

Abstract
PLRG1, an evolutionarily conserved component of the spliceosome, forms a complex with Pso4/SNEV/Prp19 and the cell division and cycle 5 homolog (CDC5L) that is involved in both pre-mRNA splicing and DNA repair. Here, we show that the inactivation of PLRG1 in mice results in embryonic lethality at 1.5 days postfertilization. Studies of heart- and neuron-specific PLRG1 knockout mice further reveal an essential role of PLRG1 in adult tissue homeostasis and the suppression of apoptosis. PLRG1-deficient mouse embryonic fibroblasts (MEFs) fail to progress through S phase upon serum stimulation and exhibit increased rates of apoptosis. PLRG1 deficiency causes enhanced p53 phosphorylation and stabilization in the presence of increased γ-H2AX immunoreactivity as an indicator of an activated DNA damage response. p53 downregulation rescues lethality in both PLRG1-deficient MEFs and zebrafish in vivo, showing that apoptosis resulting from PLRG1 deficiency is p53 dependent. Moreover, the deletion of PLRG1 results in the relocation of its interaction partner CDC5L from the nucleus to the cytoplasm without general alterations in pre-mRNA splicing. Taken together, the results of this study identify PLRG1 as a critical nuclear regulator of p53-dependent cell cycle progression and apoptosis during both embryonic development and adult tissue homeostasis.
Mammalian pleiotropic regulator (PLRG1) belongs to a highly conserved family of seven WD40 domain-containing proteins in eukaryotes (2, 4). The founding members of this protein family, PRL1 and PRL2, were identified first by T-DNA tagging in Arabidopsis thaliana, where PRL1 deficiency augments the sensitivity to growth hormones, stimulates the accumulation of sugars and starch in leaves, and inhibits root elongation (37). More recently, it has been demonstrated that the A. thaliana CDC5/PRL1 complex is essential for plant innate immunity (39).
Human PLRG1 initially was identified as a subunit of spliceosomal complexes purified from HeLa nuclear extracts by coimmunoprecipitation with CDC5L (2). Indeed, interaction between PLRG1 and CDC5L appears essential for pre-mRNA processing, as peptides inhibiting CDC5L-PLRG1 complex formation efficiently block pre-mRNA splicing (3).
The CDC5L-PLRG1 complex also was found to interact with the WRN protein, which is deficient in Werner syndrome, a rare autosomal recessive human disorder characterized by genomic instability and the premature onset of a number of age-related diseases, including cancer (1, 18, 28, 34, 53). The WRN protein has exonuclease and RecQ helicase activity, interacts with multiple proteins of the DNA replication complex, and is crucial for DNA synthesis and DNA repair (12, 30, 31, 41, 43, 53). Thus, PLRG1 offers an interesting link between the control of pre-mRNA splicing and DNA metabolism.
To elucidate the in vivo role of PLRG1 in vertebrates, we have generated conventional and conditional knockout (KO) mice for PLRG1, as well as antisense-mediated plrg1 knockdown zebrafish embryos and cell lines with an inducible inactivation of PLRG1. Taken together, our studies demonstrate a critical requirement for PLRG1 in the control of cell cycle progression and p53-dependent apoptosis during embryonic development and during adult tissue homeostasis.
MATERIALS AND METHODS
Generation of PLRG1 gene replacement vectors.
To construct a conventional PLRG1 KO allele, a PGKneo cassette was inserted into the second exon of the murine PLRG1 gene. For negative selection, a herpes simplex virus thymidine kinase (TK) gene was inserted behind the long arm of homology. The short arm of homology, consisting of a 1.2-kb fragment, was PCR amplified from genomic DNA with the primers 5′-GTAGACTAAACGGCGGCGACATG-3′ and 5′-GTGTGTGTACAGAATGCATCTGTACC-3′. The resulting fragment was cloned in the pGK12 vector after restriction digestion with BamHI and NotI. The long arm of homology, consisting of a 5-kb fragment, was PCR amplified with the primers 5′-GGCCGGCCAGGTCTTAAAGGTGCATACTCACAGGAC-3′ and 5′-CTCGAGGCATCAATGTCACCAAACCTGTAGCACT-3′ and inserted into the pGK12 vector after FseI and XhoI site digestion.
To conditionally inactivate the PLRG1 gene, exon 3 was flanked by loxP sites. Therefore, a fragment containing exon 3 was PCR amplified from mouse genomic DNA using the primers 5′-GGCGCGCCGGTCTCATCCAAAAAGGTTTTGTGT-3′ and 5′-GGCCGGCCGAATCAACTTGAGTTTTCCCTGTAG-3 and inserted between loxP sites into the pGK12 vector after AscI and FseI digestion. The short arm of homology, consisting of a 2.5-kb fragment, was PCR amplified using the primers 5′-GGCCGCGTAGACTAAACGGCGGCGACATG-3′ and 5′-CCGCGGTCAAGGGTCCAAGTGAATTAAAGAC-3′ and cloned into pGK12 after digestion with NotI and SacII. The long arm of homology, consisting of a 5-kb fragment, was PCR amplified with the primers 5′-CTCGAGCTAGCCTGTGGGGAGACCATCT-3′ and 5′-GTTTAAACAAACACCCTCTCACGAGTGGGG-3 and inserted into the pGK12 vector after XhoI and PmeI digestion.
Verification of recombination events by DNA hybridization.
Genomic DNA was extracted from embryonic stem (ES) cells using cell lysis buffer containing 10 mM Tris-HCl (pH 7.5), 10 mM EDTA, 10 mM NaCl, 0.5% lauroyl-sarcosine, 2 mg/ml proteinase K, followed by isopropanol precipitation and then incubation at 56°C overnight. DNA was digested using BamHI or BglI restriction enzyme (New England Biolabs). A probe for PLRG1 was amplified using primers 5′-CATTGCTGTATCGGCGCTACGTTT-3′ and 5′-CTTGGTGCTCCTTACTTGGAGGTT-3′ and for the neomycin phosphotransferase gene using primers 5′-TGAATGAACTGCAGGACGAGGCA-3′ and 5′-GCCGCCAAGCTCTTCAGCAATAT-3′, and the probes were labeled with [α32-P]dCTP using the Ladderman DNA labeling kit (TaKaRa).
Cell culture.
V6.5 F1 hybrid ES cells (14) were transfected, cultured, and selected as described previously (25). Targeting constructs were transfected in the V6.5 mouse ES line by electroporation, followed by a positive/negative selection with 250 μg/ml G418 and 2 × 10−6 M ganciclovir. Surviving clones were screened for homologous recombination by Southern blot analysis according to the strategies outlined in Fig. 1 and 3.
FIG. 1.

Conventional inactivation of the PLRG1 gene. (A) Northern RNA hybridization analysis of PLRG1 expression during embryogenesis. Upper panel, PLRG1 probe; lower panel, 18S and 28S RNA loading controls. Numbers indicate the days of gestation (ED). (B) Expression analysis of PLRG1 in tissues from adult mice. Upper panel, PLRG1 probe; lower panel, 18S and 28S RNA loading controls. dpc, days postcoitum. (C) Schematic representation of the PLRG1 gene replacement vector and its respective localization within the murine PLRG1 locus. WT, wild type. SA and LA, long and short arms of homology, respectively. (D) Verification of PLRG1 gene replacement. Left panel, Southern blot analysis of ES cell clones hybridized with the 5′ probe. Right panel, Southern blot of the same clones hybridized with the probe corresponding to the neomycin resistance gene for verification of single integration (RI, random integrant; HR, homologous recombinant).
FIG. 3.

Conditional inactivation of the PLRG1 gene. (A) Schematic representation of the targeting strategy to introduce loxP sites (triangle) into the PLRG1 locus. (B) Southern blot analysis of ES cell clones after transfection with the targeting construct shown in panel A. Blots were hybridized with the probe depicted in panel A (WT, wild-type; HR, homologous recombinant). The upper left panel shows a homologous recombinant (1). The lower left panel shows the 5.8-kb band for single integration (1, 2, and 3, homologous recombinants). The right panel shows the PCR analysis with primers flanking the 3′ loxP site confirming the cointegration of the loxP site (1 and 3, heterozygous clone; 2, WT clone).
