

Abstract
Although the roles of Jak-Stat pathways in type I and II interferon (IFN)-dependent transcriptional regulation are well established, the precise mechanisms of mRNA translation for IFN-sensitive genes remain to be defined. We examined the effects of IFNs on the phosphorylation/activation of eukaryotic translation initiation factor 4B (eIF4B). Our data show that eIF4B is phosphorylated on Ser422 during treatment of sensitive cells with alpha IFN (IFN-α) or IFN-γ. Such phosphorylation is regulated, in a cell type-specific manner, by either the p70 S6 kinase (S6K) or the p90 ribosomal protein S6K (RSK) and results in enhanced interaction of the protein with eIF3A (p170/eIF3A) and increased associated ATPase activity. Our data also demonstrate that IFN-inducible eIF4B activity and IFN-stimulated gene 15 protein (ISG15) or IFN-γ-inducible chemokine CXCL-10 protein expression are diminished in S6k1/S6k2 double-knockout mouse embryonic fibroblasts. In addition, IFN-α-inducible ISG15 protein expression is blocked by eIF4B or eIF3A knockdown, establishing a requirement for these proteins in mRNA translation/protein expression by IFNs. Importantly, the generation of IFN-dependent growth inhibitory effects on primitive leukemic progenitors is dependent on activation of the S6K/eIF4B or RSK/eIF4B pathway. Taken together, our findings establish critical roles for S6K and RSK in the induction of IFN-dependent biological effects and define a key regulatory role for eIF4B as a common mediator and integrator of IFN-generated signals from these kinases.
Extensive work over the years has established that the control of initiation of mRNA translation occurs primarily at the step at which the 40S ribosomal subunit is recruited to mRNA, to be positioned at the initiation codon (15). Most eukaryotic mRNAs contain a 5′ cap structure (m7GpppN) to which the cap-binding protein complex eukaryotic initiation factor 4F (eIF4F) is attached. eIF4F is composed of three subunits: eIF4E, the cap-binding subunit; eIF4A, a protein with RNA helicase activity that unwinds the mRNA 5′ secondary structure; and eIF4G, a scaffolding protein that associates with other IFs (15, 18, 20, 37, 54). The unphosphorylated/activated form of the translational repressor 4E-BP1 (eIF4E-binding protein 1) competes with eIF4G for binding to eIF4E and blocks cap-dependent mRNA translation (37), while such 4E-BP1-eIF4E interactions are decreased when phosphorylation of 4E-BP1 occurs by the mammalian target of rapamycin (mTOR) kinase (15, 18, 20, 37, 54). Beyond phosphorylation of 4E-BP1, mTOR regulates activation of the p70 S6 kinase (S6K), which in turn phosphorylates several substrates, including the S6 ribosomal protein (rpS6), eIF4B, and the tumor suppressor PDCD4 (13, 18, 20, 54).
Although much is known about the roles of mTOR-generated signals in the control of mRNA translation for cytokines and growth factors, the roles of various mTOR effectors in the initiation of mRNA translation in response to interferons (IFNs) remain to be precisely defined. Type I (α, β, ɛ, κ, and ω) and II (γ) IFNs constitute the first line of host antiviral defense and are key elements of the immune surveillance against malignant cells (reviewed in references 8, 39, 42, 43, and 50). Extensive work over the last decade has established key and essential roles for Jak-Stat pathways in both type I and II IFN signaling via control of transcriptional regulation of IFN-stimulated genes (ISGs) that leads to the generation of protein products that mediate the biological effects of IFNs (8, 30, 42, 43, 48, 50). There is also evidence for important contributions of other pathways, including the phosphatidylinositol 3′-kinase and the p38 mitogen-activated signaling cascades (27, 31, 41, 42, 51).
In previous work, we provided evidence that Akt and mTOR are activated in a type I and II IFN-inducible manner, suggesting the existence of IFN-activated cascades that control the initiation of mRNA translation (24, 25, 28, 29). In fact, we were recently able to directly demonstrate that mRNA translation for ISGs is defective in cells with targeted disruption of both the Akt1 and Akt2 genes (24). Thus, our data have suggested a critical role for mTOR-induced signals in the control of IFN responses. But these findings have also raised important new questions. A key outstanding issue has been the definition of the contributions of distinct mTOR effectors in the generation of the biological properties of IFNs. Addressing this issue and dissecting the roles of different components of the mTOR pathway in the induction of IFN biological responses is of importance, as it could ultimately lead to the definition of cellular targets for the design of novel antiviral or antitumor therapeutic agents.
In the current study, we provide the first evidence directly implicating eIF4B in IFN signaling. Our data demonstrate that eIF4B is a substrate for both S6K and RSK and that it is phosphorylated on Ser422 in an IFN-inducible manner. Such phosphorylation results in enhanced interaction of eIF4B with p170/eIF3A and plays an important role in mRNA translation of ISGs. In addition, our data establish a key role for RSK-mediated signals in the generation of the biological effects of IFNs, underscoring the importance of this pathway in IFN signaling.
MATERIALS AND METHODS
Constructs, cells, and reagents.
KT1 and NB4 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. Immortalized mouse embryonic fibroblasts (MEFs) from S6k1−/− S6k2−/− mice (38) and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with fetal calf serum and antibiotics. Recombinant human IFN-α was provided by Hoffman La Roche, Inc. Recombinant mouse IFN-γ was obtained from PBL Biomedical Laboratories (Piscataway, NJ). Recombinant human IFN-γ was provided by InterMune, Inc. A rabbit polyclonal antibody against mouse ISG15 has been previously described (24). Antibodies against eIF4B, rpS6, Akt, and PDK1 and antibodies against the phosphorylated forms of eIF4B, rpS6, S6K, extracellular signal-regulated kinase 1/2 (ERK1/2), and RSK were obtained from Cell Signaling Technology (Beverly, MA). An anti-p170/eIF3A antibody was from Abnova Corporation (Taipei City, Taiwan). Antibodies against ERK1/2, p70S6K and anti-RSK1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Small interfering RNAs (siRNAs) specific for RSK1, S6K, and p170/eIF3A were from Santa Cruz Biotechnology (Santa Cruz, CA). siRNA specific for eIF4B and anti-Flag antibody were from Cell Signaling Technology (Beverly, MA). Flag-tagged eIF4B, subcloned in the pCDNA3 vector (44), was provided by John Blenis (Harvard Medical School, Boston, MA). The FRAP/mTOR inhibitor, rapamycin, and the MEK1/2 inhibitor U0126 were obtained from Calbiochem, Inc. (La Jolla, CA). The CalPhos mammalian transfection kit was from Clontech (Mountain View, CA).
Immunoprecipitations and immunoblotting.
Cells were treated with 104 IU/ml of IFN-α or 5 × 103 IU/ml of IFN-γ and lysed in phosphorylation lysis buffer (PLB) as previously described (52, 53). Immunoprecipitations and immunoblotting using an enhanced chemiluminescence method were performed as previously described (52, 53).
Quantitative RT-PCR (TaqMan).
Cells were treated with 5,000 IU/ml of IFN-α or 2,500 IU/ml of IFN-γ for 6 h, and total RNA was isolated by using an RNeasy kit (Qiagen). Real-time reverse transcription-PCR (RT-PCR) for the Isg15 and IP10 genes was performed as previously described (24, 25).
Hematopoietic progenitor assays in methylcellulose.
Clonogenic assays in methylcellulose to detect leukemic CFU-blast (CFU-L) colony formation from KT1 cells were performed essentially as previously described (5). Assays for the effects of IFN-α on CFU-L colony formation from KT1 cells transfected with control siRNA or siRNAs specific for eIF4B or RSK1 were performed essentially as previously described (5, 16, 23, 33).
ATPase assays.
