 32 kDa (Fig. 3D). Moreover, flow cytometric analyses showed that deletion of T323 in the A200 constructs increased the overall GFP fluorescence to near that of the C200 or T200 constructs (data not shown). This increase in GFP fluorescence was not due to a nonspecific effect of the T323 deletion because no significant change in fluorescence was seen with C200ΔT323 or T200ΔT323 (data not shown). Furthermore, we also noted that the threefold increase in GFP fluorescence measured by flow cytometry correlated well with the anti-GFP band intensities observed by immunoblotting (Fig. 3D; and see Fig. S3 in the supplemental material). Together, these results indicate that A200-to-C/T elevates HIV-1 particle production by inactivating the A200UG upstream start codon, which helps ensure optimal translation from the canonical HIV-1 ORF start codons.
We were originally struck by the fact that three independent APOBEC3G resistance mutations occurred at the same nucleotide (11). Together with the fact that almost all HIV-1 isolates already have a pyrimidine at position 200 (www.hiv.lanl.gov), we questioned whether the origin of A200-to-C/T was nonrandom and possibly even templated. All retroviruses, including HIV-1, use a cellular tRNA to prime minus-strand strong-stop DNA synthesis during RT (reviewed in reference 10). Plus-strand strong-stop DNA synthesis is primed by the polypurine track, and it is thought to terminate when reverse transcriptase stalls at the conserved 1-methyladenine 58 (1-mA58) in the tRNA. In fact, 1-mA58 has been proposed to serve as a stop signal for reverse transcriptase (20). However, it has been argued previously that synthesis does not always stop at 1-mA58 but instead occasionally terminates at the second modified base in the tRNA, pseudouridine 55 (ψ55) (28). Thus, since position 200 is located immediately downstream of the primer biding site, it could potentially be templated by 1-mA58 in the tRNA should plus-strand strong-stop DNA synthesis go beyond 1-mA58 (Fig. 4A). Such a scenario might explain the emergence of T200, but it is hard to comprehend how a 1-methyladenine would template C200 without invoking noncanonical base-pairing schemes.
FIG. 4.

C/T200 is not templated by the tRNA during plus-strand strong-stop DNA synthesis. (A) Schematic showing how T200 could potentially be copied from the tRNA during RT if plus-strand strong-stop DNA synthesis terminates at ψ55 instead of 1-mA58. The C200 scenario is not depicted but may occur with alternative base-pairing schemes. The light gray line represents the DNA, and the black line represents the RNA. (B) Summary of the base substitution mutations found in A200-Vif+ and A200-Vif− viruses produced in the presence and absence of APOBEC3G. At least four independent proviral sequences were analyzed for each condition.
To determine whether the tRNA is capable of templating the insertion of T200 (or C200), we sequenced HIV-1 proviruses that had undergone approximately one round of replication. Such tRNA-templated directional evolution has been demonstrated for natural and man-made HIV-1 variants with an altered primer-binding site (4, 5). If the tRNA does indeed template the insertion of C/T200, then singly replicated HIV-1 proviruses should harbor this mutation. We therefore produced A200-Vif+ and A200-Vif− viruses by 293T transfection in the presence and absence of APOBEC3G and used these viruses to infect reporter T cells. To try to limit infection to a single round, we harvested genomic DNA from the infected cells 2 days postinfection. We then PCR amplified a portion of the LTR (including position 200), TOPO cloned the amplicons, and sequenced them using universal primers. We found that none of the viruses had acquired the A200-to-C/T mutation (Fig. 4B). We wanted to investigate this further by asking whether A200-to-C/T would emerge after multiple rounds of virus replication. We therefore analyzed proviral DNA sequences from Vif+ and Vif− viruses cultured for 20 days on APOBEC3G-expressing cells. While neither the Vif+ or Vif− virus had acquired A200-to-C/T, we were surprised to find that one of the Vif− viral cultures had instead acquired another mutation, T201-to-G (Fig. 4B). This base substitution mutation is particularly interesting because it disrupts the same potential upstream start codon, and like A200-to-C/T, it probably also helps optimize HIV-1 translation. Moreover, the fact that T201 mutated to G further shows that plus-strand strong-stop DNA synthesis does not frequently go beyond 1-mA58, since T201's complementary base in the tRNA is A57 (Fig. 4A). Taken together, these data indicate that A200-to-C/T is not copied frequently from the tRNA during RT. Other mechanisms, such as reverse transcriptase infidelity, likely underlie the formation of these mutations within the A200TG sequence.
Our studies demonstrate that the A200-to-C/T mutation contributes to APOBEC3G resistance by eliminating a premature AUG translation start codon. This event ensures optimal HIV-1 mRNA translation from canonical start codons, increases virus titers, and ultimately causes a reduction in APOBEC3G packaging. Diminished concentrations of APOBEC3G in virions cause a corresponding drop in the levels of G-to-A hypermutation from lethal to sublethal, which helps enable Vif-deficient virus replication in the presence of APOBEC3G (11). The fact that almost all HIV-1 isolates already have a pyrimidine at position 200 and that they rarely have an upstream start codon strongly suggests that, in general, premature start codons are unlikely to be tolerated in vivo. However, the pyrimidine at position 200 may have another essential viral function because very few isolates have a G at this position. Finally, our data strongly indicate that any method used by the virus to diminish APOBEC3G packaging will be advantageous. Although our selection experiments yielded HIV-1 variants that lowered APOBEC3G packaging by an indirect mechanism that involved enhanced translation initiation, additional experiments could easily select variants that employ alternative means to decrease APOBEC3G packaging, such as increased particle production due to elevated transcription. An extreme version of such an APOBEC3G avoidance mechanism may already be used by the related retroviruses human T-cell leukemia virus, murine leukemia virus, and Mason-Pfizer monkey virus, which appear to have evolved to simply avoid encapsidation of APOBEC3 proteins (1, 7, 8).
