

Abstract
Spi-1 and Fli-1 are ETS transcription factors recurrently deregulated in mouse erythroleukemia induced by Friend viruses. Since they share the same core DNA binding site, we investigated whether they may contribute to erythroleukemia by common mechanisms. Using inducible knockdown, we demonstrated that Fli-1 contributes to proliferation, survival, and differentiation arrest of erythroleukemic cells harboring an activated fli-1 locus. Similarly, we used inducible Fli-1 knockdown and either hexamethylenebisacetamide (HMBA)- or small interfering RNA-mediated Spi-1 knockdown to investigate their respective contributions in erythroleukemic cells harboring an activated spi-1 locus. In these cells, simple or double knockdown of both Spi-1 and Fli-1 additively contributed to induce proliferation arrest and differentiation. Transcriptome profiling revealed that virtually all transcripts affected by both Fli-1 knockdown and HMBA are affected in an additive manner. Among these additively downregulated transcripts, more than 20% encode proteins involved in ribosome biogenesis, and conserved ETS binding sites are present in their gene promoters. Through chromatin immunoprecipitation, we demonstrated the association of Spi-1 and Fli-1 on these promoters in Friend erythroleukemic cells. These data lead us to propose that the oncogenicity of Spi-1, Fli-1, and possibly other ETS transcription factors may involve their ability to stimulate ribosome biogenesis.
Friend erythroleukemia has been a powerful model for dissection of how multiple oncogenes cooperate to initiate and maintain leukemic transformation. The Friend viral complex contains a replication-defective spleen focus-forming virus (SFFV) and a replication-competent Friend murine leukemia virus (F-MuLV). It induces a multistep erythroleukemic process in susceptible mice (8, 33, 50). SFFV virus is the pathogenic component responsible for this acute erythroleukemia. During the early stage of the disease, the product of the SFFV env gene, gp55, interacts with the erythropoietin receptor (Epo-R) and constitutively activates signaling pathways allowing the proliferation of proerythroblasts still able to differentiate in the absence of Epo. During this early step, the activation of signaling pathways allowing proerythroblast proliferation is also strictly dependent on the c-Kit receptor and on the small form of the STK receptor tyrosine kinase (20, 57). The second stage of the disease is characterized by a clonal population outgrowth of leukemic proerythroblasts blocked in their differentiation and able to grow as permanent cell lines in vitro. Virtually all tumors display SFFV proviral integration upstream of the Spi-1/PU.1 gene, leading to overexpression of the normal transcription factor Spi-1/PU.1 (hereinafter called Spi-1). It is now well established that the dysregulation of Spi-1 expression is a critical event in the process of SFFV-induced erythroleukemia. Indeed, the terminal erythroid differentiation can be reinitiated in Friend tumor cells by chemical inducers such as hexamethylenebisacetamide (HMBA). This differentiation is associated with a decrease in Spi-1 levels (18, 24, 27, 51), and it can be reversed by Spi-1-enforced expression (43, 64). Similarly, enforced expression of Spi-1 together with both gp55 and constitutively activated Epo-R inhibits differentiation of avian erythroid progenitors (42). Furthermore, transgenic mice overexpressing Spi-1 spontaneously develop an erythroleukemia characterized by an Epo-dependent proliferation of proerythroblasts blocked in their differentiation (38). Spi-1 knockdown induced by RNA interference is sufficient to inhibit proliferation and restore terminal erythroid differentiation of erythroleukemic cell lines established from either SFFV-infected (4) or Spi-1 transgenic mice (47). At least one contribution of Spi-1 overexpression to erythroleukemia is through the inhibition of GATA-1 transcriptional activity (39, 45, 46, 67). Indeed, enforced expression of GATA-1 is sufficient to restore the differentiation of SFFV-infected erythroleukemic cells (13, 45, 46). Recent data have shown that Spi-1 inhibits expression of some GATA-1 target genes by binding to GATA-1 on transcriptional promoters and creating a repressive chromatin structure through the recruitment of pRB, the histone methylase SUV39h, and the heterochromatin protein HP1α (55). However, although this mechanism might explain the contribution of Spi-1 to the repression of erythroid-specific GATA-1-dependent gene transcription, the contribution of Spi-1 to the proliferation of erythroleukemic cells remains poorly understood.
The replication-competent F-MuLV itself also induces clonal erythroleukemia when injected into newborn mice. In 75% of these tumors, F-MuLV provirus integrates upstream of the Fli-1 gene, leading to Fli-1 transcription factor overexpression (9, 62). We and others have shown that enforced expression of Fli-1 in erythroleukemic cell lines is sufficient to inhibit proliferation arrest and terminal differentiation induced either by chemicals or by SCF withdrawal (54, 60, 68). Others have also shown that enforced expression of Fli-1 in avian erythroid progenitors induces their survival and proliferation and impedes their differentiation in response to Epo (3, 34, 40). These data strongly suggest that Fli-1 contributes to survival and proliferation of F-MuLV-infected erythroleukemic cells blocked in their differentiation. The antiapoptotic bcl2 gene, known as a direct target gene of Fli-1, may contribute to the survival of erythroleukemic cells that overexpress Fli-1 (34). Furthermore, several studies suggest that Fli-1 overexpression in erythroid cells interferes with Epo-R signaling (32). Our own studies have shown that Fli-1 is a direct transcriptional target for Spi-1 in SFFV-infected erythroleukemic cells (54) and that Fli-1 behaves as a functional antagonist of EKLF in transactivation assays, further suggesting that Fli-1 might also contribute to the inhibition of erythroid differentiation through the repression of EKLF target genes (53). Altogether, these data indicate that either Spi-1 or Fli-1 overexpression can induce survival and proliferation and inhibit the differentiation of erythroid precursors.
Ribosome biogenesis is a highly ordered process responsible for the production of ribosome, the central protein synthesis factory of the cell (22). This process involves many different events taking place in the nucleolus: the transcription of rRNA genes by RNA polymerases I and III, the cleavage of primary rRNA transcripts into mature rRNA, their modification by methylation or pseudouridylation at more than 150 specific positions, and their assembly with ribosomal proteins (26). All of these events require the coordinated action of more than 170 nucleolar proteins mainly involved in ribosomal rRNA synthesis, cleavage, and posttranscriptional modifications (15) with the help of many snoRNAs. According to our present knowledge, the rate of ribosome biogenesis is determined mainly by the rate of rRNA synthesis and maturation, whereas ribosomal proteins that are produced in excess are rapidly degraded by the proteasome (15). Most cancer cells are characterized by increased ribosome biogenesis, as evidenced by the frequent observation of hypertrophied nucleoli. Although it was initially considered only a consequence of the high proliferation rate of cancer cells, increasing evidence indicates that increased ribosome biogenesis can actively contribute to cell transformation (37, 49). This possibility is supported by the recent demonstration that well-known antioncogenes, like those encoding p53 and RB, as well as proto-oncogenes, like that encoding c-Myc, either repress or activate ribosome biogenesis, respectively. For example, c-Myc not only directly stimulates RNA polymerase I and III activity but also directly activates the transcription of most of the genes involved in ribosome biogenesis through its direct binding to their proximal promoters (16). Furthermore, a very recent study showed that c-Myc oncogenicity can be reversed by ribosomal protein haploinsufficiency (6). Interestingly, ribosomal protein gene promoters are also characterized by an overrepresentation of binding sites for YY1, SP1, and ETS family transcription factors (41). More recently, whole-genome chromatin immunoprecipitation (ChIP)-chip analyses with human cells revealed that many ribosome-related gene promoters are indeed bound by YY1 (63) and SP1 (44) and by three members of the ETS family (ETS1, ELF1, and GABPα) (28).
Remarkably, Spi-1 and Fli-1, which are both recurrently involved in erythroleukemia, belong to the same ETS family. As such, they recognize the same core DNA binding motif, GGAA/G, raising the intriguing possibility that their contribution to erythroleukemia might be mediated through the transcriptional deregulation of a common set of genes. In the present study, we addressed this possibility by exploring the consequences of simple or double Spi-1 and Fli-1 knockdown for the proliferation, survival, and differentiation of F-MuLV- or SFFV-induced erythroleukemic cells. We found that Spi-1 and Fli-1 knockdowns additively contribute to induce proliferation arrest and differentiation in SFFV cells. By combining transcriptome profiling and chromatin immunoprecipitation analyses, we determined that this additive effect of Spi-1 and Fli-1 is associated with the additive decrease in the expression of a common set of genes involved in ribosome biogenesis whose promoters are bound by both Spi-1 and Fli-1 in SFFV cells and by Fli-1 in F-MuLV cells.
MATERIALS AND METHODS
Cell lines, culture, and transfection.
Mouse erythroleukemic cell clone 745A, harboring SFFV proviral integration upstream of Spi-1 (54), mouse erythroleukemic cell clone NN10, harboring F-MuLV proviral integration upstream of Fli-1 (14, 54), and all of their derivatives were cultured at 37°C under 5% CO2 in a humidified incubator and in Iscove's modified Dulbecco's medium (PAA Laboratories, Les Mureaux, France) supplemented with 10% fetal calf serum (PAA Laboratories) and antibiotics. The NN10/TR and 745A/TR cell lines, stably expressing high levels of the Tetr repressor, were established by transfection of NN10 or 745A cells with the pcDNA-6TR-EF-1α vector (47) followed by blasticidin selection. NN10 clones harboring doxycycline (Dox)-inducible expression of either one of two short hairpin RNAs (shRNAs) (fli1_749 or fli1_1508) targeting two different specific regions of Fli-1 mRNA (with no homology to any other ETS mRNA) or of control shRNA (SCR) with scrambled sequence were established by transfection of NN10/TR cells with pGJ10/shfli1_749 (NN10 clone 5), pGJ10/shfli1_1508 (NN10 clones 10 and 23), or pGJ10/scr vector (NN10 clones 3B10, 3E9, and 1C6), followed by G418 selection. Similarly, 745A clones harboring Dox-inducible shRNA were established by transfection of 745A/TR cells with pGJ10/shfli1_749 (745A clone 44), pGJ10/shfli1_1508 (745A clone 1), or pGJ10/scr vector (745A clones 1A1, 1D10, and 1F8), followed by G418 selection. Clone bcl2 22 was derived from NN10 5 cells following cotransfection with a 3:1 mix of human bcl2 gene expression vector and pcDNA4/TO (Invitrogen, Cergy Pontoise, France) carrying a zeocin resistance gene, followed by zeocin selection. Transfections were performed either by lipofection using FuGENE 6 (Roche, Meylan, France) or by nucleofection using the Nucleofector Kit V and program G-16 on a Nucleofector electroporation device (Amaxa, Cologne, Germany). shRNA production was induced by adding 100 ng/ml of Dox (Clontech, Saint-Germain-en-Laye, France). Erythroid differentiation was induced by 5 mM HMBA (Sigma, Saint-Quentin Fallavier, France), and hemoglobin-containing differentiated cells were numbered by benzidine staining (54). Cell death was determined by trypan blue exclusion. Colony assays were performed in quadruplicate by seeding 50 cells/well in 500 μl of 0.8% methylcellulose in Iscove's modified Dulbecco's medium supplemented with 10% fetal calf serum in 24-well culture plates.