Cells were incubated in the presence or absence of IFN-α, and cell lysates were immunoprecipitated with an anti-eIF4B antibody or control non-immune rabbit immunoglobulin G (RIgG) using protein G-Sepharose beads. The beads were subsequently washed five times with PLB and two times with the ATPase buffer [15 mM HEPES-KOH (pH 7.5), 80 mM KCl, 2.5 mM Mg(CH3CO2)2, 1 mM dithiothreitol], and assays to determine ATPase activity in the immunoprecipitates were performed essentially as previously described (16). Values measured in anti-eIF4B immunoprecipitates were normalized by subtracting nonspecific activities detected in control RIgG immunoprecipitates.
In vitro kinase assays.
Assays to detect the IFN-dependent activation of the RSK1 kinase were performed essentially as previously described (21). For the experiments in which Flag-eIF4B was used as a substrate, 293T cells were transfected with Flag-tagged eIF4B. Overexpressed eIF4B was then immunoprecipitated with an anti-Flag antibody, and immune complexes were collected by using protein G-Sepharose beads. The immunoprecipitated eIF4B was subsequently used as a substrate in the immune complex kinases in anti-RSK1 immunoprecipitates, as in the case of troponin.
Isolation of polysomal RNA and quantitative RT-PCR.
S6k wild type and S6k1−/− S6k2−/− MEFs were incubated for 24 h in DMEM with 0.5% fetal calf serum and then left untreated or treated with mouse IFN-α for 18 h, and isolation of polysomal RNA and quantitative RT-PCR on the polysomal fractions were performed as previously described (24). Total polysomal RNA for each experimental condition was quantitated, and equal amounts of RNA were reverse-transcribed into cDNA by using an Omniscript RT kit and oligo(dT) primers (Qiagen). Real-time PCR for the Isg15 gene was conducted by using commercially available 6-carboxyfluorescein (FAM)-labeled probes and primers (Applied Biosystems), and the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene was used for normalization. mRNA amplification was determined as previously described (24, 25), and relative quantitation of mRNA levels was plotted as the level of increase compared to the result for untreated samples.
RESULTS
In initial studies, we sought to determine whether IFN-α and/or IFN-γ phosphorylates/activates eIF4B, a protein that acts as a substrate for the kinase activity of S6K in other systems (44, 49). KT1 cells were incubated for different times in the presence or absence of IFN-α, and total cell lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with an antibody against the phosphorylated form of eIF4B on Ser422. IFN-α induced strong phosphorylation of eIF4B (Fig. 1A), while there were no differences in the total eIF4B protein levels after IFN treatment (Fig. 1A). Similarly, treatment of sensitive NB4 cells (1) with IFN-γ also resulted in phosphorylation of eIF4B (Fig. 1B), suggesting that eIF4B is an element common to the signaling pathways of both type I and II IFNs.
FIG. 1.

IFN-α and IFN-γ induce phosphorylation of eIF4B. (A) Serum-starved KT1 cells were treated with IFN-α for the indicated times. Total cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of eIF4B on Ser422 or against eIF4B as indicated. (B) Serum-starved NB4 cells were treated with IFN-γ for the indicated times. Total cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of eIF4B on Ser422 or against eIF4B as indicated. (C and D) S6k1+/+ S6k2+/+ (wild type [wt]) and S6k1−/− S6k2−/− MEFs were incubated with IFN-α (C) or IFN-γ (D) for the indicated times. Total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-phospho-Ser422-eIF4B, anti-phospho-Ser240/244-rpS6, anti-eIF4B, or anti-rpS6 antibodies as indicated.
To understand the mechanisms of regulation of eIF4B phosphorylation by IFNs, we performed studies using MEFs from S6k1/S6k2 double-knockout mice (38). Both IFN-α (Fig. 1C) and IFN-γ (Fig. 1D) induced strong phosphorylation of eIF4B in S6k1+/+ S6k2+/+ MEFs, but such phosphorylation was defective in S6k1/S6k2 double-knockout MEFs (Fig. 1C and D). As expected, IFN-α-inducible (Fig. 1C) and IFN-γ-inducible (Fig. 1D) phosphorylation of rpS6 were also defective in the S6k1/S6k2 double-knockout MEFs (Fig. 1C and D), consistent with regulation of IFN-dependent phosphorylation of both eIF4B and rpS6 by the activated S6K.
As there is evidence that, beyond p70S6K, p90RSK, a kinase whose activation is regulated by the Ras/Erk pathway (3), also phosphorylates eIF4B on Ser422 in other systems (49), we sought to determine the effects of Mek/Erk inhibition on IFN-dependent phosphorylation of eIF4B. When wild-type MEFs were preincubated with the mTOR inhibitor rapamycin, we found that phosphorylation of eIF4B on Ser422 was blocked (Fig. 2A), consistent with the lack of phosphorylation seen in the S6k1/S6k2 double-knockout MEFs. On the other hand, IFN-α-dependent eIF4B phosphorylation was still detectable (Fig. 2A) in cells treated with the MEK inhibitor U0126, suggesting that the dominant kinase for eIF4B phosphorylation in MEFs is p70S6K. Similarly, rapamycin blocked the phosphorylation of the rpS6 on Ser240/244, while such phosphorylation was unaffected by U0126 (Fig. 2A). However, when similar experiments were performed in cells of hematopoietic origin, a different pattern was seen. As shown in Fig. 2B, IFN-α-dependent phosphorylation of eIF4B was not blocked by rapamycin in KT1 hematopoietic cells, but it was blocked by U0126 (Fig. 2B). On the other hand, phosphorylation of rpS6 on Ser240/244 was blocked only by rapamycin and not by U0126 (Fig. 2B). In these cells, IFN-α-treatment also resulted in phosphorylation/activation of p90RSK and Erk1/2 (Fig. 2B). The phosphorylation of both Erk1/2 and p90RSK was also blocked by MEK inhibition (Fig. 2B), suggesting that IFN-α-dependent p90RSK activation occurs downstream of the Mek/Erk pathway and leads to eIF4B phosphorylation.
FIG. 2.

Effects of mTOR and Mek/Erk inhibition on the IFN-α-inducible phosphorylation of eIF4B. (A) S6k1+/+ S6k2+/+ MEFs were pretreated for 60 min with rapamycin (15 nM) or U0126 (10 μM) and were subsequently treated with IFN-α in the continuous presence or absence of rapamycin or U0126, as indicated. The cells were lysed, and total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-phospho-Ser422-eIF4B, anti-phospho-Ser240/244-rpS6, anti-eIF4B, or anti-rpS6 antibodies as indicated. (B) Serum-starved KT1 cells were pretreated for 60 min with rapamycin or U0126 and were subsequently treated with IFN-α in the continuous presence or absence of rapamycin or U0126 as indicated. Total cell lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (C) KT1 cells were treated with IFN-α for the indicated times. After cell lysis, equal amounts of protein were immunoprecipitated with either an anti-RSK1 antibody or control non-immune RIgG. In vitro kinase assays to detect RSK activity were subsequently carried out on the immunoprecipitates, using troponin as an exogenous substrate. The blot from the kinase assay was subsequently immunoblotted with an anti-RSK1 antibody, to control for protein loading. IP, immunoprecipitation; +, present; −, absent. (D) In vitro kinase assays to detect RSK activity were subsequently carried out using recombinant eIF4B as substrate. KT1 cells were treated with IFN-α for the indicated times. After cell lysis, equal amounts of protein were immunoprecipitated with an anti-RSK1 antibody. The blot from the kinase assay was subsequently immunoblotted with an anti-RSK1 antibody and with an anti-Flag antibody to control for protein loading. IP, immunoprecipitation; +, present. (E) Serum-starved NB4 cells were pretreated for 60 min with rapamycin or U0126 as indicated. The cells were then treated with IFN-γ for the indicated times in the continuous presence or absence of rapamycin or U0126, as indicated. Total cell lysates were subsequently resolved by SDS-PAGE and immunoblotted with antibodies against the phosphorylated form of eIF4B on Ser422; the phosphorylated form of RSK on Thr359/Ser363; or eIF4B, RSK1, and GAPDH as indicated. None, no treatment with either rapamycin or U0126.