Supplementary Material
Acknowledgments
We thank H. Hutoff and N. Somia for helpful comments.
This work was supported by grants from the National Institutes of Health (R01 AI064046 to R.S.H.), the Canadian Institutes for Health Research (Doctoral Research Award to G.H.), and the Dutch Organization for Scientific Research (Chemical Sciences, NWO-CW, Top grant to B.B.).
Footnotes
Published ahead of print on 18 March 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama. 2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol. 161565-1570. [DOI] [PubMed] [Google Scholar]
- 2.Berkhout, B. 1996. Structure and function of the human immunodeficiency virus leader RNA. Prog. Nucleic Acid Res. Mol. Biol. 541-34. [DOI] [PubMed] [Google Scholar]
- 3.Conticello, S. G., R. S. Harris, and M. S. Neuberger. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 132009-2013. [DOI] [PubMed] [Google Scholar]
-
4.Das, A. T., B. Klaver, and B. Berkhout. 1995. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural
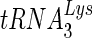 . J. Virol. 693090-3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
. J. Virol. 693090-3097. [DOI] [PMC free article] [PubMed] [Google Scholar] - 5.Das, A. T., B. Klaver, and B. Berkhout. 1997. Sequence variation of the human immunodeficiency virus primer-binding site suggests the use of an alternative tRNA(Lys) molecule in reverse transcription. J. Gen. Virol. 78837-840. [DOI] [PubMed] [Google Scholar]
- 6.Das, A. T., A. P. van Dam, B. Klaver, and B. Berkhout. 1998. Improved envelope function selected by long-term cultivation of a translation-impaired HIV-1 mutant. Virology 244552-562. [DOI] [PubMed] [Google Scholar]
- 7.Doehle, B. P., H. P. Bogerd, H. L. Wiegand, N. Jouvenet, P. D. Bieniasz, E. Hunter, and B. R. Cullen. 2006. The betaretrovirus Mason-Pfizer monkey virus selectively excludes simian APOBEC3G from virion particles. J. Virol. 8012102-12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doehle, B. P., A. Schafer, H. L. Wiegand, H. P. Bogerd, and B. R. Cullen. 2005. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J. Virol. 798201-8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gervaix, A., D. West, L. M. Leoni, D. D. Richman, F. Wong-Staal, and J. Corbeil. 1997. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. USA 944653-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gotte, M., X. Li, and M. A. Wainberg. 1999. HIV-1 reverse transcription: a brief overview focused on structure-function relationships among molecules involved in initiation of the reaction. Arch. Biochem. Biophys. 365199-210. [DOI] [PubMed] [Google Scholar]
- 11.Haché, G., K. Shindo, J. S. Albin, and R. S. Harris. 2008. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr. Biol. 18819-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113803-809. [DOI] [PubMed] [Google Scholar]
- 13.Kao, S., M. A. Khan, E. Miyagi, R. Plishka, A. Buckler-White, and K. Strebel. 2003. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 7711398-11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, B., X. Yu, K. Luo, Y. Yu, and X. F. Yu. 2004. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J. Virol. 782072-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 42499-103. [DOI] [PubMed] [Google Scholar]
- 16.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 91398-1403. [DOI] [PubMed] [Google Scholar]
- 17.Mehle, A., B. Strack, P. Ancuta, C. Zhang, M. McPike, and D. Gabuzda. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2797792-7798. [DOI] [PubMed] [Google Scholar]
- 18.Miele, G., A. Mouland, G. P. Harrison, E. Cohen, and A. M. Lever. 1996. The human immunodeficiency virus type 1 5′ packaging signal structure affects translation but does not function as an internal ribosome entry site structure. J. Virol. 70944-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Opi, S., S. Kao, R. Goila-Gaur, M. A. Khan, E. Miyagi, H. Takeuchi, and K. Strebel. 2007. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 818236-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
20.Renda, M. J., J. D. Rosenblatt, E. Klimatcheva, L. M. Demeter, R. A. Bambara, and V. Planelles. 2001. Mutation of the methylated
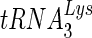 residue A58 disrupts reverse transcription and inhibits replication of human immunodeficiency virus type 1. J. Virol. 759671-9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
residue A58 disrupts reverse transcription and inhibits replication of human immunodeficiency virus type 1. J. Virol. 759671-9678. [DOI] [PMC free article] [PubMed] [Google Scholar] - 21.Schumacher, A. J., G. Haché, D. A. MacDuff, W. L. Brown, and R. S. Harris. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 822652-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz, S., B. K. Felber, and G. N. Pavlakis. 1992. Mechanism of translation of monocistronic and multicistronic human immunodeficiency virus type 1 mRNAs. Mol. Cell. Biol. 12207-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 24.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 91404-1407. [DOI] [PubMed] [Google Scholar]
- 25.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12591-601. [DOI] [PubMed] [Google Scholar]
- 26.Van Lint, C., C. A. Amella, S. Emiliani, M. John, T. Jie, and E. Verdin. 1997. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 716113-6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf, D., and S. P. Goff. 2007. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 13146-57. [DOI] [PubMed] [Google Scholar]
- 28.Wu, T., J. Guo, J. Bess, L. E. Henderson, and J. G. Levin. 1999. Molecular requirements for human immunodeficiency virus type 1 plus-strand transfer: analysis in reconstituted and endogenous reverse transcription systems. J. Virol. 734794-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L. Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 42494-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.