Plasmid constructs.
pcDNA6/TR-EF-1α (47) and pGJ10 have already been described (19). pGJ10/shfli1_749 and pGJ10/shfli1_1508 were obtained by cloning double-stranded oligonucleotides encoding shRNA directed against murine Fli-1 mRNA into BglII and NotI sites of pGJ10. Similarly, pGJ10/scr was obtained by cloning double-stranded oligonucleotides encoding shRNA with scrambled sequence into the same sites of pGJ10. An expression vector of human Bcl2 (a gift of M. Moulin, Villeurbanne) was obtained by cloning human bcl2 cDNA under the control of the cytomegalovirus promoter. Oligonucleotide sequences are given in Table S1 in the supplemental material.
Western blot analyses.
Western blot analyses were performed on total cell lysates as described previously (53, 54) using the following antibodies: anti-Fli-1 (sc-356; Santa Cruz Biotechnology, Santa Cruz, CA) (see Fig. 1, 5, and 8), anti-Fli-1 (Ab-15-289-500; ABcam, Paris, France) (see Fig. 4), anti-Spi-1 (sc-352; Santa Cruz), antiactin (MAB1501; Millipore, Saint-Quentin-en-Yvelines, France), and anti-Bcl2 (sc-7382; Santa Cruz).
FIG. 1.
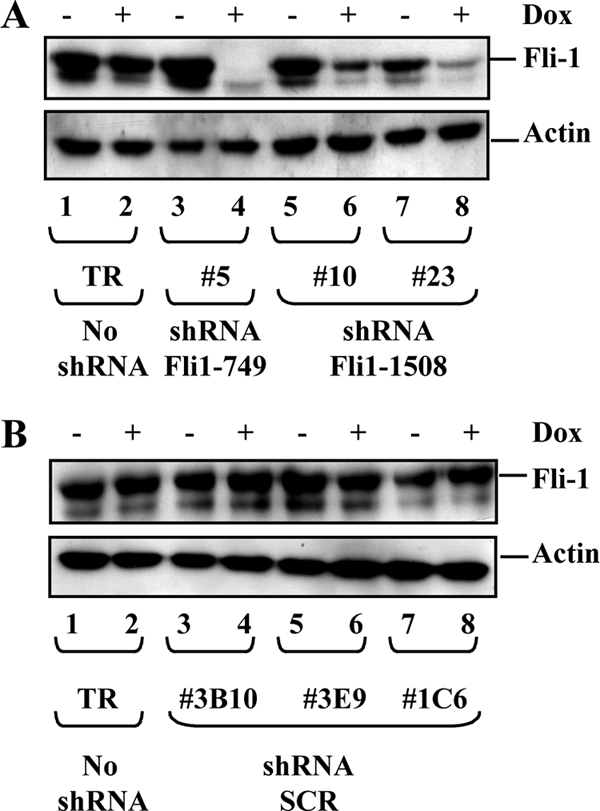
Dox-inducible Fli-1 knockdown in NN10 cells. Western blot analysis of the Fli-1 protein in individual clones after 2 days of culture in the presence or absence of Dox. (A) Parental NN10/TR cells (lanes 1 and 2) and three independent clones expressing either Fli1_749 (lanes 3 and 4) or Fli1_1508 shRNA (lanes 5 to 8). (B) Parental NN10/TR cells (lanes 1 and 2) and three randomly chosen clones expressing control shRNA (lanes 3 to 8). The two bands revealed by the Fli-1-specific antibody correspond to the major (51-kDa) and minor (48-kDa) isoforms of Fli-1. The β-actin protein is shown as a loading control.
FIG. 5.
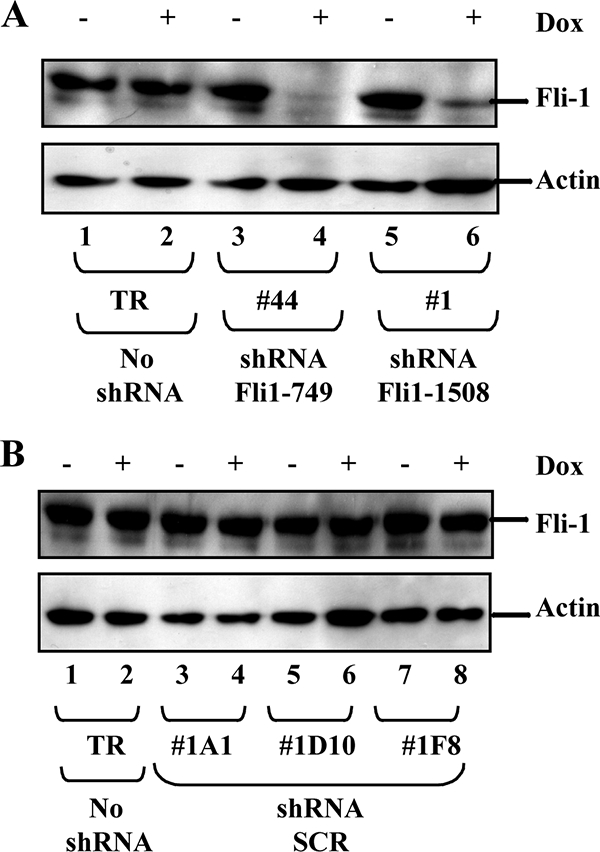
Dox-inducible fli-1 knockdown in 745A cells. Western blot analysis of the Fli-1 protein in individual clones after 2 days of culture in the presence or absence of Dox. (A) Parental 745/TR cells (lanes 1 and 2) and two clones expressing either fli1_749 (lanes 3 and 4) or fli1_1508 shRNA (lanes 7 and 8). (B) Parental cells (lanes 1 and 2) and three independent clones expressing control shRNA (lanes 3 to 8). The β-actin protein is shown as a loading control.
FIG. 8.

Additive effects of Spi-1 and Fli-1 knockdown in 745A cells. 745A/TR/shfli1_749#44 cells were grown in the presence or absence of Dox for 2 days. Cells were then left untransfected (UT) or transfected every 12 h during 2 days with either control siRNA directed against luciferase (siLUC) or siRNA directed against Spi-1 (siSPI). (A) Western blot analysis of the Fli-1 and Spi-1 proteins performed 60 h after the first siRNA transfection. β-Actin is shown as a loading control. (B) Percentages of benzidine-positive cells determined 60 h after the first siRNA transfection. (C) Clonogenicity determined as for Fig. 5A. (D) Percentages of dead cells (trypan blue positive). Results in panels B, C, and D represent means and standard deviations from three independent experiments. Asterisks indicate statistically significant differences (P < 0.05 in paired Student test).
FIG. 4.
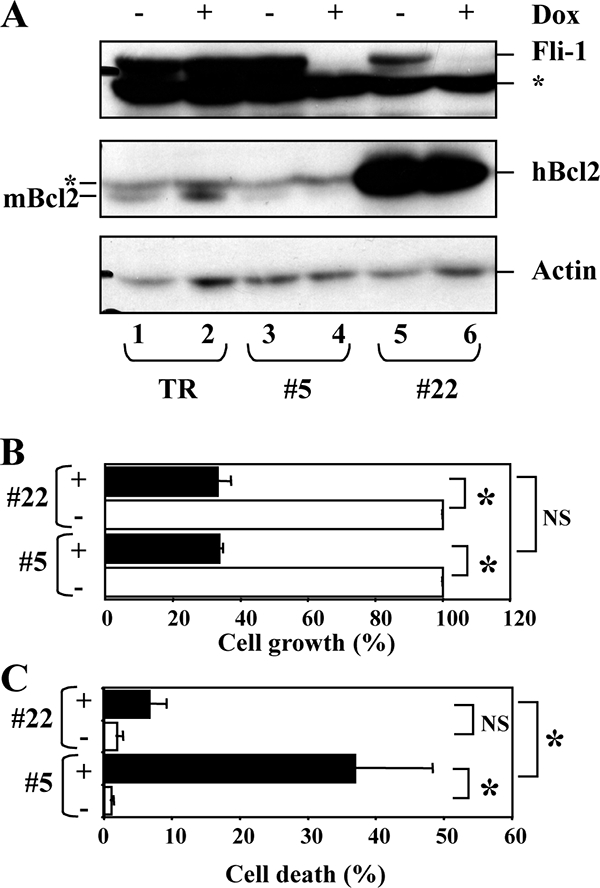
Fli-1 knockdown reduces the proliferation of NN10 cells independently of cell death induction. Clone bcl2 22 was derived from NN10/TR/shfli_749#5 by transfection with a human Bcl2 expression vector. NN10/TR/shfli_749#5 and bcl2 22 cells were grown for 5 days with or without Dox before analysis. (A) Western blot analysis of the Fli-1, human Bcl2 (hBcl2), and murine Bcl2 (mBcl2) proteins. An unspecific band revealed by the Bcl2 antibody and which comigrates with hBcl2 is indicated by an asterisk. β-Actin revealed on the same membranes is shown as a loading control. (B) Relative cell growth, expressed as the percentages of total cells (viable and dead cells) in the presence of Dox (black bars) and in its the absence (white bars). (C) Percentages of dead cells determined by trypan blue in the presence (black bars) or absence (white bars) of Dox, respectively. (Means and standard deviations from three independent experiments are shown). Significant (P < 0.05) and nonsignificant differences are indicated by asterisks and NS, respectively.