To definitively establish whether RSK is activated in an IFN-dependent manner, experiments were performed to directly examine whether the kinase domain of RSK is activated by IFN-α. For this purpose, immune complex kinase assays were carried out on anti-p90RSK immunoprecipitates from IFN-α-treated cell lysates from KT1 cells, using troponin as an exogenous substrate. As shown in Fig. 2C, troponin was strongly phosphorylated in an IFN-α-dependent manner, consistent with activation of the kinase domain of p90RSK. Immune complex kinase assays were also carried out using Flag-eIF4B as a substrate in vitro. As in the case of troponin, activation of RSK1 was also detectable when eIF4B was used (Fig. 2D), further suggesting that eIF4B is an RSK1 substrate. In a similar manner, when the activation of RSK and phosphorylation of eIF4B in response to IFN-γ were examined in the IFN-γ-sensitive NB4 hematopoietic cell line, we also found that both events were selectively blocked by MEK inhibition but not mTOR inhibition (Fig. 2E).
To further determine the mechanisms of regulation of eIF4B phosphorylation in the KT1 hematopoietic cell line, experiments were carried out in which S6K, RSK1, or RSK2 was knocked down using specific siRNAs, and the phosphorylation of eIF4B was assessed. As shown in Fig. 3A, although siRNA targeting of S6K resulted in significant blocking of S6K protein expression, phosphorylated eIF4B was still clearly detectable after IFN treatment (Fig. 3A). On the other hand, the phosphorylation of the rpS6 was partially inhibited (Fig. 3A), indicating that in these cells, the IFN-activated form of S6K is the dominant kinase for rpS6 but not eIF4B phosphorylation. When the effects of RSK1 or RSK2 inhibition on the phosphorylation of eIF4B were examined, we found that RSK1 knockdown abrogated eIF4B phosphorylation, while RSK2 knockdown had no significant effects (Fig. 3B). Thus, in contrast to what we observed in MEFs, the IFN-inducible phosphorylation of eIF4B is regulated by RSK1, but not S6K, in KT1 hematopoietic cells. On the other hand, RSK1 knockdown had no effect on the phosphorylation of eIF4B in MEFs (Fig. 3C), strongly suggesting that in such cells, eIF4B phosphorylation is exclusively mediated by S6K.
FIG. 3.

Cell type-dependent phosphorylation of eIF4B by p70S6K or RSK1 in response to IFN-α. (A) KT1 cells were transfected with either control siRNA or siRNA specifically targeting p70S6K and treated with IFN-α, as indicated. Equal amounts of total cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against p70S6K, anti-phospho-Ser422-eIF4B, anti-phospho-Ser240/244-rpS6, anti-eIF4B, or anti-rpS6. (B) KT1 cells were transfected with control siRNA or siRNA specifically targeting RSK1 or RSK2, as indicated. The cells were treated with IFN-α for the indicated times, and equal amounts of total cell lysates were analyzed by SDS-PAGE and immunoblotted with an antibody against RSK1. Equal cell lysates from the same experiments were analyzed separately by SDS-PAGE and immunoblotted with an anti-RSK2 antibody. The blot in the top panel was reprobed with antibodies against phospho-eIF4B, eIF4B, and GAPDH as indicated. (C) S6k1+/+ S6k2+/+ MEFs were transfected with either control siRNA or siRNA specifically targeting RSK1, as indicated. After treatment with IFN-α as indicated, equal amounts of total cell lysates were analyzed by SDS-PAGE and immunoblotted with antibodies against RSK1, the phosphorylated form of eIF4B on Ser422, or eIF4B, as indicated.
Previous studies have established that eIF4B interacts with p170/eIF3A and that phosphorylation of eIF4B on Ser422 enhances its affinity for and interaction with p170/eIF3A (3, 34). To determine whether IFN treatment results in the formation of p170/eIF3A-eIF4B complexes, coimmunoprecipitation studies were carried out. Serum-starved wild-type MEFs were treated with IFN-α for different times, and cell lysates were immunoprecipitated with an anti-eIF4B antibody, followed by immunoblotting with an antibody against p170/eIF3A. IFN-α treatment induced an association of p170/eIF3A with eIF4B that was detectable after 90 min of treatment of the cells and persisted at 180 min (Fig. 4A). Such IFN-α-dependent eIF4B-p170/eIF3A complex formation was not detectable in S6k1−/− S6k2−/− MEFs (Fig. 4B), consistent with a lack of IFN-α-inducible phosphorylation of eIF4B in these cells (Fig. 1C). Notably, the increased detection of p170/eIF3A-eIF4B complexes did not result from enhanced p170/eIF3A protein expression, as immunoblotting experiments did not disclose any significant changes in the levels of p170/eIF3A protein expression upon IFN-α treatment (Fig. 4C). We also examined whether p170/eIF3A knockdown indirectly modifies IFN-dependent phosphorylation of eIF4B, but as shown in Fig. 4D, such knockdown did not block eIF4B phosphorylation (Fig. 4D).
FIG. 4.

IFN-α-dependent phosphorylation of eIF4B results in its association with p170/eIF3A. (A) Serum-starved S6k1+/+ S6k2+/+ MEFs were treated with IFN-α for the indicated times. The cells were lysed, and equal amounts of cell lysates were immunoprecipitated with an anti-eIF4B polyclonal antibody or with control non-immune RIgG. Immune complexes were resolved by 5% SDS-PAGE for analysis of p170/eIF3A and eIF4B. (B) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with IFN-α for 90 min, as indicated and, after SDS-PAGE analysis, immunoprecipitates were immunoblotted with anti-p170/eIF3A or anti-eIF4B antibodies, as indicated. (C) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with IFN-α for the indicated times. Equal amounts of cell lysates were resolved by 5% or 7% SDS-PAGE and immunoblotted with antibodies against p170/eIF3A, the phosphorylated form of eIF4B on Ser 422, or eIF4B. (D) S6k1+/+ S6k2+/+ MEFs were transfected with control siRNA or p170/eIF3A siRNA and treated with IFN-α for the indicated times. Equal amounts of cell lysates were resolved by 5% or 7% SDS-PAGE and immunoblotted with antibodies against p170/eIF3A, the phosphorylated form of eIF4B on Ser 422, or eIF4B, as indicated. IP, immunoprecipitation; wt, wild-type; +, present; −, absent.
Cap-dependent binding of mRNA to the 40S ribosomal subunit requires energy derived from ATP hydrolysis (16, 17). Previous studies have established that purified eIF4B has little or no ATPase activity itself, but it can stimulate the activities of eIF4A or eIF4F (16). It has been also shown that there is a lack of direct interaction between eIF4A and eIF4B (22), although recent evidence suggests that eIF4AI and eIF4B can form a complex under certain conditions (47). Thus, it has been difficult to explain how eIF4B enhances the induction of ATP hydrolysis by eIF4A. Consistent with the results of a previous study (22), we have also failed to observe an interaction of eIF4B with eIF4A (data not shown). However, based on our data, eIF4B forms a complex with p170/eIF3A in a type I IFN-dependent manner (Fig. 4A and B), consistent with the results of previous work in other systems (49). We determined whether eIF4B-associated ATPase activity is detectable after IFN treatment of cells. Serum-starved S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEF cells were either left untreated or treated with mouse IFN-α for 30 and 90 min, and after cell lysis, equal amounts of lysates were immunoprecipitated with an anti-eIF4B antibody or control non-immune RIgG. After extensive washing, ATPase activity present in the immunoprecipitates was measured. Increases of approximately 2.6- and 3.9-fold after treatment of wild-type MEFs for 30 and 90 min, respectively, were detected (Fig. 5A). Such induction was not detectable in the S6k1/S6k2 double-knockout MEFs (Fig. 5A). It should be noted that the absolute values of ATPase activity were low in the immunoprecipitates (see Fig. 6A), yet a significant increase in activity after IFN-α treatment was observed in wild-type MEFs but not S6k1/S6k2 knockout cells (Fig. 5A). Such IFN-α-dependent ATPase activity in anti-eIF4B immunoprecipitates was blocked when p170/eIF3A was knocked down (Fig. 5B), suggesting that beyond phosphorylation of eIF4B, associated p170/eIF3A in the complex is also required for induction of ATPase activity.