HTN-Cre-mediated PLRG1 deletion in vitro.
Recombinant His-TAT-NLS-Cre (HTNC) fusion protein was expressed and purified as described previously (40). Murine embryonic fibroblasts (MEFs) were incubated for 16 h with 5 μM sterile HTN-Cre dissolved in Dulbecco's modified Eagle's medium-phosphate-buffered saline (DMEM-PBS) (8). After 16 h of incubation, cells were washed with PBS, trypsinized, and replated for the respective experiments.
Animal care.
The care of all animal was within institutional animal care committee guidelines. All animal procedures were conducted in compliance with the protocols of and approval by local government authorities (Bezirksregierung Köln, Cologne, Germany) and were in accordance with NIH guidelines. Mice were housed in groups of three to five at 22 to 24°C using a 12-h light/12-h dark cycle.
Genotyping.
The genotyping of mice was performed on DNA obtained from tail biopsies. The detection of the conventional PLRG1 KO allele was performed using a three-primer PCR strategy with the primers 5′-CCTTCTCCATATTTAGCGTGG-3, 5′-TCTCTCTGCACCCTTCTGTTA-3 (wild-type), and 5′-CCTACCGGTGGATGTGGAATGTG-3 (KO). The genotyping of conditional PLRG1 alleles was performed using primers flanking the 3′ loxP site: 5′-TGTGATGGTGGCCGTATTGAT-3 and 5′-CTGTTCCAGCTGTTCTTCACA-3′. The deletion of the loxP-flanked fragment upon Cre-mediated recombination was detected by PCR analysis using primers 5′-TGTTATGTGCAGTGCCTTTCT-3 and 5′-GTCCTCTGTCCAAGCATATTTG-3′.
Analysis of PLRG1Δ/Δ embryos.
Embryos were dissected from the uterus at the indicated gestational age, photographed, and genotyped by a seminested PCR using the following primers for the first PCR: common 5′ primer, 5′-CCTTCTCCATATTTAGCGTGG-3′; wild-type 3′ primer, 5′-TCTCTCTGCACCCTTCTGTTA-3′; and knockout-specific 3′ primer, 5′-ATTTGTCACGTCCTGCACGACGC-3′. Primers for the second PCR were the following: common 5′ primer, 5′-CCTTCTCCATATTTAGCGTGG-3′; wild-type 3′ primer, 5′-CCTCTCTTCATCCAAAGGCAC-3′; and KO-specific 3′ primer, 5′-CCTACCGGTGGATGTGGAATGTG-3′.
Generation of PLRG1Δmus and PLRG1ΔCNS mice.
Muscle-specific creatinine kinase-Cre (MCKCre) mice were mated with PLRG1flox/flox mice, and a breeding colony was maintained by mating PLRG1flox/flox with PLRG1flox/+ MCKCre mice. SynCre mice were mated with PLRG1flox/flox mice, and a breeding colony was maintained by mating PLRG1flox/flox with PLRG1flox/+ SynCre mice. SynCre was always transmitted from females, as the transgene previously has been demonstrated to result in germ line deletion if transmitted via male germ cells (44).
RNA hybridization.
Mouse adult tissue (N1334447-BC) and embryonic-stage (R1011-SG) blots were purchased from BioCat. Blots were hybridized with a radiolabeled PLRG1 probe that had been amplified from mouse liver cDNA using the primers 5′-TGAATGAACTGCAGGACGAGGCA-3′ and 5′-GCCGCCAAGCTCTTCAGCAATAT-3′ as previously described (10).
Western blotting.
Indicated tissues were dissected and homogenized in homogenization buffer with a polytron homogenizer (IKA Werke), and Western blot analyses were performed by standard methods with antibodies raised against the respective antibodies as described previously (19, 22). HTNC-treated wild-type and PLRG1flox/flox fibroblasts were directly lysed in 2× sodium dodecyl sulfate (SDS) sample buffer containing 125 mM Tris-HCl (pH 6.8), 5% SDS, 43.5% glycerol, 100 mM dithiothreitol (DTT), and 0.02% bromophenol blue and were heated at 95°C for 5 min. Primary antibodies against Bcl-2 (N-19; sc 492), Bax (N-20, sc 493), p21 (C-19; sc 397), p53 (FL-393; sc 6243), and α-tubulin (TU-02; sc 8035) were purchased from Santa Cruz, p53 (phospho S15; GTX 21431) was from GeneTex, CDC5L (612362) was from BD Transduction Laboratories, Prp19 (ab27692) was from Abcam, phospho-S139-γ-H2AX (JBW 301) was from Upstate, β-actin (1616) was from Sigma, cleaved caspase 3 (9661) was from Cell Signaling, and PLRG1 antiserum was previously described (4). Secondary antibodies were goat anti-rabbit immunoglobulin G (IgG) (whole molecule) peroxidase, anti-goat/sheep IgG (whole molecule) peroxidase (Sigma), and anti-mouse IgG (whole molecule) peroxidase (Amersham).
Cell cycle analysis using FACS analysis.
Cells were collated by trypsin digestion and fixed in ice-cold 70% ethanol in PBS for at least 2 h. Cells were stained with propidium staining solution containing 0.1% Triton X-100 (Sigma), 200 μg/ml DNase-free RNase A (Sigma), and 20 μg/ml propidium iodide (Sigma) in PBS. Cells (25,000) were analyzed by fluorescence-activated cell sorter (FACS) (FACSCalibur; Becton-Dickinson Biosciences Immunocytometry Systems), and the proportion of cells in the G0/G1, G2/M, and S phases was measured. FACS analyses were performed in three independent experiments.
[3H]thymidine incorporation.
The assessment of cell proliferation and [3H]thymidine incorporation was performed as previously described (33).
Analysis of apoptosis.
To assess apoptosis, a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (DeadEnd fluoromteric TUNEL system; Promega) was used. HTNC-treated wild-type and PLRG1flox/flox fibroblasts were cultivated on glass coverslips in a 6-well plate for 3 days after Cre treatment. Thereafter, cells were fixed in 4% formaldehyde for 25 min at 4°C. TUNEL assays were performed according to the manufacturer's guidelines. The percentage of apoptotic cells was calculated as the number of TUNEL-positive cells per 4′,6′-diamidino-2-phenylindole (DAPI)-stained nucleus. To determine the amount of apoptotic cells, an annexin V assay (fluorescein isothiocyanate annexin V apoptosis detection kit I; BD Pharmingen) was used. The assays were performed according to the manufacturer's guidelines.
RT-PCR and quantitative real-time PCR.
RNA from MEFs was isolated using the RNeasy mini kit (Qiagen) according to the manufacturer's protocol. The DNase treatment of RNA was performed prior to reverse transcription-PCR (RT-PCR) using the RQ1 RNase-free DNase I endonuclease according to the manufacturer's guidelines (Promega). Two hundred nanograms of total RNA per reaction mixture was used for cDNA synthesis using the one-step RT qPCR Master Mix from Eurogentec. One microliter of cDNA was used for PCR with exon-spanning primers for cyclin D1, cyclin E, and p53. Quantitative real-time PCR was performed using the qPCR Master Mix Plus without UNG kit from Eurogentec with a TaqMan Principles ABI Prism 7700 sequence detection system for mRNAs Ddit3, Foxo1, Foxo3a, Notch1, TGF-β1, and Tuba1. The relative expression of mRNAs was determined using standard curves based on MEF cDNA. Samples were adjusted for total RNA content by TATA-binding protein RNA quantitative PCR. Calculations were performed by a comparative method (2−ΔΔCT).
Immunohistochemistry.