Quantitative reverse transcription (RT)-PCR.
Total RNA was extracted using the Rneasy PLUS minikit (Qiagen, Courtaboeuf, France). Total RNA (100 to 500 ng) was retrotranscribed in duplicate using the Quantitect reverse transcription kit (Qiagen) or Moloney murine leukemia virus reverse transcriptase by random priming. Quantitative PCR was performed using the QuantiTect SYBR green PCR kit (Qiagen) on a LightCycler LC480 PCR system (Roche) and oligonucleotides indicated in Table S1 in the supplemental material. mRNA-specific signals were normalized to that of 18S rRNA (Tables 1 and 2) or β-actin mRNA (see Fig. 8 and 9).
TABLE 1.
Phenotype induced by Fli-1 knockdown in NN10 cellsa
| shRNA used | Cell clone | Relative no. of cells, day 4
|
% Dead cells, day 3
|
% Benzidine-positive cells, day 5
|
Fold change in mRNA (+Dox/−Dox), day 2
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| −Dox | +Dox | −Dox | +Dox | −Dox | +Dox | Bcl2 | Alas2 | Ahsp | α-globin | ||
| None | TR | 100 | 96.4 (17.6) | <3 | <3 | 0 | 0 | 1.16 (0.12) | 1.2 | 1.4 | 0.9 (0.78) |
| SCR | 3B10 | 100 | 110 | <3 | <3 | 0 | 0 | ND | ND | ND | ND |
| 3E9 | 100 | 87 | <3 | <3 | 0 | 0 | ND | ND | ND | ND | |
| 1C6 | 100 | 98 | <3 | <3 | 0 | 0 | ND | ND | ND | ND | |
| Fli1_749 | 5 | 100 | 30.8 (13.4) | 1 (1.1) | 13.2 (7.3) | 0 | 6.4 (5.3) | 0.34 (0.22) | 3.2 (0.4) | 2.7 (0.6) | 12.5 (5) |
| 10 | 100 | 61 | <3 | 15.7 | 0 | 4.8 | ND | ND | ND | ND | |
| 23 | 100 | 73 | <3 | 11.8 | 0 | 2.8 | ND | ND | ND | ND | |
Means and standard deviations (in parentheses) from three independent experiments. ND, not determined; −Dox, no treatment; +Dox, Dox treatment. Statistically significant Dox effects are indicated in bold.
TABLE 2.
Phenotype induced by Fli-1 knockdown in 745A cellsa
| shRNA used | Cell clone | Relative no. of colonies
|
% Dead cells, day 4 in HMBA
|
% Benzidine-positive cells, day 2 in HMBA
|
Fold change in mRNA (+Dox/−Dox), day 2 in HMBA
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| −Dox | +Dox | −Dox | +Dox | − | +Dox | Bcl2 | Alas2 | Ahsp | α−globin | ||
| None | TR | 100 | 98 (11.7) | 3.1 (0.9) | 3.4 (0.7) | 10.3 (1.1) | 7.9 (0.9) | 1 (0.1) | 0.82 (0.20) | 0.87 (0.28) | 1 (0.3) |
| SCR | 1A1 | 100 | 94.8 (26.3) | <3 | <3 | 11 (2.1) | 5.7 (4.6) | ND | ND | ND | ND |
| 1D10 | 100 | 112.0 (28.9) | <3 | <3 | 8.6 (1.7) | 7.1 (3.1) | ND | ND | ND | ND | |
| 1F8 | 100 | 90.0 (30.2) | <3 | <3 | 7.4 (4.9) | 5.9 (2.9) | ND | ND | ND | ND | |
| Fli1_749 | 44 | 100 | 54.1 (6.1) | 4.2 (1.7) | 38.0 (9.2) | 9.2 (4.5) | 28.2 (10.5) | 1,1 (0,1) | 4.9 (2.1) | 3.4 (1.1) | 2.3 (0.7) |
| 1 | 100 | 73.8 (7.7) | 4.2 (1.7) | 28.8 (5.7) | 16.8 (1.5) | 40.5 (9.9) | 1,2 (0,2) | ND | ND | 2,7 (0,1) | |
Means and standard deviations (in parentheses) from three independent experiments. ND, not determined; −Dox, no treatment; +Dox, Dox treatment. Statistically significant Dox effects are indicated in bold.
FIG. 9.

Spi-1 and Fli-1 directly activate a common set of genes involved in ribosome biogenesis in 745A cells. 745A/TR and 745A/TR/shfli1_749#44 cells were treated with Dox or left untreated for 2 days, and differentiation was induced by adding 5 mM HMBA for 1 day before performing quantitative RT-PCR analyses of the indicated gene transcripts (45S corresponds to 45S rRNA precursor). (A) Relative levels of transcripts determined in 745A/TR cells (means and standard deviations from three independent experiments). (B) Same as panel A but for 745A/TR/shfli1_749#44 cells. (C) Spi-1 chromatin occupancy of the indicated gene promoters determined by chromatin immunoprecipitation on untreated 745A/TR/shfli1_749#44 cells using Spi-1-specific or control Ubc9 antibody. Gapdh promoter and α-globin gene 129k and 153k regions (2) were used as negative controls. Results are expressed as relative proportions of the input chromatin which has been precipitated by antibodies standardized to the background levels determined on the Gapdh gene promoter (means and standard deviations from three independent experiments). (D) Fli-1 chromatin occupancy of the indicated gene promoters, determined as in panel C by chromatin immunoprecipitation on untreated 745A/TR/shfli1_749#44 cells using Fli-1 antibody.
ChIP.
For ChIP assays, 4 × 107 cells were fixed for 15 min at room temperature by adding 1% of formaldehyde directly to the culture medium. Fixation was stopped by adding 0.125 M glycine, followed by two washes in cold phosphate-buffered saline (PBS). Cells were resuspended in lysis buffer (50 mM Tris-HCl [pH 8.1], 10 mM EDTA, 1% sodium dodecyl sulfate [SDS] containing a 1/20 volume of EDTA free protease inhibitor cocktail [Roche]), and chromatin fragmentation was performed on ice by sonication (Vibra Cell 72405 sonicator; Bioblock, Illkirch, France). The mean length of fragmented DNA was controlled at this step (around 300 bp). Sonicated chromatin preparations (corresponding to 8 × 106 cells) were first diluted in 10 volumes of dilution buffer (16.7 mM Tris [pH 8.0], 1.2 mM EDTA, 167 mM NaCl, 1.1% Triton X-100, 0.01% SDS containing a 1/20 volume of EDTA free protease inhibitors cocktail [Roche]) and precleared by adding 60 μl of salmon sperm DNA-protein A agarose beads (Upstate, Lake Placid, NY) for 1 h at 4°C on a rolling wheel. Immunoprecipitations were performed overnight at 4°C on a rolling wheel using 3 μg of antibody (antibodies used were anti-Fli-1 [ab-15-289-500 {ABcam} or sc-356 {Santa-Cruz}], anti-Spi-1 [sc-352 {Santa Cruz}], and anti-UBC9 [sc-10759 {Santa Cruz}]). Twenty microliters of protein A agarose beads were added to 2 ml of immunoprecipitations, and the incubation was pursued for 90 min at 4°C. Beads were washed six times (2 min each on ice) in the following buffers: twice in 500 μl of low-salt buffer (20 mM Tris [pH 8.0], 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100), once in 1 ml of high-salt buffer (10 mM Tris [pH 8.0], 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100), once in 1 ml of LiCl buffer (10 mM Tris [pH 8.0], 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.25 M LiCl), and twice in 1 ml of TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA). Chromatin samples were eluted in a final volume of 500 μl by two successive incubations of the beads (15 min at room temperature) in 250 μl of elution buffer (100 mM NaHCO3, 1% SDS). The cross-link was reversed by 4 h of incubation at 65°C in the presence of 200 mM NaCl. DNA was finally treated by proteinase K (1 h at 45°C), extracted with phenol-chloroform, and precipitated. Immunoprecipitated and input DNA (prepared from aliquots taken before the immunoprecipitation step) were quantified by real-time PCR using a SYBR green PCR kit and the oligonucleotides indicated in Table S1 in the supplemental material.
Flow cytometry analyses.
Proportions of apoptotic cells were determined by flow cytometry performed on a FACSCalibur flow cytometer (Becton-Dickinson, Le Pont deClaix, France) after double labeling using an annexin V-fluorescein isothiocyanate kit (AbCys, Paris, France) and following the recommendations of the manufacturer. For cell cycle analyses, 106 cells were washed in PBS and fixed for at least 30 min at 4°C in PBS-ethanol (70%). After another PBS wash, RNA was digested for 30 min at room temperature by 50 μg/ml RNase A (Sigma), and propidium iodide (50 μg/ml; Sigma) was added before fluorescence-activated cell sorting analysis.
siRNA transfection.
745A/TR/shfli1_749#44 cells (2 × 105) were transfected with 80 pmol of a double-stranded RNA oligonucleotide targeting Spi-1 (Dharmacon) or a control double-stranded RNA oligonucleotide targeting the luciferase mRNA (Eurogentec, Angers, France) using Oligofectamine (Invitrogen) according to the manufacturer's instructions. Transfections were repeated every 12 h during 2 days. Small interfering RNA (siRNA) sequences are indicated in Table S1 in the supplemental material.