FIG. 5.
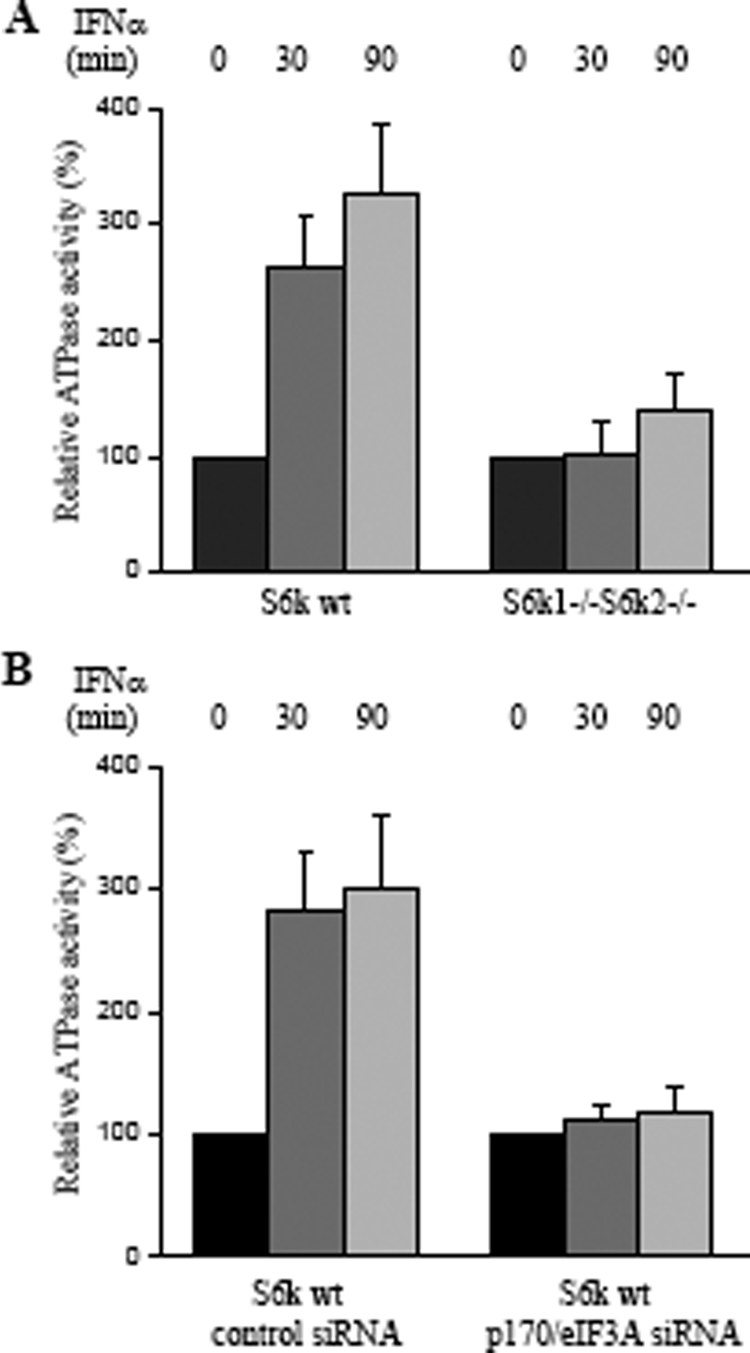
Induction of IFN-α-dependent ATPase activity in association with eIF4B. Data shown represent means ± standard errors of the results of three independent experiments. (A) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with IFN-α as indicated, and ATP hydrolysis reactions using [γ-32P]ATP were performed. Data are expressed as relative ATPase activity (% activity for control over untreated cells for each cell type). One hundred percent was set as 3 pmol of ATP hydrolyzed. (B) S6k1+/+ S6k2+/+ MEFs transfected with either control siRNA or p170/eIF3A siRNA were treated with IFN-α, as indicated. ATP hydrolysis reactions using [γ-32P]ATP were performed. Data are expressed as relative ATPase activities (% activity for control over untreated cells). One hundred percent was set as 3 pmol of ATP hydrolyzed. wt, wild-type.
FIG. 6.

Kinetic analysis of ATP hydrolysis in anti-eIF4B immunoprecipitates. (A) eIF4B-associated activities from S6k1+/+ S6k2+/+ MEFs that were either not treated (gray squares) or treated with IFN-α for 90 min (black circles) are shown. (B) eIF4B-associated activities from S6k1−/− S6k2−/− MEFs that were either not treated (gray squares) or treated with IFN-α for 90 min (black circles) are shown. Data shown in panels A and B represent means ± standard errors of the results of five independent experiments. (C) eIF4B-associated activities from S6k1+/+ S6k2+/+ MEFs transfected with control siRNA that were either not treated (gray squares) or treated with IFN-α for 90 min (black circles) are shown. (D) eIF4B-associated activities from S6k1+/+ S6k2+/+ MEFs transfected with p170/eIF3A siRNA that were either not treated (gray squares) or treated with IFN-α for 90 min (black circles) are shown. Data shown in panels C and D represent means ± standard errors of the results of four independent experiments. V, velocity; wt, wild-type.
The kinetic parameters for IFN-inducible eIF4B-associated ATPase activity were determined and are shown in Fig. 6 and Table 1. A 15-min incubation time was chosen to maximize Pi release with relatively low background. There was clear induction of IFN-dependent activity in S6k wild type MEFs (Fig. 6A) but not S6k1/S6k2 double-knockout MEFs (Fig. 6B). The KmATP for eIF4B-associated activity in S6k wild type MEFs treated with IFN-α was significantly lower than the KmATP for eIF4B-associated activity from S6k wild type MEFs not treated with IFN-α or from S6k1/S6k2 double-knockout cells either treated or not treated with IFN-α (Table 1). A kinetic analysis of ATPase was also performed in anti-eIF4B immunoprecipitates from wild-type MEFs transfected with control siRNA (Fig. 6C) or siRNA targeting p170/eIF3A (Fig. 6D). Clearly, IFN-dependent activity was detectable in control siRNA-transfected MEFs (Fig. 6C) but not p170/eIF3A siRNA-transfected MEFs (Fig. 6D). The increases of kcat/KmATP of approximately fivefold for anti-eIF4B immunoprecipitates from S6k wild type MEFs treated with IFN-α and ∼fourfold from wild-type MEFs transfected with control siRNA treated with IFN-α were primarily due to the reductions of KmATP and stimulation of the catalytic step. The kinetic data were not significantly altered in anti-eIF4B immunoprecipitates from wild-type MEFs transfected with siRNA targeting p170/eIF3A and either treated or not treated with IFN-α (Table 1).