Brains were dissected from 2- to 3-day-old control and PLRG1ΔCNS mice and snap-frozen in tissue-freezing medium (Jung tissue-freezing medium; Leica Microsystems). Sections were performed on a cryostat. Brain slices were either stained with hemolysin and eosin (H&E) or used for TUNEL assays. For PLRG1Δmus mice, hearts were dissected from 3-week-old control and PLRG1Δmus mice, frozen in tissue-freezing medium (Jung tissue-freezing medium; Leica Microsystems), and sectioned on a cryostat. Sections were stained with H&E or processed for TUNEL assays.
RNAi.
The RNA interference (RNAi)-mediated knockdown of endogenous PLRG1 and p53 was performed using lipofection. PLRG1 small interfering RNA (siRNA) was purchased from Ambion (no. 16704), and p53 siRNA was purchased from Santa Cruz (sc-44219 and sc-29436). si-CONTROL from Dharmacon (D-001210-01) was used as the control siRNA. These siRNA duplexes (25 nM) were introduced into MEFs using Lipofectamine 2000 (Invitrogen) by following the manufacturer's guidelines. Forty-eight hours after transfection, cells were harvested and used for TUNEL assay and immunoblotting.
Nuclear extraction.
Cells (1 × 106) were resuspended in 15 μl buffer A containing 10 mM HEPES (pH 7.6), 10 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, and 1 tablet of proteinase inhibitor (Complete mini; Roche) and incubated for 10 min at 4°C. NP-40 was added to a final concentration of 1% and incubated at 4°C for 1 min. Cells were immediately collected by centrifugation at 13,000 rpm at 4°C for 1 min. The supernatant represented the cytoplasmic fraction. The pellet was washed with buffer A and resuspended in 10 μl buffer B containing 50 mM HEPES (pH 7.8), 50 mM KCl, 300 mM NaCl, 0.5 mM DTT, 0.1 mM EDTA, 10% glycerol, and 1 tablet of proteinase inhibitor (Complete mini; Roche). The pellet was incubated at 4°C on a full-speed shaker for 1 h. After incubation, the suspension was centrifuged at 13,000 rpm at 4°C for 1 h, and the supernatant contained the nuclear fraction.
Immunofluorescence.
MEFs were cultivated on glass coverslips in a 6-well plate for 2 days after Cre treatment. Thereafter, cells were fixed and permeabilized with 0.5% Triton X-100. Subsequently, fixed and permeabilized fibroblasts were blocked with 3% bovine serum albumin in PBS for 30 min at room temperature. Samples were incubated for 1 h at room temperature with the respective antibodies according to the manufacturer's guidelines. Samples then were washed with PBS and incubated for 45 min at room temperature with the respective 1:500-diluted secondary antibody (Alexa-Fluor 555 and 546; Invitrogen). Cells were stained with DAPI in Vectashield to visualize nuclei. The percentage of γ-H2AX-positive cells was calculated per DAPI-stained nucleus.
Targeted gene knockdown in zebrafish.
Embryos were obtained through natural crosses and staged as previously described (24). plrg1 morpholino-oligonucleotide (MO) (5′-AGTGCTTCTGCACGTCCTCGGTCAT-3′) and wrn MO (5′-ATTTAGGTAGAGTCCTGTCACCCAT-3′), targeting coding nucleotides 1 to 25 of plrg1 or wrn transcript, respectively, and p53 MO (5′-AGAATTGATTTTGCCGACCTCCTCT-3′) (42) were purchased from GeneTools (Philomath, OR), diluted in Danieau's buffer, and injected into zebrafish embryos at the one-cell stage at indicated concentrations as previously described (36). Acridine orange stainings of apoptotic cells were carried out as previously described (17). Photos were taken on a Zeiss Axiophot microscope with a Hamamadsu Orca charge-coupled-device camera, and fluorescent and bright-field images were superimposed using Openlab software (Improvision). Whole-mount in situ hybridization was performed as previously described (20).
Statistical methods.
Data were analyzed for statistical significance using a two-tailed unpaired Student's t test.
RESULTS
Embryonic lethality in PLRG1-deficient mice.
To gain first insights into the role of mammalian PLRG1, we determined the PLRG1 expression pattern in mice. Northern blot analysis of mRNA isolated during murine embryogenesis and from various organs of adult mice showed PLRG1 to be expressed throughout murine embryogenesis, peaking between days 12 and 15 postfertilization (Fig. 1A) in all adult organs analyzed, with high levels in thymus, testis, kidney, and spleen (Fig. 1B). This suggests a widespread role of PLRG1 in basic cellular processes.
We next generated mice with a targeted inactivation of the PLRG1 gene (Fig. 1C, D). By analyzing the offspring of heterozygous PLRG1Δ/+ intercrosses at different developmental stages, we could retrieve homozygous PLRG1Δ/Δ embryos only up to embryonic day 1.5 (E1.5) but not at later stages (Table 1). At E1.5, PLRG1-deficient embryos displayed cell lysis (n = 9/9), while wild-type and PLRG1Δ/+ embryos exhibited normal two-cell stage morphology (Fig. 2). In sum, these results reveal a crucial role for PLRG1 during the first cell division stage of embryogenesis.
TABLE 1.
Genotype analysis of PLRG1Δ/+ intercross offspring
| Day | No. of PLRG1Δ/+ × PLRG1Δ/+ mice with genotype:
|
Total | ||
|---|---|---|---|---|
| PLRG1+/+ | PLRG1Δ/+ | PLRG1Δ/Δ | ||
| E0.5 | 3 | 11 | 3 | 17 |
| E1.5 | 6 | 15 | 9 | 30 |
| E2.5 | 3 | 10 | 0 | 13 |
| E3.5 | 11 | 15 | 0 | 26 |
| E12.5 | 3 | 15 | 0 | 18 |
| E18.5 | 15 | 32 | 0 | 47 |
| P21a | 33 | 58 | 0 | 91 |
P21, postnatal day 21.
FIG. 2.
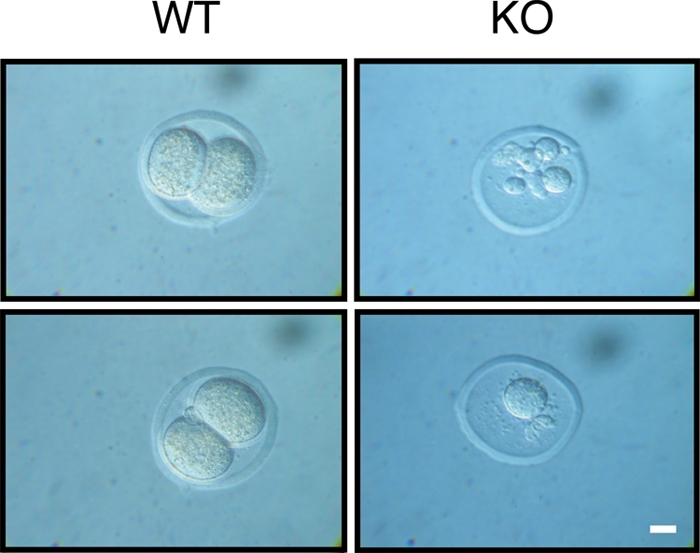
Embryonic lethality in PLRG1-deficient mice. Shown is a representative morphology of wild-type (WT) and PLRG1-KO (KO) embryos at E1.5. Wild-type embryos show a normal two-cell stage, whereas KO embryos are degenerated, showing failed division and fragmented nuclei. Scale bar, 10 μm.
PLRG1 deficiency in MEFs causes cell cycle arrest and apoptosis.