Whole-transcriptome profiling and analysis.
745A/TR/shfli1_749#44 cells were grown for 2 days in the presence or absence of Dox to induce Fli-1 knockdown and then for one additional day in the presence of HMBA (still in the presence or absence of Dox) to induce erythroid differentiation associated with Spi-1 knockdown. Whole-transcriptome profiles under these four different conditions were then determined by two-color microarrays experiments using the same universal mouse RNA reference (Clontech) and murine cDNA microarrays representing 17106 mouse unigene clusters (IGBMC, Strasbourg, France). Normalized data were analyzed using the GeneSpring GX 7.3 software program (Agilent, CA) to select all transcripts (879) that were statistically downregulated (P < 0.05) by at least twofold in response to HMBA in the presence of Dox. Among this list, 128 transcripts were found to be statistically (P < 0.05) affected by Dox by at least 1.3-fold, and 117 of these 128 transcripts (corresponding to the list given in Table S3 in the supplemental material) were also downregulated by Dox, thus indicating the additive effect of Dox and HMBA. Symmetrically, we identified 560 transcripts that are significantly (P < 0.05) upregulated by HMBA in the presence of Dox. Among this list, 164 were significantly affected by Dox by at least 1.3-fold, and most of them (146 transcripts, corresponding to the list given in Table S4 in the supplemental material) were also upregulated by Dox, indicating the additive effect of HMBA and Dox. Transcripts that appear several times in Tables S3 and S4 in the supplemental material correspond to transcripts for which several probes were present on the microarrays. Gene set enrichment analysis was performed using the software program GSEA v2.0 (56).
RESULTS
Fli-1 knockdown in F-MuLV-infected erythroleukemic cells.
To address the role of Fli-1 in F-MuLV-infected erythroleukemic cells, we decided to knock down Fli-1 in the NN10 erythroleukemic cell line using Dox-inducible shRNA expression. NN10/TR cells expressing high levels of the Tetr repressor were first established and then transfected with vectors allowing Dox-inducible expression of either one of two shRNAs (fli1_749 or fli1_1508) targeting two different regions of Fli-1 mRNA or of control shRNA (SCR) with scrambled sequence. Fli-1 levels were analyzed by Western blotting in individual clones grown for 2 days in the presence or absence of Dox (Fig. 1). Clone 5 (expressing shRNA fli1_749), which displayed a 95% reduction of Fli-1 in response to Dox (Fig. 1A, lanes 3 and 4), and clones 10 and 23 (expressing shRNA fli1_1508), with smaller Fli-1 reductions (65% and 85%, respectively; Fig. 1B, lanes 5 to 8), were retained for further studies. Importantly, Fli-1 levels did not change significantly in response to Dox either in parental NN10/TR cells (Fig. 1A and B, lanes 1 and 2) or in three randomly chosen clones expressing control shRNA (Fig. 1B, lanes 3 to 8).
Dox did not affect the growth rate, viability, or differentiation status of parental NN10/TR cells (Fig. 2, right). In contrast, the growth rate of clone 5 was strongly reduced in the presence of Dox in association with an increase in the percentage of dead cells (trypan blue positive) and differentiated cells (benzidine positive) (Fig. 2, left). After 5 days in the presence of Dox, the total number of cells in clone 5 cultures was reduced more than fivefold, which included >35% of dead cells and around 6% of differentiated cells. Confirming the induction of erythroid differentiation, the levels of several erythroid-specific transcripts, such as Alas2, Ahsp, and α-globin, were increased at as early as 2 days of Dox treatment (3.2-, 2.7-, and 12.5-fold increases, respectively) (Table 1). No Dox effect on proliferation, survival, or differentiation was observed in the three clones expressing control shRNA (Table 1). In contrast, similar but slightly less pronounced Dox effects were observed in clones 10 and 23 (Table 1), displaying a smaller Fli-1 reduction (Fig. 1B).
FIG. 2.
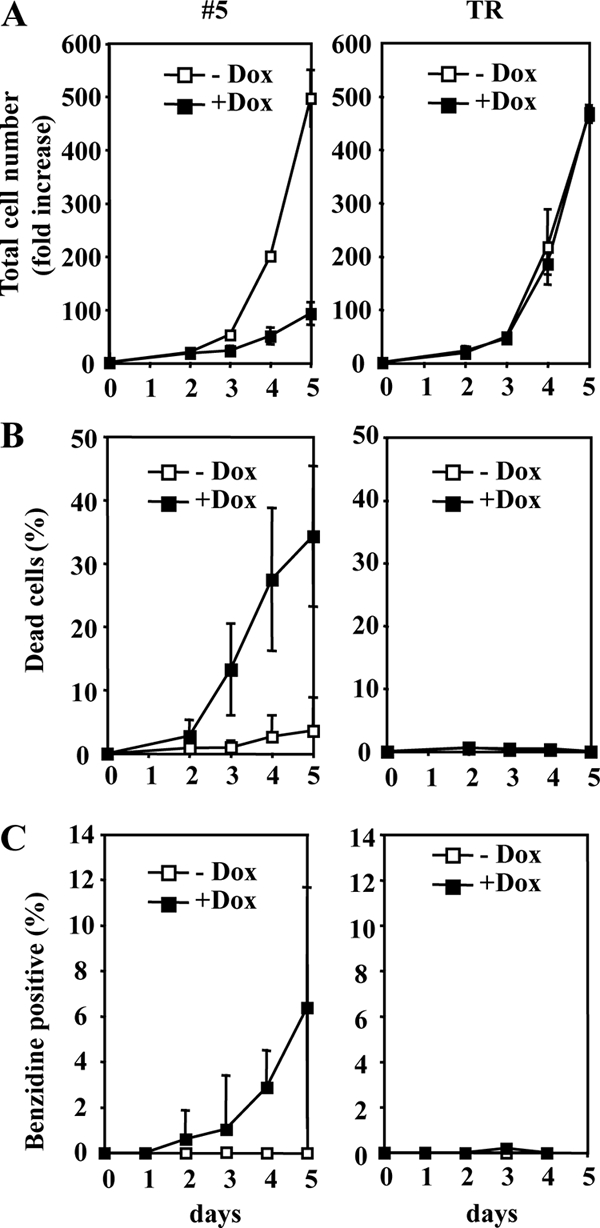
Fli-1 knockdown reduces proliferation and survival and induces differentiation of NN10 cells. NN10/TR and NN10/TR/shfli1_749#5 cells were treated with Dox or not treated for 5 days. The evolution of the increase in the total cell number (viable and dead cells) (A), the percentages of dead cells determined by Trypan blue exclusion (B), and the percentages of hemoglobin-containing differentiated cells determined by benzidine staining (C) are shown (means and standard deviations from three independent experiments are indicated).
We determined that increased cell death was due to apoptosis, as evidenced by an increased proportion of hypodiploid cells (sub-G1) (Fig. 3A) and annexin-positive/propidium-negative cells (Fig. 3B). Fli-1 has been described (34) as a direct transcriptional activator of the bcl2 gene in F-MuLV-induced erythroleukemic cells. As expected, bcl2 transcripts in clone 5 decreased by threefold at as early as 2 days of Dox treatment (Table 1). This prompted us to enforce Bcl2 expression in clone 5 to reduce cell death in the presence of Dox and to determine if the growth rate was still affected. We thus selected clone 22, which expresses high levels of the exogenous human Bcl2 protein when Fli-1 expression is reduced in the presence of Dox (Fig. 4A). Despite a strong reduction of Dox-induced mortality in clone 22 (Fig. 4C), Dox-induced cell growth reduction remains similar to that of clone 5 (Fig. 4B). This result indicates that effects of Dox on cell proliferation and cell death occur independently. Altogether, these data demonstrate that Fli-1 knockdown in NN10 cells independently reduces cell proliferation and viability and concomitantly allows a small proportion of cells to reinitiate terminal erythroid differentiation.
FIG. 3.
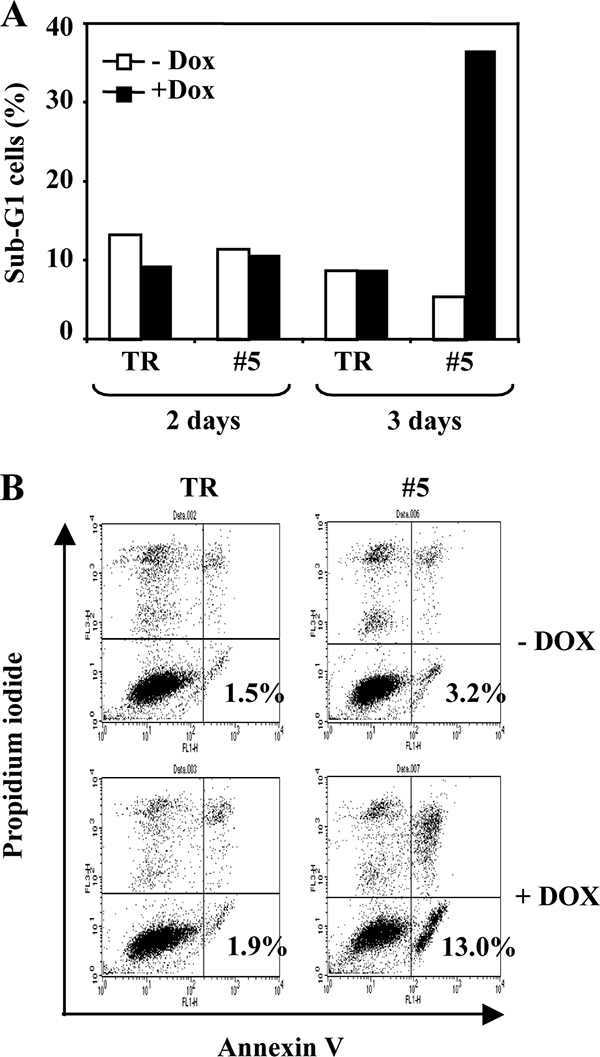
Fli-1 knockdown induces apoptosis of NN10 cells. NN10/TR and NN10/TR/shfli1_749#5 cells were treated with Dox or not treated for 3 days. After 2 and 3 days, the percentages of hypodiploid cells (sub-G1) were determined by flow cytometry after propidium iodide labeling (A). After 3 days, cells were labeled with annexin V and propidium iodide and the percentage of annexin V-positive/propidium-negative apoptotic cells was determined by flow cytometry (B). Typical results from two different experiments are shown.