TABLE 1.
eIF4B-associated ATPase activity in S6K wild type and S6k1−/− S6k2−/− MEFsa
| Type of MEF and treatment | KmATP (μM) | kcat (min−1) | kcat/KmATP (μM−1 min−1) |
|---|---|---|---|
| S6K wild type | 63.7 ± 11.8 | 795 ± 39.8 | 12.5 |
| S6K wild type treated with IFN-α | 16.7 ± 4.5 | 1108 ± 51.4 | 66.3 |
| S6k1−/− S6k2−/− | 52.6 ± 5.4 | 675 ± 12.1 | 12.8 |
| S6k1−/− S6k2−/− treated with IFN-α | 83.3 ± 6.8 | 1038 ± 61.7 | 12.5 |
| S6K wild type + control siRNA | 43.5 ± 6.7 | 523 ± 17.0 | 12.0 |
| S6K wild type + control siRNA, treated with IFN-α | 19.6 ± 5.9 | 949 ± 40.5 | 48.4 |
| S6K wild type + p170 siRNA | 55.6 ± 11.0 | 586 ± 17.0 | 10.5 |
| S6K wild type + p170 siRNA, treated with IFN-α | 47.6 ± 12.5 | 575 ± 37.0 | 12.1 |
Kinetic parameters for ATP hydrolysis by immunoprecipitated eIF4B from the indicated MEFs or after the indicated treatments. Kinetic constants were derived from the experiments whose results are presented in Fig. 6. Means ± standard errors are shown for KmATP and kcat values.
Altogether, these data suggested that the IFN-α-inducible ATPase activity detected in association with eIF4B requires both phosphorylation of eIF4B on Ser422 and its interaction with p170/eIF3A. In subsequent experiments, we determined whether the engagement of S6K1/-2 and eIF4B in IFN signaling plays roles in the expression of proteins known to regulate generation of IFN responses. We first determined the role of S6K1/-2 kinases in the expression of ISG15, a type I IFN-induced protein product that mediates IFN responses by regulating ISGylation (40, 45, 55), and on the expression of CXCL-10, an IFN-γ-inducible chemokine that promotes apoptosis (55). As expected, there was strong induction of CXCL-10 (Fig. 7A and B) or ISG15 (Fig. 7C and D) in response to treatment of wild-type MEFs with IFN-γ or IFN-α, respectively. However, such expression was decreased in the S6k1/-2 double-knockout MEFs (Fig. 7A, B, C, and D). Such defective CXCL-10 and ISG15 expression in the knockout MEFs was not associated with defects in IFN-dependent transcription of the Cxcl10 (Fig. 7E) or Isg15 (Fig. 7F) genes, strongly suggesting that it results from defective mRNA translation. Importantly, in experiments in which p170/eIF3A (Fig. 8A) or eIF4B (Fig. 8C) was knocked down by specific siRNAs in wild-type MEFs, ISG15 expression was also significantly diminished, indicating that intact p170/eIF3A and functional eIF4B complexes are required for IFN-inducible mRNA translation of the Isg15 gene.
FIG. 7.

Requirement of S6K activity for IFN-dependent CXCL-10 and ISG15 protein expression. (A) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with mouse IFN-γ for the indicated times. Cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against CXCL-10 (IP10) or GAPDH. (B) The signals from the experiment whose results are shown in panel A were quantified by densitometry and used to calculate the intensity of expression of CXCL-10 relative to that of GAPDH. Data are expressed as means of ratios of CXCL-10 to GAPDH for each experimental condition and represent means ± standard errors of the results of three experiments. (C) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with mouse IFN-α for the indicated times. Cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against ISG15 or GAPDH. (D) The signals from the experiment whose results are shown in panel C were quantified by densitometry and used to calculate the intensity of expression of ISG15 relative to that of GAPDH. Data are expressed as means of ratios of ISG15 to GAPDH for each experimental condition and represent means ± standard errors of the results of three experiments. (E) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with mouse IFN-γ for 6 h at 37°C. Expression of mRNA for the Cxcl10 (Ip10) gene was assessed by quantitative real-time RT-PCR. The GAPDH transcript was used for normalization. Data are expressed as the factor of increase in expression over that in samples not treated with IFN-γ and represent means ± standard errors of the results from three experiments. (F) S6k1+/+ S6k2+/+ and S6k1−/− S6k2−/− MEFs were treated with mouse IFN-α for 6 h at 37°C. Expression of mRNA for the Isg15 gene was assessed by quantitative real-time RT-PCR. The GAPDH transcript was used for normalization. Data are expressed as the factor of increase in expression over that in samples not treated with IFN-α and represent means ± standard errors of the results from three experiments. wt, wild-type.
FIG. 8.

Requirement for eIF4B and eIF3A for IFN-α-dependent ISG15 expression. (A) MEFs were transfected with either control siRNA or p170/eIF3A siRNA, as indicated. The cells were subsequently treated or not treated with mouse IFN-α for 24 h, as indicated. Equal amounts of total cell lysates were resolved by 7.5% and 12% SDS-PAGE and immunoblotted with antibodies against p170/eIF3A, ISG15, or GAPDH, as indicated. (B) The signals for ISG15 and GAPDH from the experiment whose results are shown in panel A and two additional experiments were quantitated by densitometry and used to calculate the intensity of expression of ISG15 relative to that of GAPDH. Data are expressed as means of ratios of ISG15 to GAPDH for each experimental condition and represent means ± standard errors of the results of three experiments. (C) MEFs were transfected with either control siRNA or eIF4B-siRNA, as indicated. The cells were subsequently treated or not treated with mouse IFN-α for 24 h, as indicated. Equal amounts of total cell lysates were resolved by SDS-PAGE and immunoblotted with antibodies against eIF4B, ISG15, or GAPDH, as indicated. (D) The signals for ISG15 and GAPDH from the experiment whose results are shown in panel C and two additional experiments were quantitated by densitometry and used to calculate the intensity of expression of ISG15 relative to that of GAPDH. Data are expressed as means of ratios of ISG15 to GAPDH for each experimental condition and represent means ± standard errors of the results of three experiments.
In subsequent experiments, we sought to determine directly whether mRNA translation for ISG15 is defective in cells with targeted disruption of the S6k1 and S6k2 genes. For this purpose, polysomes from S6K wild type and S6k1−/− S6k2−/− MEFs that were either untreated or treated with mouse IFN-α were isolated. The polysomal profiles from S6K wild type or S6k1−/− S6k2−/− MEFs, before and after IFN-α treatment, are shown in Fig. 9A. There was an abundance of Isg15 mRNA in polysomes isolated from IFN-α-treated S6k1+/+ k2+/+ but not in S6k1−/− S6k2−/− MEFs (Fig. 9B), definitively demonstrating that Isg15 mRNA translation is defective in the absence of p70S6K activity.
FIG. 9.

Activation of p70S6K is essential for type I IFN-regulated Isg15 mRNA translation. (A) S6K wild type and S6k1−/− S6k2−/− MEFs were either left untreated (UT) or treated with mouse IFN-α for 18 h. Cell lysates were separated on a 10-to-50% sucrose gradient, and optical density (O.D.) at 254 nm was recorded. The optical density at 254 nm is shown as a function of gradient depth for each treatment. (B) Polysomal fractions were collected as indicated for panel A, and RNA was isolated. Quantitative real-time RT-PCR assays to determine Isg15 mRNA expression in polysomal fractions were conducted, using Gapdh for normalization. Data are expressed as the factor of increase over results for samples not treated with IFN-α and represent means ± standard deviations of the results of two independent experiments. wt, wild-type.
We subsequently evaluated whether eIF4B plays a role in the generation of the antileukemic effects of IFN-α. For this purpose, the effects of knockdown of eIF4B or RSK1 in the generation of the suppressive effects of IFN-α on primitive leukemic progenitors from KT1 hematopoietic cells were assessed by clonogenic assays in methylcellulose. CFU-blast (CFU-L) colony formation from KT1 cells transfected with control siRNA or siRNA specific for eIF4B or RSK1 was determined in the presence or absence of IFN-α. As expected, IFN-α treatment suppressed the growth of KT1-derived CFU-L (Fig. 10). This suppression was reversible by either RSK1 or eIF4B knockdown (Fig. 10), strongly suggesting that the engagement of this pathway is essential for the generation of the suppressive effects of IFN-α on primary leukemic precursors.