To circumvent the limitations arising from the early embryonic lethality of PLRG1-deficient embryos, we generated mice with the conditional, Cre-loxP-mediated inactivation of the PLRG1 gene, introducing loxP sites to flank exon 3 of the PLRG1 gene (Fig. 3A, B). To investigate the cellular defects caused by the loss of PLRG1 function in more detail, we established MEFs from control and PLRG1flox/flox embryos. Subsequent Cre treatment (40) resulted in the efficient excision and removal of exon 3 from the PLRG1flox/flox locus, leading to the absence of immunodetectable PLRG1 protein (Fig. 4A). Moreover, Cre treatment resulted in the immediate cessation of cell proliferation in PLRG1flox/flox MEFs, which is in line with the defects during the first cell cycle in conventional PLRG1 KO mice, whereas Cre treatment had no effect on cell proliferation in wild-type MEFs (Fig. 4B; also see Fig. S1 in the supplemental material). Taken together, these experiments clearly demonstrate that PLRG1 is essential for cell proliferation.
FIG. 4.

PLRG1 deficiency in MEFs causes cell cycle arrest. (A) Upper panel, genomic PCR analysis of wild-type (WT; 1 and 2) and PLRG1flox/flox MEFs (3) after treatment with cell-permeable Cre protein. Lower panel, Western blot analysis of PLRG1flox/flox MEFs before (1) and after (2) Cre treatment. (B) Growth rates of viable untreated or Cre-treated wild-type and PLRG1flox/flox (lox/lox) MEFs. Circles, wild-type MEFs; squares, PLRG1flox/flox MEFs; closed symbols, without Cre; open symbols, with Cre. Data represent means ± standard errors of the means of three independent experiments each performed in duplicate. (C) FACS analysis of serum (fetal calf serum [FCS])-stimulated cell cycle progression in untreated or Cre-treated wild-type and PLRG1flox/flox MEFs. Left panel, different cell cycle phases of untreated or Cre-treated wild-type and PLRG1flox/flox MEFs [lox/lox −Cre and lox/lox +Cre (KO)] before serum stimulation (−FCS) and 24 h after serum stimulation (+FCS) (the total number of cells counted for each condition was 25,000). Right panel, the percentage of the respective MEFs in different cell cycle phases before and after serum stimulation. Values represent means ± standard errors of the means from three independent experiments (*, P < 0.05). (D) [3H]thymidine incorporation after 0, 16, and 24 h of serum stimulation in untreated PLRG1flox/flox (open bars) and Cre-treated PLRG1flox/flox (closed bars) MEFs. Values represent means ± standard errors of the means from three independent experiments (*, P < 0.05).
To further define the role of PLRG1 in the control of cell proliferation, an analysis of serum-stimulated cell cycle progression was performed in wild-type and PLRG1flox/flox MEFs that either were left untreated or Cre treated by FACS analysis. Compared to levels for MEFs growing with serum (Fig. 4C), serum deprivation caused an accumulation of both wild-type and PLRG1-deficient cells in G1 phase. However, not all cells finished mitosis, indicating that the populations were not fully synchronized (Fig. 4C). Twenty-four hours after the addition of serum, wild-type cells left untreated or treated with cell-permeable, recombinant Cre, as well as untreated PLRG1flox/flox MEFs cells, displayed a significant reduction in the number of cells in G1 phase, while more cells were in S and M phase. In contrast, only Cre-treated PLRG1flox/flox MEFs did not respond to the serum, and the numbers of cells in G1 phase remained unaltered (Fig. 4C). Failed S-phase progression was further revealed by a lack of [3H]thymidine incorporation into the DNA of serum-stimulated PLRG1-deficient MEFs (Fig. 4D). These data demonstrate that PLRG1 is required for serum-stimulated cell cycle progression in murine cells.
To further characterize the nature of the cell cycle dysregulation caused by the PLRG1 mutation, we carried out Western blot analyses for Ser-15-phosphorylated p53, the stabilized and active version of the tumor suppressor p53 (35, 45, 50) that promotes cell cycle arrest and/or apoptosis (21, 48). This analysis revealed increased levels of p53 expression and phosphorylation in the absence of PLRG1 (Fig. 5A). Consistently with p53 stabilization in the absence of PLRG1, these MEFs also displayed increased apoptosis as assessed by TUNEL assay, accompanied by higher levels of the proapoptotic p53 target Bax and of proteolytically activated caspase 3, the key executing protease of apoptosis (15, 29) (Fig. 5B). Moreover, annexin V stainings confirmed increased apoptosis in the absence of PLRG1 (Fig. 5C, D). In summary, these experiments indicate that PLRG1 deficiency blocks cell cycle progression and stimulates apoptosis, possibly as a consequence of increased p53 levels and phosphorylation.
FIG. 5.

PLRG1 deficiency results in increased apoptosis. (A) Western blot analysis of untreated PLRG1flox/flox (Ctrl) and Cre-treated PLRG1flox/flox (KO) cells. MEFs were probed with the respective antibodies. β-Actin served as the loading control. (B) TUNEL analysis of control (Ctrl) and PLRG1-deficient MEFs. The percentage of apoptotic cells in control and PLRG1-deficient MEFs is shown. Values represent means ± standard errors of the means from three independent experiments (***, P < 0.001). Right panel, expression of Bax, Bcl-2, and active cleaved caspase 3 of control (Ctrl) and PLRG1-deleted (KO) MEFs. β-Actin served as the loading control. (C) Annexin V analysis of untreated (WT −Cre) or Cre-treated wild-type (WT +Cre) and PLRG1flox/flox [lox/lox −Cre and lox/lox +Cre (KO)] MEFs (the total number of cells counted for each condition was 25,000). (D) The percentage of annexin V-positive cells of untreated PLRG1flox/flox (Ctrl) and Cre-treated PLRG1flox/flox (KO) MEFs. Values represent means ± standard errors of the means from three independent experiments (***, P < 0.001). (E) Analysis of γ-H2AX phosphorylation in untreated (Ctrl) and Cre-treated PLRG1flox/flox MEFs (KO). Photomicrographs of phospho-S139-γ-H2AX-stained untreated (Ctrl) and PLRG1-deficient MEFs (KO) are shown. Scale bar, 100 μm. (F) Percentage of γ-H2AX-positive cells in untreated (Ctrl) and PLRG1-deficient MEFs (KO). Values represent means ± standard errors of the means from three independent experiments (***, P < 0.001). (G) Analysis of the colocalization of γ-H2AX and 53BP1 in control (Ctrl) and PLRG1-deficient MEFs (KO). Scale bar, 10 μm.
p53-dependent cell cycle arrest and/or apoptosis can result from different cellular insults, such as replication stress or DNA damage. Given the recent demonstration that PLRG1 interacts with the Werner helicase, we next stained control and PLRG1-deficient cells for γ-H2AX, a phosphorylated histone, as an indicator of DNA double-strand breaks and arrested replication forks. While in control cells γ-H2AX immunoreactivity was barely detectable, PLRG1-deficient cells exhibited massive accumulation of γ-H2AX immunoreactivity (Fig. 5E, F). Moreover, PLRG1 deficiency induced the nuclear accumulation of p53 binding protein 1 (53BP1) as a further marker of an activated DNA damage response that colocalized with γ-H2AX (Fig. 5G). Taking these results together, the deletion of PLRG1 induces a DNA damage response, resulting in p53 stabilization.
Cytoplasmic relocalization of CDC5L in PLRG1-deficient cells.
Since PLRG1-deficient cells underwent apoptosis and revealed increased γ-H2AX immunoreactivity as a marker of an activated DNA damage response, and since it has been demonstrated that some of the biological activities of PLRG1, including the control of pre-mRNA splicing and DNA repair, appear to depend on the formation of the nuclear complex, we next analyzed the expression and subcellular localization of CDC5L and Prp19 in the presence and absence of PLRG1. Western blot analysis revealed no difference in the total cellular content of CDC5L in control and PLRG1-deficient MEFs (Fig. 6A). However, in striking contrast to control cells, where CDC5L was almost exclusively detectable in the nuclear fraction, CDC5L was detectable only in the cytoplasm of PLRG1-deficient cells (Fig. 6A). Furthermore, immunocytochemistry with CDC5L antibodies revealed an almost exclusive nuclear staining pattern for CDC5L in control cells, while PLRG1-deficient cells exhibited a faint and diffuse cytoplasmic staining (Fig. 6A). In contrast to the cytoplasmic relocalization of CDC5L in PLRG1-deficient cells, Prp19 expression and subcellular localization remained unchanged in the absence of PLRG1 (Fig. 6B). Taken together, these experiments indicate that PLRG1 serves as a critical nuclear scaffold to retain CDC5L in the nucleus and to allow for the functional assembly of the nuclear Prp19/CDC5L/PLRG1 complex.