Fli-1 knockdown in SFFV-infected erythroleukemic cells.
To address the role of Fli-1 in SFFV-infected erythroleukemic cells, we decided to knock down Fli-1 in the 745A erythroleukemic cell line. As described above, 745A/TR cells expressing high levels of the Tetr repressor were transfected with vectors allowing Dox-inducible expression of either one of the two shRNAs targeting Fli-1 mRNA or control shRNA. We identified clone 44 (expressing shRNA fli1_749) and clone 1 (expressing shRNA fli1_1508), which display a 90% and 80% decrease of Fli-1 protein levels in the presence of Dox (Fig. 5A, lanes 3 to 6), respectively. Such a decrease in the Fli-1 protein was observed neither in parental 745A/TR cells (Fig. 5A and B, lanes 1 and 2) nor in three randomly chosen clones expressing control shRNA (Fig. 5B, lanes 3 to 8). Dox did not induce cell death or differentiation and did not change significantly the growth rate of clones 44 and 1 under standard liquid culture conditions (data not shown). However, in semisolid medium with Dox, clones 44 and 1 generated colonies in reduced number, of smaller size (45% and 25% reduction, respectively; Fig. 6A and Table 2), and composed of undifferentiated cells. The number and size of colonies generated by clone 745/TR or clones expressing control shRNA were not affected (Fig. 6A and Table 2). The absence of spontaneous erythroid differentiation in Dox-treated clones 44 and 1 most probably reflects the dominant effect of endogenous Spi-1, such as inhibition of GATA-1 (55), but does not exclude a putative contribution of Fli-1 to the inhibition of differentiation. We previously observed that whereas Spi-1 expression was abolished, Fli-1 expression was moderately reduced during HMBA-induced differentiation of 745A cells (54). Thus, we investigated the effect of Fli-1 knockdown in clones 44 and 1 grown for 2 days in the presence or absence of Dox before HMBA addition in culture medium. The kinetic of differentiation was examined through G1 arrest (Fig. 6B) and an increase in benzidine-positive differentiated cells (Fig. 6C). After only 2 days in HMBA, Dox-induced Fli-1 knockdown causes an increase in G1-blocked and benzidine-positive cells. Accelerated differentiation was further confirmed by a marked increase in several erythroid transcripts, such as Alas2, Ahsp, or α-globin (Table 2). Concomitantly, cell viability was markedly and specifically affected by Dox, as deduced from the progressive increase in the number of dead cells, reaching 30% and 40% after 4 days in the presence of both HMBA and Dox (Fig. 6D). This cell death increase was due to apoptosis, as indicated by the increased number of hypodiploid cells (Fig. 7A) and propidium iodide-negative/annexin-positive cells (Fig. 7B). These effects were not observed either in 745/TR cells or in clones expressing control shRNA. Altogether, these results indicated that Fli-1 knockdown reduces cell clonogenicity, induces apoptosis, and accelerates the HMBA-induced differentiation of 745A erythroleukemic cells.
FIG. 6.

Fli-1 knockdown reduces the clonogenicity and increases the differentiation and death of 745A cells in the presence of HMBA. 745A/TR, 745A/TR/shfli1_749#44, and 745A/TR/shfli1_1508#1 cells were treated with Dox or not treated for 2 days and reseeded either in semisolid medium or in liquid medium supplemented with 5 mM HMBA to induce their differentiation. (A) Relative numbers of clones obtained in semisolid medium; (B) evolution of the percentage of viable cells in G1 phase; (C) evolution of the percentage of hemoglobin-containing (benzidine-positive) differentiated cells; (D) evolution of the percentage of dead cells (trypan blue positive). Means and standard deviations of each parameter from three independent experiments are shown. Statistically significant differences (P < 0.05 in paired Student test) are indicated by asterisks.
FIG. 7.
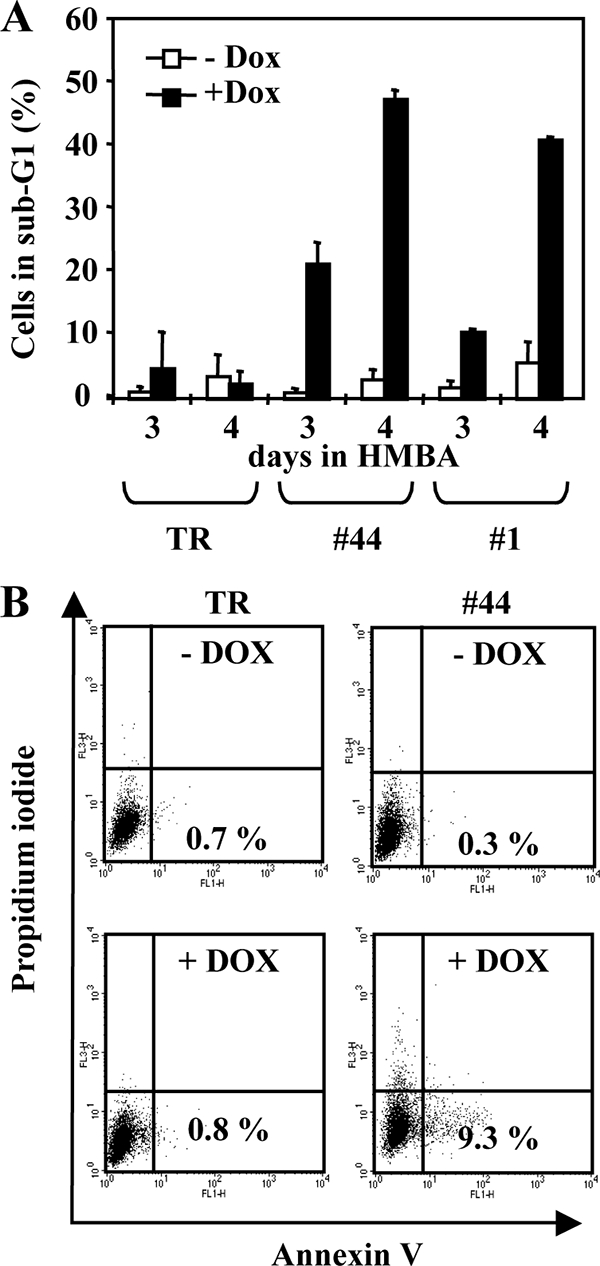
Fli-1 knockdown induces apoptosis in 745A cells. 745A/TR (TR), 745A/TR/shfli1_749#44 (#44), and 745A/TR/shfli-2#1 (#1) cells were treated with Dox or not treated for 2 days, and erythroid differentiation was then induced by adding 5 mM HMBA for 3 or 4 days before analysis. (A) Percentages of hypodiploid cells (sub-G1) determined by flow cytometry after propidium iodide labeling (means and standard deviations from three independent experiments). (B) Annexin V/propidium iodide flow cytometer diagrams obtained 2 days after the addition of HMBA. The percentages of annexin V-positive and propidium iodide-negative apoptotic cells are indicated (typical results from two different experiments).
Additive effects of Spi-1 and Fli-1 knockdown in SFFV-infected erythroleukemic cells.
Since HMBA suppresses Spi-1 expression, the additive effects of Fli-1 knockdown and HMBA on proliferation arrest and differentiation of clone #44 and clone 1 cells could result from the combined loss of Spi-1 and Fli-1. To test this possibility, we combined Dox-inducible Fli-1 knockdown with siRNA-mediated Spi-1 knockdown. Clone 44 cells were transfected two times a day for 2 days with Spi-1 siRNA in the presence or absence of Dox and analyzed 60 h after the first transfection (Fig. 8). Spi-1 siRNA transfection suppressed most Spi-1 protein expression (Fig. 8A, lane 3) and induced the differentiation of around 10% of the cells (Fig. 8B). Simultaneous knockdown of Spi-1 and Fli-1 expression (Fig. 8A, lane 6) increased the percentage of differentiated cells up to 25%. Clonogenicity was markedly reduced by Spi-1 knockdown and almost completely suppressed by concomitant Fli-1 knockdown induced by Dox (Fig. 8C). Finally, a threefold increase in cell death could be observed after Spi-1 and Fli-1 double knockdown compared to results with Spi-1 or Fli-1 simple knockdown. None of these effects could be observed following transfection with control siRNA (Fig. 8D). These results indicated that losses of Spi-1 and Fli-1 contribute in an additive manner to the proliferation arrest and differentiation of 745A cells, whereas cell death is induced only in the absence of both factors.
Spi-1 and Fli-1 directly activate a common set of genes involved in ribosome biogenesis.
Spi-1 and Fli-1 recognize similar core DNA binding sites, suggesting the interesting possibility that they may deregulate a common set of genes. This prompted us to look for a set of common target genes which could at least partially explain the additive effects of their loss of expression on the proliferation and differentiation of clone 44. To address this question, we used microarrays harboring 17106 murine cDNAs to perform a gene expression profiling analysis with clone 44 cells grown for 2 days in the presence or absence of Dox (allowing Fli-1 downregulation) and then for 1 day in the presence or absence of HMBA (allowing Spi-1 downregulation). From this analysis, we identified two groups of transcripts whose expression was increased or decreased by at least twofold by HMBA and further significantly increased or decreased in the same way by Dox (see Tables S4 and S3, respectively, in the supplemental material).