FIG. 10.

Roles of pathways that regulate eIF4B phosphorylation/activation in the generation of the biological effects of IFN-α. KT1 cells were transfected with either control siRNA or siRNA specifically targeting RSK1 or eIF4B, as indicated. The cells were subsequently incubated in methylcellulose in the presence or absence of IFN-α, and CFU-L colony formation was assessed. Data are expressed as percent control colony formation for each condition and represent means ± standard errors of the results of five experiments.
DISCUSSION
The important biological effects of IFNs in vitro and in vivo have triggered extensive research activity over the years, aimed at understanding the mechanisms by which these cytokines generate signals for the production of proteins that mediate their biological effects. The functional diversity of the IFN system, involving multiple different IFN subtypes, likely reflects adaptations of the innate immune response through evolution and underscores the relevance and importance of IFNs as the first line of defense against viruses and as key elements in the immunosurveillance against cancer. Beyond the characterization of signals that control IFN-dependent transcriptional regulation, efforts in recent years have been focused on identifying the mechanisms by which the regulation of mRNA translation by IFNs occurs. It is well established that IFNs inhibit different viral RNAs and that this is one of the mechanisms by which they generate antiviral responses (reviewed in reference 26). At the same time, IFNs also induce mRNA translation for their target genes, but the precise mechanisms by which such events occur have not been elucidated.
There is now a significant amount of accumulated experimental evidence indicating a central and essential role for IFN-activated mTOR pathways in the control of ISG mRNA translation. Initial studies had demonstrated that type I (29) and II (28) IFNs induce activation of mTOR, followed by phosphorylation/activation of S6K and rpS6 and phosphorylation/deactivation of the translational repressor 4E-BP1. Subsequently, there has been emerging evidence that activation of the mTOR pathway by IFNs plays important roles in the control of IFN responses. It was recently shown that MEFs with targeted disruption of the genes coding for negative upstream (TSC2) or downstream (4E-BP1) effectors of the mTOR pathway are more sensitive to the antiviral effects of IFN-α than parental MEFs (25). The regulatory actions of such mTOR effectors in the generation of antiviral responses were also found to correlate with effects on key IFN-α- or IFN-γ-inducible proteins, such as ISG15 or CXCL-10, in the absence of any regulatory effects on transcriptional activation of their respective encoding genes (25). Moreover, it was recently demonstrated that the activation of Akt by either type I or type II IFNs is essential for the engagement of mTOR and its effectors (24). Importantly, these studies established that IFN-dependent mRNA translation for certain ISGs is defective in Akt1/Akt2 double-knockout cells, as evidenced by the decreased presence of mRNA for such genes in polysomal RNA fractions (24). Such findings have provided strong and definitive evidence that the engagement of mTOR and its effectors downstream of the IFN-activated form of Akt complements the function of IFN-activated Jak-Stat pathways, leading to mRNA translation and, ultimately, the expression of IFN-regulated gene products. Consistent with this, both Stat-mediated (30, 43) and Akt/mTOR-mediated (25, 24) signals appear to be essential for the ability of IFNs to inhibit viral replication and generate an antiviral state in target cells.
Despite the emerging evidence for an important role of the Akt/mTOR pathway in the induction of the biological effects of IFNs, the contributions and specific functions of distinct downstream mTOR effectors and regulators in the control of IFN responses remain to be precisely defined. Interestingly, a recent study demonstrated that S6K1/S6K2 are required for interferon regulatory factor 7 activation in dendritic cells and that in S6k1/S6k2 knockout dendritic cells there is defective IFN-α/-β production (4). In the present study, we examined whether IFNs engage eIF4B, an eIF belonging to the eIF4 family of IFs (15, 19), and the functional relevance of such engagement in IFN signaling. eIF4B has been previously shown to play important roles in the initiation of mRNA translation in other systems by promoting eIF4F activity via its ability to enhance the function of the eIF4A RNA helicase (15, 19, 46, 49). Our data demonstrate that both type I and II IFNs induce phosphorylation of eIF4B on Ser422, an event required for its activation. Consistent with the previously described dual regulation of such phosphorylation by either RSK or S6K in other systems (49), we found that IFN-α- or IFN-γ-dependent phosphorylation of the protein is mediated by either S6K or RSK1 in a cell type-dependent manner. It should be noted that although the involvement of p70S6K in IFN signaling in different cell types (28, 29, 36) had been previously established, the involvement of p90RSK1 has remained unknown. Our findings provide the first evidence for an involvement of this kinase in IFN signaling, demonstrating that it undergoes phosphorylation on Thr359/Ser363 during IFN treatment and that its kinase domain is induced. Our data also establish that such activation occurs downstream of IFN-dependent MEK/Erk activation (9), providing evidence for an additional functional role for Erk in IFN signaling, as a regulator of RSK activation.
In studies to define the functional relevance of eIF4B in IFN signaling, we found the presence of associated IFN-α-dependent ATPase activity in eIF4B complexes. Such ATPase activity was diminished in S6k1/S6k2 knockout cells, suggesting a requirement for phosphorylation of the protein on serine 422. ATPase activity was also suppressed in cells in which p170/eIF3A was knocked down by RNA interference, indicating that an interaction of eIF4B with p170/eIF3A is required for the process. These data raise the possibility that eIF4B, via its association with p170/eIF3A and/or other proteins, plays a role in the generation of energy required for the initiation of cap-dependent mRNA translation for ISGs. It should be noted that previous studies have established that eIF4B can increase eIF4A ATPase and helicase activities (2, 16). It is therefore possible that the energy derived from eIF4B-facilitated ATP hydrolysis in response to IFNs physically promotes unwinding of the RNA secondary structure, but this remains to be directly established in further studies. Interestingly, our data suggest an unexpected and potentially important role for p170/eIF3A in mRNA translation for ISGs. The role of this subunit of eIF3 in mRNA translation has been generally unclear. It has been previously suggested that p170/eIF3A may not be an essential contributor in the function of the eIF3 complex during the initiation of mRNA translation (6). On the other hand, other studies have shown that p170/eIF3A participates in the control of translation for a subset of mRNAs (10, 12, 32). Our findings raise the possibility of a unique function of p170/eIF3A, playing an essential role in facilitating eIF4B-associated ATPase activity during the initiation of mRNA translation by IFNs. siRNA-mediated knockdown of either eIF4B or p170/eIF3A resulted in decreased ISG15 protein expression, further supporting such a concept. In addition, in experiments in which mRNA expression for the Isg15 gene was determined directly in polysomal fractions, we found defective expression of this gene, establishing an essential role for S6K/eIF4B in mRNA translation for ISGs. Thus, mTOR-mediated signals regulating both S6K/eIF4B and 4E-BP1 (25) appear to be required for optimal mRNA translation of ISGs.
The potential involvement of p170/eIF3A in IFN signaling is of considerable interest. There is prior evidence that p170/eIF3A is overexpressed in certain malignancies and may be involved in the regulation of cell cycle progression and proliferation (6, 11). Notably, inhibiting p170/eIF3A expression has been associated with arrest of the growth of lung and breast carcinoma cells (12). It is well established that IFNs induce arrest of the growth of various types of malignant cells (35). The fact that p170/eIF3A is engaged in IFN signaling during its association with eIF4B raises the intriguing possibility that such engagement deprives growth factor-regulated and other promitogenic cellular pathways that promote neoplastic transformation of eIF4B. A similar competitive engagement by IFNs may occur for other elements of the pathway, as suggested by our data demonstrating that siRNA-mediated knockdown of eIF4B or RSK1 reverses the antileukemic effects of IFN-α on primitive leukemic (CFU-L) progenitors. It is also possible that eIF4B activity is required for the generation of protein products that may mediate antileukemic activities in response to IFNs, while at the same time its engagement accompanying IFN receptor activation may sequester it away from other cellular pathways that promote leukemogenesis. Notably, other studies have shown that mTOR-mediated signals mediate the induction of apoptosis by IFNs in other tumor cell types (14, 51). Although the precise mechanisms by which such effects occur are not well understood, it is possible that such competition between IFN-dependent growth-suppressive signals and opposing promitogenic signals may exist for other elements of the mTOR pathway as well.