FIG. 6.

Interaction of PLRG1 and CDC5L. (A) Upper panel, Western blot analysis of immunoreactive CDC5L in nuclear, cytosolic, and whole-cell extracts (WCE) of control (Ctrl) and PLRG1-deficient (KO) MEFs. Similar results were obtained in three independent experiments. β-Actin and lamin A/C served as loading controls. Lower panel, immunofluorescent detection of CDC5L localization in control (Ctrl) and PLRG1-deficient (KO) MEFs. Bar, 10 μm. (B) Western blot analysis of immunoreactive Prp19 in nuclear, cytosolic, and whole-cell extracts (WCE) of control (Ctrl) and PLRG1-deficient (KO) MEFs. Similar results were obtained in three independent experiments. β-Actin and lamin A/C served as loading controls.
Previous mass spectrometry analyses of CDC5L-associated proteins have identified numerous spliceosomal components, including ASF/SF2, hnRNP-G, SAP145, and U2A, that are associated with PLRG1, and subsequent work has demonstrated a critical role for the PLRG1-CDC5L complex in the control of pre-mRNA splicing (2). Moreover, in yeast, the CDC5L homologue Prp46p regulates cell cycle progression via the control of tubulin splicing (11). Given the cytoplasmic relocalization of CDC5L in the absence of PLRG1, we next addressed the expression of tubulin in control and PLRG1-deficient MEFs. Our analysis revealed no detectable pre-mRNA accumulation and even increased the mRNA expression of tubulin in PLRG1-deficient cells compared to that of controls (see Fig. S2 in the supplemental material). Moreover, spliced mRNAs of nine other intron-containing genes that are essential for cell cycle progression or other fundamental cellular processes were found to be normally expressed in PLRG1-deficient MEFs (see Fig. S2 in the supplemental material). Taken together, these data indicate that the cytoplasmic relocalization of CDC5L in the absence of PLRG1 does not result in general impairment in pre-mRNA splicing.
Conditional inactivation of PLRG1 in heart cells and neurons causes apoptosis.
To further investigate the role of PLRG1 in postnatal tissue homeostasis, we generated mice lacking PLRG1 specifically in skeletal and heart muscle (PLRG1Δmus). To this end, PLRG1flox/+ mice were crossed with mice expressing the Cre recombinase under the control of the MCK promoter (10). In contrast to the conventional KO mice, PLRG1Δmus mice were detected at the expected Mendelian frequency at birth (data not shown). DNA analysis from individual tissues of control and PLRG1Δmus mice at postnatal day 5 revealed Cre-mediated recombination only in the heart (Fig. 7A). Presumably, Cre expression and activity in the skeletal muscle of MCKCre mice is initiated during later stages of development.
FIG. 7.

Conditional inactivation of PLRG1 in heart cells causes apoptosis. (A) Heart-restricted PLRG1 deficiency in PLRG1Δmus mice. PCR analysis on genomic DNA isolated from different organs of 5-day-old PLRG1flox/flox and PLRG1Δmus mice. The panel shows the specific deletion of PLRG1 in the heart using primers flanking exon 3. The excision of exon 3 is indicated by the PCR amplification of a 1-kb DNA fragment. PCR analysis with primers flanking the 3′ loxP site was used as a loading control (lanes: 1, brain; 2, skeletal muscle; 3, heart; 4, liver; 5, spleen). (B) Survival rates of control (open circles) (n = 40) and PLRG1Δmus (closed circles) (n = 14) mice. (C) Autopsy of postnatal day 24 wild-type (Ctrl) and PLRG1Δmus mice. Note the enlarged heart for PLRG1Δmus compared to that of the wild type. (D) Histological analysis of control (Ctrl) and PLRG1Δmus heart tissue at postnatal day 24. Upper and lower panels, H&E- and TUNEL-stained control and PLRG1Δmus heart sections, respectively. Note the clearly visible increased cardiomyocyte apoptosis in PLRG1Δmus hearts (lower). Scale bar, 100 μm. (E) Percentage of TUNEL-positive cells in hearts of PLRG1flox/flox (ctrl) and PLRG1Δmus mice. Values represent means ± standard errors of the means from three independent experiments (*, P < 0.05). (F) Western blot analysis of total cellular protein extracts from control (Ctrl) and PLRG1Δmus hearts probed with the respective antibodies. α-Tubulin served as a loading control. (G) Analysis of γ-H2AX phosphorylation in hearts of PLRG1flox/flox (Ctrl) and PLRG1Δmus mice. Scale bar, 20 μm. (H) Percentage of γ-H2AX positive cells in hearts of PLRG1flox/flox (Ctrl) and PLRG1Δmus mice. Values represent means ± standard errors of the means from three independent experiments (***, P < 0.001).
At 28 postnatal days, the survival rate of PLRG1Δmus mice was reduced to 5% (Fig. 7B), while it was 80% for their siblings, indicating that PLRG1 deficiency in heart muscle also is lethal (Table 2). Histological analysis at postnatal day 24 demonstrated a massive dilatation and atrophy of both ventricles in PLRG1Δmus mice, reflecting the pathology of severe dilated cardiomyopathy (Fig. 7C). This phenotype results from the dramatically increased apoptosis of mutant cardiomyocytes, as revealed by a significant increase of TUNEL-positive cells in mutant hearts, leading to reduced ejection force and subsequent blood retention in both the left and right ventricle, resulting in the macroscopic aspect of largely dilated hearts (Fig. 7C to E), which is consistent with the phenotype of other mouse models with increased cardiomyocyte death (51). In addition, levels of the transcriptional p53 target and the proapoptotic factor Bax were increased, while levels of the antiapoptotic factor Bcl-2 were lower (Fig. 7F) in PLRG1-deficient hearts. In sum, the data suggest that increased cardiomyocyte apoptosis causes the severe thinning of ventricle walls. Consistently with the results obtained in PLRG1-deficient MEFs, PLRG1-deficient hearts also exhibited massively increased γ-H2AX immunoreactivity as a marker of an activated DNA damage response in the hearts of these mice in vivo (Fig. 7G, H).
TABLE 2.
Genotype analysis of mice obtained from breedings of PLRG1flox/flox mice with either PLRG1flox/+ SynCre or PLRG1flox/+ MCKCre mice
| Postnatal day | No. of mice with genotype:
|
Total | |||||
|---|---|---|---|---|---|---|---|
| PLRG1flox/+ | PLRG1flox/flox | PLRG1flox/+ SynCre | PLRG1flox/+ MCKCre | PLRG1°CNS | PLRG1°mus | ||
| 21 | 8 | 8 | 13 | 0 | 29 | ||
| 28 | 10 | 19 | 16 | 0 | 45 | ||
We also generated PLRG1ΔCNS mice with neuron-restricted PLRG1 deficiency in the central nervous system (CNS) by intercrossing PLRG1flox/flox mice with mice expressing Cre recombinase under the control of the synapsin promoter (55). Genomic PCR analysis confirmed the CNS-restricted recombination of the PLRG1 gene in PLRG1ΔCNS mice (Fig. 8A). Compared to control mice (88%), PLRG1ΔCNS mice displayed strongly reduced survival rates (25%) at 3 days after birth, while no mutant survivors were found at day 9 (Fig. 8B). TUNEL staining revealed a dramatic increase in the number of apoptotic neurons in the dentate gyrus of mutant mice (Fig. 8C) (55). In addition, brain extracts of PLRG1ΔCNS mice showed increased levels of p53 and p53 Ser-15 phosphorylation and decreased levels of the antiapoptotic protein Bcl-2, while Bax levels remained unaltered (Fig. 8D). Thus, the absence of PLRG1 in the CNS leads to an apoptotic response in the hippocampus that is similar to that found in heart muscle or MEFs.