Consistent with the additive contribution of Spi-1 and Fli-1 knockdown to the stimulation of differentiation of clone 44, several transcripts characteristic of erythroid differentiation, such as alas 2, α-globin, ftl1, and fech, were increased by both HMBA and Dox treatment (see Table S4 in the supplemental material). Besides this erythroid signature, we found that a strikingly high proportion of transcripts (22%) which were significantly decreased by both HMBA and Dox treatment corresponded to genes encoding proteins involved in ribosome biogenesis (see Table S3 in the supplemental material). These genes include, notably, two genes (Myc and Rpo1-3) involved in rRNA transcription, eight genes (Ddx21, Imp4, Nhp2l1, Nip7, Nol6, Nolc1, Npm1, and Rcl1) involved in different steps of rRNA maturation, one ribosomal protein gene (Rpl18), and eight snoRNA host genes involved in posttrancriptional rRNA modifications (Cct6a, Gnb2L1, Hspa8, Snord22, Eif4g1, Snrpb, Mbd2, and Tcp1). These results prompted us to list all ribosome-related genes present on our microarrays and to perform gene set enrichment analysis in response to HMBA and Dox. This analysis allowed us to identify two sets of genes that are indeed significantly enriched among genes that are the most downregulated in response to HMBA and Dox (93 genes involved in different steps of rRNA transcription and maturation and 35 H/ACA box snoRNA host genes) (see Fig S1 in the supplemental material). Most members of two other sets of genes (32 C/D box snoRNA host genes and 66 genes encoding ribosomal proteins) are also downregulated in response to either HMBA or Dox, although in that case the enrichment is not statistically significant (see Fig S1 in the supplemental material).
Nine of the ribosome-related genes individually identified by microarray analysis as putative common target genes of Spi-1 and Fli-1 were randomly selected for further investigation. Quantitative RT-PCR analyses confirmed that the transcript levels from all of these ribosome-related genes decreased in an additive manner in response to HMBA and Dox in clone 44 cells (Fig. 9B) whereas they decreased in response to HMBA only in control 745A/TR cells (Fig. 9A). Interestingly, each one of these genes displays at least one highly conserved putative ETS binding site (GGAA) located less than 100 bp from the known transcription initiation site (data not shown). We thus investigated whether Spi-1 and Fli-1 interact directly with these gene promoter regions in erythroleukemic cells through chromatin immunoprecipitation assays using Spi-1 or Fli-1 antibody. Except for the Npm1 gene, significant enrichments indicating Spi-1 (Fig. 9C) and Fli-1 (Fig. 9D) occupancy in clone 44 cells were identified for all of them. Further RT-PCR and chromatin immunoprecipitation analyses performed on five additional snoRNA host genes confirmed their additive downregulation in response to HMBA and Dox and both Spi-1 and Fli-1 occupancy in the proximal promoter region of three of them (Fig. 10). Similar analyses were then performed in clone 5 to determine if Fli-1 might be involved in the direct activation of the same set of genes in F-MuLV-infected erythroleukemic cells harboring a fli-1-activated locus. Except for Rpl18, Npm1, or Nolc1, the transcript levels for all other ribosome-related genes analyzed were significantly decreased in response to Dox in clone 5 but not in parental NN10/TR cells (Fig. 11A). Control Gata1, p45 Nfe2, and Scl gene transcripts were not affected by Dox treatment either in clone 5 or in parental NN10/TR cells. Furthermore, except for Npm1, as in clone 44, significant enrichments indicating Fli-1 occupancy at all other gene promoters were clearly detected in clone 5 (Fig. 11B). Taken together, these data indicate that in correlation with their contribution to proliferation and inhibition of differentiation in Friend erythroleukemic cells, Spi-1 and Fli-1 directly activate a significant number of common target genes involved in ribosome biogenesis through their direct binding to highly conserved ETS binding sites located in the proximal promoters of these genes.
FIG. 10.
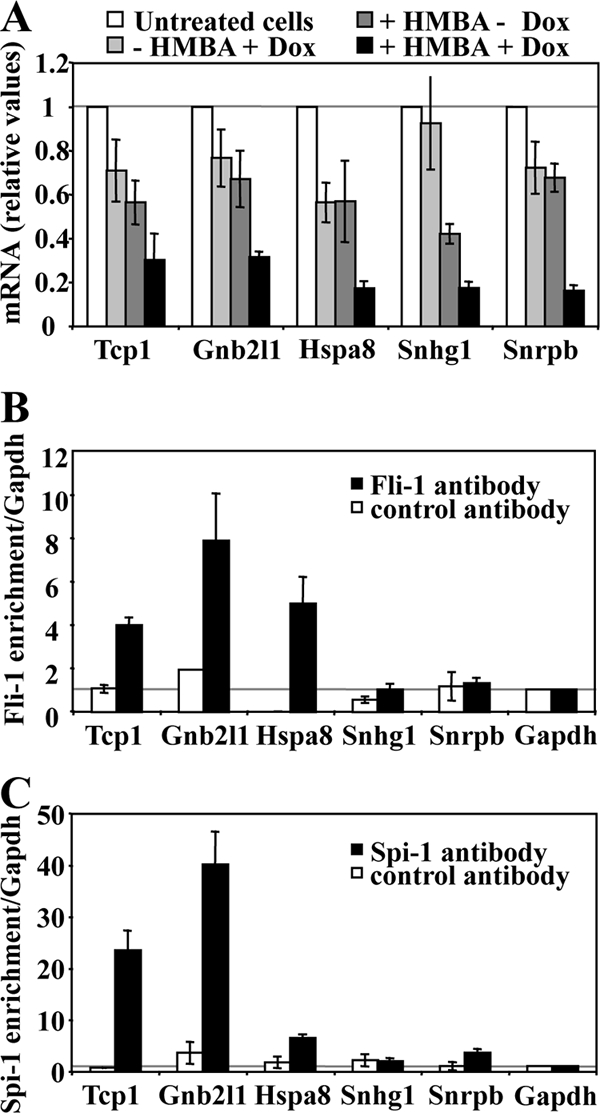
Spi-1 and Fli-1 directly activate several snoRNA host genes in 745A cells. 745A/TR/shfli_749#44 cells were treated with Dox for 2 days or left untreated, and differentiation was induced by adding 5 mM HMBA for 1 day before performing quantitative RT-PCR analyses of the indicated gene transcripts. (A) Relative levels of transcripts determined in 745A/TR/shfli_749#44 cells (means and standard deviations from three independent experiments). (B) Fli-1 chromatin occupancy of the indicated gene promoters, determined by chromatin immunoprecipitation on untreated 745A/TR/shfli_749#44 cells using Fli-1-specific (black bars) or control Ubc9 antibody. The Gapdh promoter was used as a negative control. Results are expressed as relative proportions of the input chromatin which has been precipitated by antibodies standardized to the background levels determined on the Gapdh gene promoter (means and standard deviations from three independent experiments). (C) Spi-1 chromatin occupancy of the indicated gene promoters, determined as in panel B by chromatin immunoprecipitation on untreated 745A/TR/shfli_749#44 cells using Spi-1 antibody.
FIG. 11.

Fli-1 directly activates the same set of genes involved in ribosome biogenesis in NN10 cells as do Spi-1 and Fli-1 in 745A cells. NN10/TR and NN10/TR/shfli1_749#5 cells were treated with Dox or not treated for 2 days before quantitative RT-PCR analyses of the indicated gene transcripts were performed. (A) Dox-induced n-fold changes in the indicated gene transcripts levels in NN10/TR (white bars) or NN10/TR/shfli1_749#5 (black bars) cells (means and standard deviations from three different experiments). (B) Fli-1 chromatin occupancy of the indicated gene promoters, determined by chromatin immunoprecipitation on untreated NN10/TR/shfli1_749#5 cells. See the legend for Fig. 8C and D (typical result from two different experiments are shown).
DISCUSSION
We showed in this study that Fli-1 knockdown induced proliferation arrest, massive apoptosis, and limited differentiation in F-MuLV-infected erythroleukemic cells. While Fli-1 knockdown had a very limited effect in SFFV-infected cells in the presence of Spi-1, Fli-1 knockdown markedly enhanced proliferation arrest and differentiation induced by Spi-1 knockdown. Most importantly, this additive effect of Spi-1 and Fli-1 knockdown was correlated with an additive negative effect on the expression of many genes involved in ribosome biogenesis whose promoters are occupied by both Spi-1 and Fli-1 in SFFV cells and by Fli-1 in F-MuLV cells.
The association of decreased bcl2 expression with cell death induced by Fli-1 knockdown in F-MuLV cells is in agreement with results in previous studies (34). Although viability of F-MuLV cells could be rescued by enforced expression of Bcl2, further investigations are still required to determine if Fli-1-mediated expression of Bcl2 is really required for their survival. More surprisingly, Spi-1 knockdown did not induce cell death in SFFV cells, contrasting with apoptosis induced in Spi-1 transgenic cell lines (47). This apparent discrepancy might be explained by different Epo-R signaling due to the gp55 protein being expressed only in SFFV cells. Our other observation that cell death occurred only after both Spi-1 and Fli-1 knockdown indicates that Spi-1 and Fli-1 are redundantly involved in SFFV cell survival. However, contrary to what could be expected from our results with F-MuLV cells, we found no evidence for bcl2 gene regulation by either Spi-1 or Fli-1 in SFFV cells (data not shown).
Spi-1 and Fli-1 directly activate common target genes involved in ribosome biogenesis.
Our conclusion that Spi-1 and Fli-1 directly activate common ribosome-related target genes is based on several concordant observations: (i) the expression of these genes decreased in response to Fli-1 knockdown in F-MuLV cells and decreased in an additive manner in response to Spi-1 and Fli-1 knockdown in SFFV cells, (ii) as previously noticed by others for ribosomal protein genes (41), most of these genes display highly conserved consensus ETS binding sites in their promoter regions, and (iii) with very few exceptions, these promoter regions are bound by Fli-1 in F-MuLV cells and by both Spi-1 and Fli-1 in SFFV cells. We also determined using transient expression assays that mouse imp4 gene promoter activity is reduced by 40% by the mutation of a conserved ETS binding site located at position −45 (data not shown). A few other transcription factors, such as YY1, SP1, and mainly c-Myc, have already been identified as direct transcriptional activators of ribosome-related genes (41). Interestingly, c-Myc expression also decreased in an additive manner in response to Spi-1 and Fli-1 knockdown (see Table S3 in the supplemental material), and this decrease in c-Myc might therefore also contribute, with Spi-1 and Fli-1 knockdown itself, to the decrease in ribosome-related gene expression. According to this possibility, Spi-1, Fli-1, and c-Myc seem to belong to the same regulatory network, allowing the stimulation of genes involved in ribosome biogenesis.