Altogether, our studies establish that S6K/eIF4B- or RSK/eIF4B-generated signals downstream of mTOR are important mediators of mRNA translation in response to type I and II IFNs and participate in the generation of the antiproliferative effects of IFNs. Based on this and other recent studies, a new role for mTOR-activated pathways appears to be emerging, associated with biochemical events that generate signals important for the induction of biological responses induced by IFNs. Taken together with a recent study by Colina et al. demonstrating that the mTOR-regulated translational repressor 4E-BP1 controls interferon regulatory factor 7 expression and IFN-α/IFN-β production (7), our data support a model in which mTOR-mediated signals for cap-dependent translation lead to a positive-feedback regulatory loop that controls IFN production and the generation of IFN biological responses (26). Further work in this area will be required to definitively characterize the similarities and differences in the utilization of the mTOR pathways of IFNs and growth factors that exhibit opposing biological effects. Such studies are required to further our understanding of the events required to establish signaling specificity for mRNA translation and could conceivably lead to the development of novel therapeutic agents for the treatment of viral infections and malignancies.
Acknowledgments
This work was supported by NIH grants CA77816, CA100579, and CA121192; a Merit Review grant from the Department of Veterans Affairs; and Canadian Institutes of Health Research grant MOP15094.
Footnotes
Published ahead of print on 16 March 2009.
REFERENCES
- 1.Alsayed, Y., S. Uddin, S. Ahmad, B. Majchrzak, B. J. Druker, E. N. Fish, and L. C. Platanias. 2000. IFN-gamma activates the C3G/Rap1 signaling pathway. J. Immunol. 1641800-1806. [DOI] [PubMed] [Google Scholar]
- 2.Bi, X., J. Ren, and D. J. Goss. 2000. Wheat germ translation initiation factor eIF4B affects eIF4A and eIFiso4F helicase activity by increasing the ATP binding affinity of eIF4A. Biochemistry 395758-5765. [DOI] [PubMed] [Google Scholar]
- 3.Blenis, J. 1993. Signal transduction via the MAP kinases: proceed at your own RSK. Proc. Natl. Acad. Sci. USA 905889-5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao, W., S. Manicassamy, H. Tang, S. P. Kasturi, A. Pirani, N. Murthy, and B. Pulendran. 2008. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 91157-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carayol, N., E. Katsoulidis, A. Sassano, J. K. Altman, B. J. Druker, and L. C. Platanias. 2008. Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J. Biol. Chem. 2838601-8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhuri, J., A. Chakrabarti, and U. Maitra. 1997. Biochemical characterization of mammalian translation initiation factor 3 (eIF3). Molecular cloning reveals that p110 subunit is the mammalian homologue of Saccharomyces cerevisiae protein Prt1. J. Biol. Chem. 27230975-30983. [DOI] [PubMed] [Google Scholar]
- 7.Colina, R., M. Costa-Mattioli, R. J. Dowling, M. Jaramillo, L. H. Tai, C. J. Breitbach, Y. Martineau, O. Larsson, L. Rong, Y. V. Svitkin, A. P. Makrigiannis, J. C. Bell, and N. Sonenberg. 2008. Translational control of the innate immune response through IRF-7. Nature 452323-328. [DOI] [PubMed] [Google Scholar]
- 8.Darnell, J. E., Jr., I. M. Kerr, and G. R. Stark. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 2641415-1420. [DOI] [PubMed] [Google Scholar]
- 9.David, M., E. Petricoin III, C. Benjamin, R. Pine, M. J. Weber, and A. C. Larner. 1995. Requirement for MAP kinase (ERK2) activity in interferon alpha- and interferon beta-stimulated gene expression through STAT proteins. Science 2691721-1723. [DOI] [PubMed] [Google Scholar]
- 10.Dong, Z., and J. T. Zhang. 2003. EIF3 p170, a mediator of mimosine effect on protein synthesis and cell cycle progression. Mol. Biol. Cell 143942-3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong, Z., and J. T. Zhang. 2006. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit. Rev. Oncol. Hematol. 59169-180. [DOI] [PubMed] [Google Scholar]
- 12.Dong, Z., L. H. Liu, B. Han, R. Pincheira, and J. T. Zhang. 2004. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene 233790-3801. [DOI] [PubMed] [Google Scholar]
- 13.Dorrello, N. V., A. Peschiaroli, D. Guardavaccaro, N. H. Colburn, N. E. Sherman, and M. Pagano. 2006. S6K1- and ßTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314467-471. [DOI] [PubMed] [Google Scholar]
- 14.Fang, P., V. Hwa, and R. G. Rosenfeld. 2006. Interferon-gamma-induced dephosphorylation of STAT3 and apoptosis are dependent on the mTOR pathway. Exp. Cell Res. 3121229-1239. [DOI] [PubMed] [Google Scholar]
- 15.Gingras, A. C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68913-963. [DOI] [PubMed] [Google Scholar]
- 16.Grifo, J. A., R. D. Abramson, C. A. Satler, and W. C. Merrick. 1984. RNA-stimulated ATPase activity of eukaryotic initiation factors. J. Biol. Chem. 2598648-8654. [PubMed] [Google Scholar]
- 17.Grifo, J. A., S. M. Tahara, J. P. Leis, M. A. Morgan, A. J. Shatkin, and W. C. Merrick. 1982. Characterization of eukaryotic initiation factor 4A, a protein involved in ATP-dependent binding of globin mRNA. J. Biol. Chem. 2575246-5252. [PubMed] [Google Scholar]
- 18.Hay, N., and N. Sonenberg. 2004. Upstream and downstream of mTOR. Genes Dev. 181926-1945. [DOI] [PubMed] [Google Scholar]
- 19.Hernández, G., and P. Vazquez-Pianzola. 2005. Functional diversity of the eukaryotic translation initiation factors belonging to eIF4 families. Mech. Dev. 122865-876. [DOI] [PubMed] [Google Scholar]
- 20.Hershey, J. W. 1991. Translational control in mammalian cells. Annu. Rev. Biochem. 60717-755. [DOI] [PubMed] [Google Scholar]
- 21.Itoh, S., B. Ding, C. P. Bains, N. Wang, Y. Takeishi, T. Jalili, G. L. King, R. A. Walsh, C. Yan, and J. Abe. 2005. Role of p90 ribosomal S6 kinase (p90RSK) in reactive oxygen species and protein kinase C beta (PKC-beta)-mediated cardiac troponin I phosphorylation. J. Biol. Chem. 28024135-24142. [DOI] [PubMed] [Google Scholar]
- 22.Jaramillo, M., T. E. Dever, W. C. Merrick, and N. Sonenberg. 1991. RNA unwinding in translation: assembly of helicase complex intermediates comprising eukaryotic initiation factors eIF-4F and eIF-4B. Mol. Cell. Biol. 115992-5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsoulidis, E., Y. Li, P. Yoon, A. Sassano, J. Altman, P. Kannan-Thulasiraman, L. Balasubramanian, S. Parmar, J. Varga, M. S. Tallman, A. Verma, and L. C. Platanias. 2005. Role of the p38 mitogen-activated protein kinase pathway in cytokine-mediated hematopoietic suppression in myelodysplastic syndromes. Cancer Res. 659029-9037. [DOI] [PubMed] [Google Scholar]