FIG. 8.

PLRG1 deficiency in the CNS reveals increased apoptosis. (A) Analysis of neuron-restricted recombination of the PLRG1flox/flox allele. The panel shows the specific deletion of PLRG1 in the brain of PLRG1ΔCNS mice. Using a primer pair flanking exon 3, the appearance of a product of 1 kb indicated the excision of exon 3. PCR analysis with primers flanking the 3′ loxP site was used as a loading control (lanes: 1, brain; 2, skeletal muscle; 3, heart; 4, liver; 5, spleen). (B) Survival rates of control (n = 25) and PLRG1ΔCNS (n = 12) mice at days 1, 3, and 9. (C) Immunohistochemical analysis of brains dissected from 3-day-old control (Ctrl) and PLRG1ΔCNS mice. Shown is DAPI (top; nucleus) and TUNEL (bottom) staining for control (Ctrl) and PLRG1ΔCNS tissue of the same brain sections. Scale bars, 100 μm (top) and 50 μm (bottom). (D) Western blot analysis of total cellular protein extracts from control (Ctrl) and PLRG1ΔCNS brain tissue probed with respective antibodies. β-Actin served as a loading control.
Apoptosis caused by PLRG1 deficiency is p53 dependent.
To study whether the increase in p53 activity causes the cell cycle and cell death defects in PLRG1-deficient cells, we carried out combined PLRG1-p53 loss-of-function experiments. For this purpose, wild-type MEFs were transfected with siRNA oligonucleotides directed against PLRG1 or p53 only or against PLRG1 along with p53. Western blot analyses revealed the successful siRNA-mediated silencing of PLRG1 and p53 (Fig. 9A). As for PLRG1-deficient MEFs, the siRNA-mediated knockdown of PLRG1 resulted in dramatically increased apoptosis in the presence of increased p53 expression and phosphorylation, as well as increased Bax and p21 expression (Fig. 9A, B). In contrast, the simultaneous downregulation of both PLRG1 and p53 expression restored cell survival, while Bax and p21 expression dropped to the same levels as those of the controls (Fig. 9A, B).
FIG. 9.

Apoptosis resulting from PLRG1 deficiency is p53 dependent. (A) Western blot analysis of MEFs left untreated or treated with control, PLRG1, p53, and PLRG1/p53 siRNA oligonucleotides and probed with the respective antibodies. β-Actin served as a loading control. (B) Determination of apoptosis by TUNEL assay in MEFs. The percentage of TUNEL-positive cells in MEFs that were left untransfected or were transfected with control, PLRG1, p53, and PLRG1/p53 siRNA oligonucleotides are shown. Values represent means ± standard errors of the means from three independent experiments (***, P < 0.001). (C) Effect of MO-mediated plrg1 knockdown in zebrafish. Upper panel, morphological consequence of plrg1 knockdown. The upper row of images are from phase-contrast microscopy, and the lower row of images show the acridine orange staining of apoptotic cells. Lower panel, summary of the occurrence of different degrees of morphological changes and the degree of apoptosis resulting from the injection of plrg1 MO or the simultaneous injection of plrg1/p53 MO at the indicated concentrations. Note the rescue of the plrg1 MO-induced phenotype by p53 MO.
To investigate whether p53 upregulation also accounts for the effect of PLRG1 deficiency in vivo, we suppressed both genes in zebrafish embryos. By searching zebrafish genomic databases we identified a gene (GenBank accession number NM_213440) with a deduced amino acid sequence that is 77.9% identical to that of mouse PLRG1, indicating that it is the zebrafish plrg1 ortholog. Whole-mount in situ hybridization revealed the ubiquitous distribution of zebrafish plrg1 transcripts during all investigated stages, i.e., from the one-cell stage to the larval stages, indicating that the gene product is both maternally and zygotically derived (http://zfin.org). The inactivation of zebrafish plrg1 with an antisense MO targeting the plrg1 translation initiation site led to dose-dependent severe and widespread apoptosis that already was apparent at early segmentation stages (12 h postfertilization) (Fig. 9C). Morphologically, the phenotype of most severely affected embryos was slightly stronger than that of zebrafish plrg1 mutants (6) (http://zfin.org). This indicates that in mutants, defects are partly rescued by maternally supplied plrg1 transcripts, which, however, are inactivated along with the zygotic transcripts by MO injection. However, MO treatment does not affect maternally derived Plrg1 protein, which would explain why the defects of zebrafish morphants develop later than those of PLRG1 mouse mutants. Importantly, the concomitant inactivation of zebrafish p53 in plrg1 morphants via the coinjection of plrg1 and p53 MOs (26) led to a significant attenuation of apoptosis and morphological defects (Fig. 9C), adding further in vivo evidence that apoptosis resulting from plrg1 deficiency is indeed p53 dependent.
DISCUSSION
Our experiments reveal an essential role for PLRG1 in the development of the preimplantation mouse embryo and the early zebrafish embryo. The death of PLRG1-deficient mouse embryos occurs during the first cell cycle, which is earlier than that of mutants in most other thus-far-described house-keeping genes, pointing to a pivotal role in regulating basic cellular processes. It further indicates a very short half-life of PLRG1 mRNA and protein, as maternal PLRG1 from the oocyte apparently cannot compensate for the lack of zygotic gene products even during the first day of embryogenesis. This notion is consistent with the fact that in PLRG1flox/flox MEFs, the amount of immunodetectable PLRG1 already is largely reduced after overnight incubation with the cell-permeable Cre protein (A. Kleinridders and J. C. Brüning, unpublished data).
PLRG1 initially was identified as a subunit of spliceosomal complexes (2). Also, in yeast it has been demonstrated that the PLRG-1 orthologue Prp46p is essential for pre-mRNA splicing (5). Moreover, prp46 mutants exhibit a defect in cell cycle progression. While PLRG1 deficiency in mice results in a failure to progress through the cell cycle, including S phase, the inactivation of yeast Prp46p results in impaired mitosis with a G2/M block (5). Biochemical and genetic studies demonstrate that Prp46p interacts with other components of the spliceosome, such as Prp45p, Prp19 (Pso4), and Cef1p/Cdc5p (38). Mutations of these spliceosomal components result in pleiotropic defects, including temperature sensitivity, enhanced sensitivity to mutagens and radiation, and the accumulation of pre-mRNA (38). Although the molecular mechanisms resulting in cell cycle defects as a consequence of the deletion of spliceosomal components have not been fully characterized, previous experiments in yeast demonstrated that defective pre-mRNA splicing directly blocks mitosis. Thus, the altered splicing of a single intron from the TUB1 α-tubulin gene caused by the CDC5 mutation results in the G2/M cell cycle defect in fission yeast (11). Moreover, besides the direct effect of the impaired splicing of critical cell cycle regulators, stalled splicing may lead to the rehybridization of nascent transcripts with the DNA template and the introduction of DNA breaks and genomic instability that ultimately leads to cell death (27).