Remarkably, ribosome-related target genes of Spi-1 and Fli-1 appear to be involved in all different steps of ribosome biogenesis, including rRNA synthesis, maturation, and posttranscriptional modifications, as well as ribosomal protein synthesis. Accordingly, a significant impact on ribosome biogenesis can be expected from Spi-1 and Fli-1 knockdown. Indeed, previous studies already reported a decrease in ribosomal DNA gene transcription in HMBA-treated SFFV cells (21). In agreement with these studies, we observed a marked decrease in the size of nucleoli (data not shown) and in the level of 45S rRNA precursors in SFFV cells in response to HMBA treatment or Fli-1 knockdown (Fig. 9A and B). We suggest that this decrease in ribosomal DNA gene transcription might be at least partially explained by the decreased expression of the RNA polymerase I subunit RPO1-3, but this remains to be formally established.
Can the decrease in ribosome-related gene expression contribute to the proliferation arrest and differentiation induced by Spi-1 and Fli-1 knockdown?
The decrease in ribosome-related gene expression could be the simple consequence of the terminal differentiation process allowed by Spi-1 and Fli-1 knockdown. This terminal differentiation process itself is potentially mediated by the deregulation of many Spi-1 and Fli-1 target genes. Among interesting candidates, we noticed the additive decrease in the expression of c-Myc (see Table S3 in the supplemental material) and the additive increase of Btg2 (35) (see Table S4 in the supplemental material), known as a prooncogene and an antioncogene, respectively. Furthermore, according to the known Gata-1/Spi-1 (55) and Fli-1/Eklf (23, 53) functional antagonisms, the derepression of critical Gata-1 and/or Eklf target genes most probably also contributes to the induction of terminal differentiation. However, independently of these possibilities, the identification of ribosome-related genes as being direct target genes representing more than 20% of common target genes of Spi-1 and Fli-1 strongly favors the other possibility that their decreased expression may at least partially contribute to the effect of Spi-1 and Fli-1 knockdown on differentiation and/or proliferation arrest. Indeed, increasing evidence indicates that ribosome biogenesis per se is actively involved in cell cycle control either through the control of protein translation (16, 37, 49) or through the p53 pathway (25, 36). For example, in several situations known as situations of ribosomal or nucleolar stress, free ribosomal proteins, such as RPS7 (12), RPL5, RPL11, or RPL23, directly interact with MDM2, leading to the stabilization of p53 and/or derepression of p73 activity and cell cycle arrest (17, 29, 59, 66). This stress pathway can be activated by many different chemicals affecting nucleolar organization, such as low doses of actinomycin D, specifically inhibiting RNA polymerase I activity, or 5-fluorouracyl (59). Interestingly, low doses of actinomycin D have already been reported to induce proliferation arrest and differentiation of SFFV cells (61). In our work, we found that not only treatment with actinomycin D but also 5-fluorouracyl treatment nicely reproduced Spi-1 and Fli-1 knockdown in 745A cells and Fli-1 knockdown in NN10 cells by inducing proliferation arrest and differentiation in both cases (see Table S2 in the supplemental material). Intriguingly, the fact that P53 is inactivated in 745A and NN10 cells (7) would preclude the possibility of a nucleolar stress response in these cells. However, several other proteins, such as p73 or FOXO3a, which is known to be very important in terminal erythroid differentiation (5), are also regulated by MDM2 (65) and remain interesting signaling candidates in response to nucleolar stress potentially induced by Spi-1 and Fli-1 knockdown. Alternatively, we cannot presently exclude the possibility that Spi-1 and Fli-1 knockdown also contribute to terminal differentiation by altering protein translation through the production either of a reduced number or of altered ribosomes. All these possibilities justify further investigations of the incidence of Spi-1 and Fli-1 expression levels on ribosome biogenesis not only in established erythroleukemic cells but also in native erythrocytic progenitors in order to fully understand how their involvement in ribosome-related gene activation may contribute, as already reported for c-Myc (6, 16), to their oncogenic potential.
Control of ribosome biogenesis as a common redundant and ancestral function of ETS transcription factors.
Our present study is reminiscent of a recent genome-wide analysis of promoter occupancy which showed that three other ETS proteins (Ets1, Elf1, and Gabpα) occupy a common set of gene promoters in the human Jurkat lymphoid cell line (28). Strikingly, the promoter regions of most ribosome-related target genes of Spi-1 and Fli-1 identified in the present study were also identified as being bound by at least one of these three other ETS factors. These convergent results strengthen the emerging concept that transcriptional regulation of genes involved in ribosome biogenesis is most probably a generic property shared by many members of the ETS family (28). Interestingly, many ETS factors are oncogenic (52). For example, prostate cancers are frequently associated with deregulated expression of ETS factors (1, 31, 48), and the active contribution of at least ETS2 (11) and ERG (30, 58) to the transformed phenotype has been clearly established. Furthermore, we found that promoters of several ribosome-related genes are also bound by ETS1 or ETS2 in the two prostate cancer cell lines PC3 and DU145, respectively (data not shown). Together, these data raise the interesting possibility that, as for c-Myc, deregulation of ribosome biogenesis might be a common mechanism contributing to the oncogenic potential of several ETS factors.
Ribosome biogenesis is a highly sophisticated process involving many genes, suggesting a high degree of coordinated expression and a strong selection pressure on their transcriptional regulation (10). This strong selection pressure is highlighted by the strikingly high conservation of ETS DNA binding sites that we and others (28) noticed in the proximal promoters of genes involved in ribosome biogenesis. We suggest that the ancestral ETS gene was already involved in this function and that this ancestral function has been conserved in most successive members which appeared later during evolution by duplication of the ancestral ETS gene. This could confer the interesting property of current ETS factors to coordinate such ancestral housekeeping functions with other more recently acquired tissue-specific functions.
Supplementary Material
Acknowledgments
We thank Colette Gonnet for excellent technical assistance in DNA cloning, Laure Granger for DNA sequencing facilities, Céline Keime and Aurélie Landreau for their valuable help in the use of GeneSpring software on the PRABI (Pôle Rhône-Alpin de Bioinformatique) platform, and Edouard Bertrand for his help in the identification of snoRNA host genes.
This work was supported by grants from the CNRS and Université Lyon 1 and by specific grants from the following associations: Fondation de France (no. 2003005020), Ligue contre le Cancer (Labelisation 2005, CIT program, and salaries to G.G., S.B., and G.J.), and the Société Française d'Hématologie (salary to G.J.). F.M., J.-J.D., F.M.-G., and C.G. are members of the INSERM (Institut National de la Santé et de la Recherche Médicale). B.G. and J.S. are members of the CNRS (Centre National de la Recherche Scientifique).