- 24.Kaur, S., A. Sassano, B. Dolniak, S. Joshi, B. Majchrzak-Kita, D. P. Baker, N. Hay, E. N. Fish, and L. C. Platanias. 2008. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 1054808-4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaur, S., L. Lal, A. Sassano, B. Majchrzak-Kita, M. Srikanth, D. P. Baker, E. Petroulakis, N. Hay, N. Sonenberg, E. N. Fish, and L. C. Platanias. 2007. Regulatory effects of mammalian target of rapamycin-activated pathways in type I and II interferon signaling. J. Biol. Chem. 2821757-1768. [DOI] [PubMed] [Google Scholar]
- 26.Kaur, S., E. Katsoulidis, and L. C. Platanias. 2008. Akt and mRNA translation by interferons. Cell Cycle 72112-2116. [DOI] [PubMed] [Google Scholar]
- 27.Kaur, S., S. Uddin, and L. C. Platanias. 2005. The PI 3′ kinase pathway in interferon signaling. J. Interferon Cytokine Res. 25780-787. [DOI] [PubMed] [Google Scholar]
- 28.Lekmine, F., A. Sassano, S. Uddin, J. Smith, B. Majchrzak, S. M. Brachmann, N. Hay, E. N. Fish, and L. C. Platanias. 2004. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp. Cell Res. 295173-182. [DOI] [PubMed] [Google Scholar]
- 29.Lekmine, F., S. Uddin, A. Sassano, S. Parmar, S. M. Brachmann, B. Majchrzak, N. Sonenberg, N. Hay, E. N. Fish, and L. C. Platanias. 2003. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J. Biol. Chem. 27827772-27780. [DOI] [PubMed] [Google Scholar]
- 30.Levy, D. E., and J. E. Darnell, Jr. 2002. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3651-662. [DOI] [PubMed] [Google Scholar]
- 31.Li, Y., A. Sassano, B. Majchrzak, D. K. Deb, D. E. Levy, M. Gaestel, A. R. Nebreda, E. N. Fish, and L. C. Platanias. 2004. Role of p38alpha Map kinase in type I interferon signaling. J. Biol. Chem. 279970-979. [DOI] [PubMed] [Google Scholar]
- 32.Liu, Z., Z. Dong, Z. Yang, Q. Chen, Y. Pan, Y. Yang, P. Cui, X. Zhang, and J. T. Zhang. 2007. Role of eIF3a (eIF3 p170) in intestinal cell differentiation and its association with early development. Differentiation 75652-661. [DOI] [PubMed] [Google Scholar]
- 33.Mayer, I. A., A. Verma, I. M. Grumbach, S. Uddin, F. Lekmine, F. Ravandi, B. Majchrzak, S. Fujita, E. N. Fish, and L. C. Platanias. 2001. The p38 Map kinase pathway mediates the growth inhibitory effects of IFNα in BCR-ABL expressing cells. J. Biol. Chem. 27628570-28577. [DOI] [PubMed] [Google Scholar]
- 34.Methot, N., M. S. Song, and N. Sonenberg. 1996. A region rich in aspartic acid, arginine, tyrosine, and glycine (DRYG) mediates eukaryotic initiation factor 4B (eIF4B) self-association and interaction with eIF3. Mol. Cell. Biol. 165328-5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parmar, S., and L. C. Platanias. 2003. Interferons: mechanisms of action and clinical applications. Curr. Opin. Oncol. 15431-439. [DOI] [PubMed] [Google Scholar]
- 36.Parmar, S., J. Smith, A. Sassano, S. Uddin, E. Katsoulidis, B. Majchrzak, S. Kambhampati, E. A. Eklund, M. S. Tallman, E. N. Fish, and L. C. Platanias. 2005. Differential regulation of the p70 S6 kinase pathway by interferon alpha and imatinib mesylate (STI571) in chronic myelogenous leukemia cells. Blood 1062436-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pause, A., G. J. Belsham, A. C. Gingras, O. Donzé, T. A. Lin, J. C. Lawrence, Jr., and N. Sonenberg. 1994. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371762-767. [DOI] [PubMed] [Google Scholar]
- 38.Pende, M., S. H. Um, V. Mieulet, M. Sticker, V. L. Goss, J. Mestan, M. Mueller, S. Fumagalli, S. C. Kozma, and G. Thomas. 2004. S6K1−/−/S6K2−/− mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 243112-3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pestka, S., J. A. Langer, K. C. Zoon, and C. E. Samuel. 1987. Interferons and their actions. Annu. Rev. Biochem. 56727-777. [DOI] [PubMed] [Google Scholar]
- 40.Pitha-Rowe, I. F., and P. M. Pitha. 2007. Viral defense, carcinogenesis and ISG15: novel roles for an old ISG. Cytokine Growth Factor Rev. 18409-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platanias, L. C. 2003. The p38 mitogen-activated protein kinase pathway and its role in interferon signaling. Pharmacol. Ther. 98129-142. [DOI] [PubMed] [Google Scholar]
- 42.Platanias, L. C. 2005. Mechanisms of type-I- and type-II-interferon-mediated signaling. Nat. Rev. Immunol. 5375-386. [DOI] [PubMed] [Google Scholar]
- 43.Platanias, L. C., and E. N. Fish. 1999. Signaling pathways activated by interferons. Exp. Hematol. 271583-1592. [DOI] [PubMed] [Google Scholar]
- 44.Raught, B., F. Peiretti, A. C. Gingras, M. Livingstone, D. Shahbazian, G. L. Mayeur, R. D. Polakiewicz, N. Sonenberg, and J. W. Hershey. 2004. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO. J. 231761-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ritchie, K. J., and D. E. Zhang. 2004. ISG15: the immunological kin of ubiquitin Semin. Cell Dev. Biol. 15237-246. [DOI] [PubMed] [Google Scholar]
- 46.Rozen, F., I. Edery, K. Meerovitch, T. E. Dever, W. C. Merrick, and N. Sonenberg. 1990. Bidirectional RNA helicase activity of eukaryotic translation initiation factors 4A and 4F. Mol. Cell. Biol. 101134-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rozovsky, N., A. C. Butterworth, and M. J. Moore. 2008. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA 142136-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schindler, C., D. E. Levy, and T. Decker. 2007. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 28220059-20063. [DOI] [PubMed] [Google Scholar]
- 49.Shahbazian, D., P. P. Roux, V. Mieulet, M. S. Cohen, B. Raught, J. Taunton, J. W. Hershey, J. Blenis, M. Pende, and N. Sonenberg. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO. J. 252781-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67227-264. [DOI] [PubMed] [Google Scholar]
- 51.Thyrell, L., L. Hjortsberg, V. Arulampalam, T. Panaretakis, S. Uhles, M. Dagnell, B. Zhivotovsky, I. Leibiger, D. Grandér, and K. Pokrovskaja. 2004. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J. Biol. Chem. 27924152-24162. [DOI] [PubMed] [Google Scholar]
- 52.Uddin, S., B. Majchrzak, J. Woodson, P. Arunkumar, Y. Alsayed, R. Pine, P. R. Young, E. N. Fish, and L. C. Platanias. 1999. Activation of the p38 mitogen-activated protein kinase by type I interferons. J. Biol. Chem. 27430127-30131. [DOI] [PubMed] [Google Scholar]
- 53.Uddin, S., L. Yenush, X. J. Sun, M. E. Sweet, M. F. White, and L. C. Platanias. 1995. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3′-kinase. J. Biol. Chem. 27015938-15941. [DOI] [PubMed] [Google Scholar]
- 54.Wullschleger, S., R. Loewith, and M. N. Hall. 2006. mTOR signaling in growth and metabolism. Cell 124471-484. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, H. M., J. Yuan, P. Cheung, D. Chau, B. W. Wong, B. M. McManus, and D. Yang. 2005. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol. Cell. Biol. 256247-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]