Also in mammals, PLRG1 interacts with the SNEV/Pso4/prp19 protein (2, 32). Interestingly, the disruption of SNEV in mice also results in early embryonic lethality at E3.5 (16). Similarly, the KO of basic splicing factors like SRp20 (23) or the hnRNP complex component C (52) leads to embryonic death, although it does so at later stages of development than KO of PLRG1 (E3.5 or E6.5, respectively, compared to E1.5 for KO of PLRG1). Again, this might be due to a shorter half-life of the PLRG1 mRNA and/or protein. However, the earlier lethality of PLRG1-deficient mice compared to that of mice lacking essential components of the spliceosome might point toward additional roles of PLRG1 that are independent of pre-mRNA processing and splicing. This is further supported by findings that the splicing of multiple investigated RNAs, including those of critical cell cycle regulators such as p53, cyclin D1, and cyclin E1, as well as tubulin, appeared normal in PLRG1-deficient MEFs (see Fig. S2 in the supplemental material). The unaltered expression of numerous intron-containing genes in the absence of PLRG1, when CDC5L translocates to the cytoplasm, is surprising in light of the previous demonstration that inhibiting PLRG1-CDC5L interaction interferes with pre-mRNA splicing (3). Although our experiments clearly cannot rule out that PLRG1 is required for the control of the pre-mRNA splicing of other specific genes, they indicate that the general impairment of pre-mRNA likely does not account for the observed phenotype in PLRG1-deficient cells. Further experiments clearly will have to address (i) the molecular basis for the PLRG1-controled subcellular localization of CDC5L and (ii) the functional consequences of the cytoplasmic relocalization of CDC5L.
Moreover, also for SNEV, an indispensable, more direct role during DNA repair has been discussed as a possible alternative explanation for the early lethality of mutant mice (16). The recently discovered interaction of the CDC5L-PLRG1 complex with the Werner (WRN) helicase adds further support for the discussed essential role of PLRG1 during DNA metabolism, as WRN binds different components of the DNA replication machinery and is involved in the regression of replication forks after replication arrest or DNA damage during S phase (41) (46). Similarly, deletions of important DNA metabolism genes such as ATR, Chk1, NBS1, BRCA1, BRCA2, and Rad51 lead to early embryonic lethality similar to that of PLRG1 mutants (9, 13, 47, 49, 54).
To study a possible function of PLRG1 during DNA metabolism, we performed more-detailed analyses in PLRG1-deficient MEFs (in vitro) and after tissue-specific conditional PLRG1 inactivation (in vivo). Indeed, in PLRG1-deficient cells and tissues, numbers of γ-H2AX foci at sites of an activated DNA damage response are dramatically increased. Moreover, FACS analysis revealed a normal distribution of mutant cells throughout the different cell cycle phases, indicating that cells become arrested in all phases rather than accumulating at a particular transition. This further supports a role of PLRG1 during DNA metabolism that is not restricted to particular phases of the cell cycle, such as DNA repair. Also consistent with a role of PLRG1 in genomic integrity is the finding that PLRG1-deficient cells displayed increased levels of p53 protein, which is stabilized upon phosphorylation by DNA damage-sensing kinases such as Chk1 (7).
Thus, the further characterization of the functional interactions between PLRG1, CDC5L, SNEV, WRN, and possible additional partners will help to decipher the regulatory links between common components of splicing and DNA metabolism and may yet uncover functions in age-related diseases and malignant transformation.
Supplementary Material
Acknowledgments
We thank Paul Ajuh and Angus Lamond for generously providing the PLRG1 antiserum and Gisela Schmall for secretarial assistance. We thank Christian Kubisch and Thomas Wunderlich for helpful advice and discussion during the course of the experiments.
This work was supported by grants from the Deutsche Forschungsgemeinschaft to J.C.B. (Br 1492/7; SFB635) and C.K. (SFB635).
Footnotes
Published ahead of print on 23 March 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Agrelo, R., W. H. Cheng, F. Setien, S. Ropero, J. Espada, M. F. Fraga, M. Herranz, M. F. Paz, M. Sanchez-Cespedes, M. J. Artiga, D. Guerrero, A. Castells, C. von Kobbe, V. A. Bohr, and M. Esteller. 2006. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc. Natl. Acad. Sci. USA 1038822-8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ajuh, P., B. Kuster, K. Panov, J. C. Zomerdijk, M. Mann, and A. I. Lamond. 2000. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 196569-6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajuh, P., and A. I. Lamond. 2003. Identification of peptide inhibitors of pre-mRNA splicing derived from the essential interaction domains of CDC5L and PLRG1. Nucleic Acids Res. 316104-6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ajuh, P., J. Sleeman, J. Chusainow, and A. I. Lamond. 2001. A direct interaction between the carboxyl-terminal region of CDC5L and the WD40 domain of PLRG1 is essential for pre-mRNA splicing. J. Biol. Chem. 27642370-42381. [DOI] [PubMed] [Google Scholar]
- 5.Albers, M., A. Diment, M. Muraru, C. S. Russell, and J. D. Beggs. 2003. Identification and characterization of Prp45p and Prp46p, essential pre-mRNA splicing factors. RNA 9138-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amsterdam, A., R. M. Nissen, Z. Sun, E. C. Swindell, S. Farrington, and N. Hopkins. 2004. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 10112792-12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartek, J., and J. Lukas. 2003. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3421-429. [DOI] [PubMed] [Google Scholar]
- 8.Baumgartl, J., S. Baudler, M. Scherner, V. Babaev, L. Makowski, J. Suttles, M. McDuffie, K. Tobe, T. Kadowaki, S. Fazio, C. R. Kahn, G. S. Hotamisligil, W. Krone, M. Linton, and J. C. Bruning. 2006. Myeloid lineage cell-restricted insulin resistance protects apolipoprotein E-deficient mice against atherosclerosis. Cell Metab. 3247-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown, E. J., and D. Baltimore. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14397-402. [PMC free article] [PubMed] [Google Scholar]
- 10.Brüning, J. C., M. D. Michael, J. N. Winnay, T. Hayashi, D. Horsch, D. Accili, L. J. Goodyear, and C. R. Kahn. 1998. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2559-569. [DOI] [PubMed] [Google Scholar]
- 11.Burns, C. G., R. Ohi, S. Mehta, E. T. O'Toole, M. Winey, T. A. Clark, C. W. Sugnet, M. Ares, Jr., and K. L. Gould. 2002. Removal of a single alpha-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in Saccharomyces cerevisiae. Mol. Cell. Biol. 22801-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng, W. H., M. Muftuoglu, and V. A. Bohr. 2007. Werner syndrome protein: functions in the response to DNA damage and replication stress in S-phase. Exp. Gerontol. 42871-878. [DOI] [PubMed] [Google Scholar]
- 13.Deng, C. X., and S. G. Brodie. 2001. Knockout mouse models and mammary tumorigenesis. Semin. Cancer Biol. 11387-394. [DOI] [PubMed] [Google Scholar]
- 14.Eggan, K., H. Akutsu, J. Loring, L. Jackson-Grusby, M. Klemm, W. M. Rideout III, R. Yanagimachi, and R. Jaenisch. 2001. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc. Natl. Acad. Sci. USA 986209-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandes-Alnemri, T., G. Litwack, and E. S. Alnemri. 1994. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J. Biol. Chem. 26930761-30764. [PubMed] [Google Scholar]
- 16.Fortschegger, K., B. Wagner, R. Voglauer, H. Katinger, M. Sibilia, and J. Grillari. 2007. Early embryonic lethality of mice lacking the essential protein SNEV. Mol. Cell. Biol. 273123-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furutani-Seiki, M., Y. J. Jiang, M. Brand, C. P. Heisenberg, C. Houart, D. Beuchle, F. J. van Eeden, M. Granato, P. Haffter, M. Hammerschmidt, D. A. Kane, R. N. Kelsh, M. C. Mullins, J. Odenthal, and C. Nusslein-Volhard. 1996. Neural degeneration mutants in the zebrafish, Danio rerio. Development 123229-239. [DOI] [PubMed] [Google Scholar]
- 18.Futami, K., Y. Ishikawa, M. Goto, Y. Furuichi, and M. Sugimoto. 2008. Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells. Cancer Sci. 99843-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gropp, E., M. Shanabrough, E. Borok, A. W. Xu, R. Janoschek, T. Buch, L. Plum, N. Balthasar, B. Hampel, A. Waisman, G. S. Barsh, T. L. Horvath, and J. C. Bruning. 2005. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 81289-1291. [DOI] [PubMed] [Google Scholar]
- 20.Hammerschmidt, M., F. Pelegri, M. C. Mullins, D. A. Kane, F. J. M. van Eeden, M. Granato, M. Brand, M. Furutani-Seiki, P. Haffter, C.-P. Heisenberg, Y.-J. Jiang, R. N. Kelsh, J. Odenthal, R. M. Warga, and C. Nüsslein-Volhard. 1996. dino and mercedes, two genes regulating dorsal development in the zebrafish embryo. Development 12395-102. [DOI] [PubMed] [Google Scholar]
- 21.Haupt, S., M. Berger, Z. Goldberg, and Y. Haupt. 2003. Apoptosis—the p53 network. J. Cell Sci. 1164077-4085. [DOI] [PubMed] [Google Scholar]
- 22.Janoschek, R., L. Plum, L. Koch, H. Munzberg, S. Diano, M. Shanabrough, W. Muller, T. L. Horvath, and J. C. Bruning. 2006. gp130 signaling in proopiomelanocortin neurons mediates the acute anorectic response to centrally applied ciliary neurotrophic factor. Proc. Natl. Acad. Sci. USA 10310707-10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jumaa, H., G. Wei, and P. J. Nielsen. 1999. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr. Biol. 9899-902. [DOI] [PubMed] [Google Scholar]
- 24.Kimmel, C. B., W. W. Ballard, S. R. Kimmel, B. Ullmann, and T. F. Schilling. 1995. Stages of embryonic development of the zebrafish. Dev. Dynamics 203253-310. [DOI] [PubMed] [Google Scholar]
- 25.Kühn, R., and R. M. Torres. 2002. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 180175-204. [DOI] [PubMed] [Google Scholar]
- 26.Langheinrich, U., E. Hennen, G. Stott, and G. Vacun. 2002. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr. Biol. 122023-2028. [DOI] [PubMed] [Google Scholar]
- 27.Li, X., and J. L. Manley. 2006. Cotranscriptional processes and their influence on genome stability. Genes Dev. 201838-1847. [DOI] [PubMed] [Google Scholar]
- 28.Lombard, D. B., and L. Guarente. 1996. Cloning the gene for Werner syndrome: a disease with many symptoms of premature aging. Trends Genet. 12283-286. [DOI] [PubMed] [Google Scholar]
- 29.MacFarlane, M., K. Cain, X. M. Sun, E. S. Alnemri, and G. M. Cohen. 1997. Processing/activation of at least four interleukin-1beta converting enzyme-like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J. Cell Biol. 137469-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machwe, A., L. Xiao, J. Groden, and D. K. Orren. 2006. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry 4513939-13946. [DOI] [PubMed] [Google Scholar]
- 31.Machwe, A., L. Xiao, R. G. Lloyd, E. Bolt, and D. K. Orren. 2007. Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 355729-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova, O. V., E. M. Makarov, H. Urlaub, C. L. Will, M. Gentzel, M. Wilm, and R. Luhrmann. 2004. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 232381-2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merkwirth, C., S. Dargazanli, T. Tatsuta, S. Geimer, B. Lower, F. T. Wunderlich, J. C. von Kleist-Retzow, A. Waisman, B. Westermann, and T. Langer. 2008. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22476-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata, K., A. Hatamochi, H. Shinkai, Y. Ishikawa, N. Kawaguchi, and M. Goto. 1999. A case of Werner's syndrome associated with osteosarcoma. J. Dermatol. 26682-686. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa, K., Y. Taya, K. Tamai, and M. Yamaizumi. 1999. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol. Cell. Biol. 192828-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nasevicius, A., and S. C. Ekker. 2000. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26216-220. [DOI] [PubMed] [Google Scholar]
- 37.Németh, K., K. Salchert, P. Putnoky, R. Bhalerao, Z. Koncz-Kalman, B. Stankovic-Stangeland, L. Bako, J. Mathur, L. Okresz, S. Stabel, P. Geigenberger, M. Stitt, G. P. Redei, J. Schell, and C. Koncz. 1998. Pleiotropic control of glucose and hormone responses by PRL1, a nuclear WD protein, in Arabidopsis. Genes Dev. 123059-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohi, M. D., and K. L. Gould. 2002. Characterization of interactions among the Cef1p-Prp19p-associated splicing complex. RNA 8798-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palma, K., Q. Zhao, Y. T. Cheng, D. Bi, J. Monaghan, W. Cheng, Y. Zhang, and X. Li. 2007. Regulation of plant innate immunity by three proteins in a complex conserved across the plant and animal kingdoms. Genes Dev. 211484-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peitz, M., K. Pfannkuche, K. Rajewsky, and F. Edenhofer. 2002. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. USA 994489-4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pichierri, P., A. Franchitto, P. Mosesso, and F. Palitti. 2001. Werner's syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Biol. Cell 122412-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plaster, N., C. Sonntag, C. E. Busse, and M. Hammerschmidt. 2006. p53 deficiency rescues apoptosis and differentiation of multiple cell types in zebrafish flathead mutants deficient for zygotic DNA polymerase d1. Cell Death. Diff. 13223-235. [DOI] [PubMed] [Google Scholar]
- 43.Rao, V. A., C. Conti, J. Guirouilh-Barbat, A. Nakamura, Z. H. Miao, S. L. Davies, B. Sacca, I. D. Hickson, A. Bensimon, and Y. Pommier. 2007. Endogenous gamma-H2AX-ATM-Chk2 checkpoint activation in Bloom's syndrome helicase deficient cells is related to DNA replication arrested forks. Mol. Cancer Res. 5713-724. [DOI] [PubMed] [Google Scholar]
- 44.Rempe, D., G. Vangeison, J. Hamilton, Y. Li, M. Jepson, and H. J. Federoff. 2006. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis 4444-49. [DOI] [PubMed] [Google Scholar]
- 45.She, Q. B., N. Chen, and Z. Dong. 2000. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 27520444-20449. [DOI] [PubMed] [Google Scholar]
- 46.Sidorova, J. M., N. Li, A. Folch, and R. J. Monnat, Jr. 2008. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7796-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takai, H., K. Tominaga, N. Motoyama, Y. A. Minamishima, H. Nagahama, T. Tsukiyama, K. Ikeda, K. Nakayama, and M. Nakanishi. 2000. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 141439-1447. [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor, W. R., and G. R. Stark. 2001. Regulation of the G2/M transition by p53. Oncogene 201803-1815. [DOI] [PubMed] [Google Scholar]
- 49.Tsuzuki, T., Y. Fujii, K. Sakumi, Y. Tominaga, K. Nakao, M. Sekiguchi, A. Matsushiro, Y. Yoshimura, and T. Morita. 1996. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 936236-6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vega, F. M., A. Sevilla, and P. A. Lazo. 2004. p53 stabilization and accumulation induced by human vaccinia-related kinase 1. Mol. Cell. Biol. 2410366-10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang, J., H. Wilhelmsson, C. Graff, H. Li, A. Oldfors, P. Rustin, J. C. Bruning, C. R. Kahn, D. A. Clayton, G. S. Barsh, P. Thoren, and N. G. Larsson. 1999. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 21133-137. [DOI] [PubMed] [Google Scholar]
- 52.Williamson, D. J., S. Banik-Maiti, J. DeGregori, and H. E. Ruley. 2000. hnRNP C is required for postimplantation mouse development but is dispensable for cell viability. Mol. Cell. Biol. 204094-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, N., R. Kaur, X. Lu, X. Shen, L. Li, and R. J. Legerski. 2005. The Pso4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 28040559-40567. [DOI] [PubMed] [Google Scholar]
- 54.Zhu, J., S. Petersen, L. Tessarollo, and A. Nussenzweig. 2001. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 11105-109. [DOI] [PubMed] [Google Scholar]
- 55.Zhu, Y., M. I. Romero, P. Ghosh, Z. Ye, P. Charnay, E. J. Rushing, J. D. Marth, and L. F. Parada. 2001. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 15859-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.