Footnotes
Published ahead of print on 16 March 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Alipov, G., T. Nakayama, M. Ito, K. Kawai, S. Naito, M. Nakashima, D. Niino, and I. Sekine. 2005. Overexpression of Ets-1 proto-oncogene in latent and clinical prostatic carcinomas. Histopathology 46202-208. [DOI] [PubMed] [Google Scholar]
- 2.Anguita, E., J. Hughes, C. Heyworth, G. A. Blobel, W. G. Wood, and D. R. Higgs. 2004. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 232841-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ano, S., R. Pereira, M. Pironin, I. Lesault, C. Milley, I. Lebigot, C. T. Quang, and J. Ghysdael. 2004. Erythroblast transformation by FLI-1 depends upon its specific DNA binding and transcriptional activation properties. J. Biol. Chem. 2792993-3002. [DOI] [PubMed] [Google Scholar]
- 4.Atar, O., and B. Z. Levi. 2005. PU.1 silencing leads to terminal differentiation of erythroleukemia cells. Biochem. Biophys. Res. Commun. 3291288-1292. [DOI] [PubMed] [Google Scholar]
- 5.Bakker, W. J., T. B. van Dijk, M. Parren-van Amelsvoort, A. Kolbus, K. Yamamoto, P. Steinlein, R. G. Verhaak, T. W. Mak, H. Beug, B. Lowenberg, and M. von Lindern. 2007. Differential regulation of Foxo3a target genes in erythropoiesis. Mol. Cell. Biol. 273839-3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barna, M., A. Pusic, O. Zollo, M. Costa, N. Kondrashov, E. Rego, P. H. Rao, and D. Ruggero. 2008. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456971-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnache, S., F. Wendling, C. Lacombe, N. Denis, M. Titeux, W. Vainchenker, and F. Moreau-Gachelin. 1998. Spi-1 transgenic mice develop a clonal erythroleukemia which does not depend on p53 mutation. Oncogene 162989-2995. [DOI] [PubMed] [Google Scholar]
- 8.Ben-David, Y., and A. Bernstein. 1991. Friend virus-induced erythroleukemia and the multistage nature of cancer. Cell 66831-834. [DOI] [PubMed] [Google Scholar]
- 9.Ben-David, Y., E. B. Giddens, and A. Bernstein. 1990. Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc. Natl. Acad. Sci. USA 871332-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown, S. J., M. D. Cole, and A. J. Erives. 2008. Evolution of the holozoan ribosome biogenesis regulon. BMC Genomics 9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carbone, G. M., S. Napoli, A. Valentini, F. Cavalli, D. K. Watson, and C. V. Catapano. 2004. Triplex DNA-mediated downregulation of Ets2 expression results in growth inhibition and apoptosis in human prostate cancer cells. Nucleic Acids Res. 324358-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, D., Z. Zhang, M. Li, W. Wang, Y. Li, E. R. Rayburn, D. L. Hill, H. Wang, and R. Zhang. 2007. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene 265029-5037. [DOI] [PubMed] [Google Scholar]
- 13.Choe, K. S., F. Radparvar, I. Matushansky, N. Rekhtman, X. Han, and A. I. Skoultchi. 2003. Reversal of tumorigenicity and the block to differentiation in erythroleukemia cells by GATA-1. Cancer Res. 636363-6369. [PubMed] [Google Scholar]
- 14.Choppin, J., N. Casadevall, C. Lacombe, F. Wendling, E. Goldwasser, R. Berger, P. Tambourin, and B. Varet. 1985. Production of erythropoietin by cloned malignant murine erythroid cells. Exp. Hematol. 13610-615. [PubMed] [Google Scholar]
- 15.Coute, Y., J. A. Burgess, J. J. Diaz, C. Chichester, F. Lisacek, A. Greco, and J. C. Sanchez. 2006. Deciphering the human nucleolar proteome. Mass Spectrom. Rev. 25215-234. [DOI] [PubMed] [Google Scholar]
- 16.Dai, M. S., and H. Lu. 2008. Crosstalk between c-Myc and ribosome in ribosomal biogenesis and cancer. J. Cell Biochem. 105670-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai, M. S., S. X. Zeng, Y. Jin, X. X. Sun, L. David, and H. Lu. 2004. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 247654-7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delgado, M. D., M. Hallier, P. Meneceur, A. Tavitian, and F. Moreau-Gachelin. 1994. Inhibition of Friend cells proliferation by spi-1 antisense oligodeoxynucleotides. Oncogene 91723-1727. [PubMed] [Google Scholar]
- 19.Demers, C., C. P. Chaturvedi, J. A. Ranish, G. Juban, P. Lai, F. Morle, R. Aebersold, F. J. Dilworth, M. Groudine, and M. Brand. 2007. Activator-mediated recruitment of the MLL2 methyltransferase complex to the beta-globin locus. Mol. Cell 27573-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finkelstein, L. D., P. A. Ney, Q. P. Liu, R. F. Paulson, and P. H. Correll. 2002. sf-Stk kinase activity and the Grb2 binding site are required for Epo-independent growth of primary erythroblasts infected with Friend virus. Oncogene 213562-3570. [DOI] [PubMed] [Google Scholar]
- 21.Fraser, P. J., and P. J. Curtis. 1987. Specific pattern of gene expression during induction of mouse erythroleukemia cells. Genes Dev. 1855-861. [DOI] [PubMed] [Google Scholar]
- 22.Fromont-Racine, M., B. Senger, C. Saveanu, and F. Fasiolo. 2003. Ribosome assembly in eukaryotes. Gene 31317-42. [DOI] [PubMed] [Google Scholar]
- 23.Frontelo, P., D. Manwani, M. Galdass, H. Karsunky, F. Lohmann, P. G. Gallagher, and J. J. Bieker. 2007. Novel role for EKLF in megakaryocyte lineage commitment. Blood 1103871-3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galson, D. L., J. O. Hensold, T. R. Bishop, M. Schalling, A. D. D'Andrea, C. Jones, P. E. Auron, and D. E. Housman. 1993. Mouse beta-globin DNA-binding protein B1 is identical to a proto-oncogene, the transcription factor Spi-1/PU. 1, and is restricted in expression to hematopoietic cells and the testis. Mol. Cell. Biol. 132929-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilkes, D. M., L. Chen, and J. Chen. 2006. MDMX regulation of p53 response to ribosomal stress. EMBO J. 255614-5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henras, A. K., J. Soudet, M. Gerus, S. Lebaron, M. Caizergues-Ferrer, A. Mougin, and Y. Henry. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol. Life Sci. 652334-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hensold, J. O., C. A. Stratton, D. Barth, and D. L. Galson. 1996. Expression of the transcription factor, Spi-1 (PU. 1), in differentiating murine erythroleukemia cells is regulated post-transcriptionally. Evidence for differential stability of transcription factor mRNAs following inducer exposure. J. Biol. Chem. 1713385-3391. [DOI] [PubMed] [Google Scholar]
- 28.Hollenhorst, P. C., A. A. Shah, C. Hopkins, and B. J. Graves. 2007. Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes Dev. 211882-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horn, H. F., and K. H. Vousden. 2008. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 275774-5784. [DOI] [PubMed] [Google Scholar]
- 30.Klezovitch, O., M. Risk, I. Coleman, J. M. Lucas, M. Null, L. D. True, P. S. Nelson, and V. Vasioukhin. 2008. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. USA 1052105-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar-Sinha, C., S. A. Tomlins, and A. M. Chinnaiyan. 2008. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 8497-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebigot, I., P. Gardellin, L. Lefebvre, H. Beug, J. Ghysdael, and C. T. Quang. 2003. Up-regulation of SLAP in FLI-1-transformed erythroblasts interferes with EpoR signaling. Blood 1024555-4562. [DOI] [PubMed] [Google Scholar]
- 33.Lee, C. R., D. Cervi, A. H. Truong, Y. J. Li, A. Sarkar, and Y. Ben-David. 2003. Friend virus-induced erythroleukemias: a unique and well-defined mouse model for the development of leukemia. Anticancer Res. 232159-2166. [PubMed] [Google Scholar]
- 34.Lesault, I., C. T. Quang, J. Frampton, and J. Ghysdael. 2002. Direct regulation of BCL-2 by FLI-1 is involved in the survival of FLI-1-transformed erythroblasts. EMBO J. 21694-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim, I. K. 2006. TIS21 (/BTG2/PC3) as a link between ageing and cancer: cell cycle regulator and endogenous cell death molecule. J. Cancer Res. Clin. Oncol. 132417-426. [DOI] [PubMed] [Google Scholar]
- 36.Lindstrom, M. S., C. Deisenroth, and Y. Zhang. 2007. Putting a finger on growth surveillance: insight into MDM2 zinc finger-ribosomal protein interactions. Cell Cycle 6434-437. [DOI] [PubMed] [Google Scholar]
- 37.Montanaro, L., D. Trere, and M. Derenzini. 2008. Nucleolus, ribosomes, and cancer Am. J. Pathol. 173301-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreau-Gachelin, F., F. Wendling, T. Molina, N. Denis, M. Titeux, G. Grimber, P. Briand, W. Vainchenker, and A. Tavitian. 1996. Spi-1/PU.1 transgenic mice develop multistep erythroleukemias. Mol. Cell. Biol. 162453-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nerlov, C., E. Querfurth, H. Kulessa, and T. Graf. 2000. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 952543-2551. [PubMed] [Google Scholar]
- 40.Pereira, R., C. T. Quang, I. Lesault, H. Holznig, H. Beug, and J. Ghysdael. 1999. FLI-1 inhibits differentiation and induces proliferation of primary erythroblasts. Oncogene 181597-1608. [DOI] [PubMed] [Google Scholar]
- 41.Perry, R. P. 2005. The architecture of mammalian ribosomal protein promoters. BMC Evol. Biol. 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quang, C. T., O. Wessely, M. Pironin, H. Beug, and J. Ghysdael. 1997. Cooperation of Spi-1/PU.1 with an activated erythropoietin receptor inhibits apoptosis and Epo-dependent differentiation in primary erythroblasts and induces their Kit ligand-dependent proliferation. EMBO J. 165639-5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao, G., N. Rekhtman, G. Cheng, T. Krasikov, and A. I. Skoultchi. 1997. Deregulated expression of the PU.1 transcription factor blocks murine erythroleukemia cell terminal differentiation. Oncogene 14123-131. [DOI] [PubMed] [Google Scholar]
- 44.Reed, B. D., A. E. Charos, A. M. Szekely, S. M. Weissman, and M. Snyder. 2008. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet. 4e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rekhtman, N., K. S. Choe, I. Matushansky, S. Murray, T. Stopka, and A. I. Skoultchi. 2003. PU.1 and pRB interact and cooperate to repress GATA-1 and block erythroid differentiation. Mol. Cell. Biol. 237460-7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rekhtman, N., F. Radparvar, T. Evans, and A. I. Skoultchi. 1999. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 131398-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rimmele, P., O. Kosmider, P. Mayeux, F. Moreau-Gachelin, and C. Guillouf. 2007. Spi-1/PU.1 participates in erythroleukemogenesis by inhibiting apoptosis in cooperation with Epo signaling and by blocking erythroid differentiation. Blood 1093007-3014. [DOI] [PubMed] [Google Scholar]
- 48.Rostad, K., M. Mannelqvist, O. J. Halvorsen, A. M. Oyan, T. H. Bo, L. Stordrange, S. Olsen, S. A. Haukaas, B. Lin, L. Hood, I. Jonassen, L. A. Akslen, and K. H. Kalland. 2007. ERG upregulation and related ETS transcription factors in prostate cancer. Int. J. Oncol. 3019-32. [PubMed] [Google Scholar]
- 49.Ruggero, D., and P. P. Pandolfi. 2003. Does the ribosome translate cancer? Nat. Rev. Cancer 3179-192. [DOI] [PubMed] [Google Scholar]
- 50.Ruscetti, S. K. 1999. Deregulation of erythropoiesis by the Friend spleen focus-forming virus. Int. J. Biochem. Cell Biol. 311089-1109. [DOI] [PubMed] [Google Scholar]
- 51.Schuetze, S., R. Paul, B. C. Gliniak, and D. Kabat. 1992. Role of the PU.1 transcription factor in controlling differentiation of Friend erythroleukemia cells. Mol. Cell. Biol. 122967-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seth, A., and D. K. Watson. 2005. ETS transcription factors and their emerging roles in human cancer. Eur. J. Cancer 412462-2478. [DOI] [PubMed] [Google Scholar]
- 53.Starck, J., N. Cohet, C. Gonnet, S. Sarrazin, Z. Doubeikovskaia, A. Doubeikovski, A. Verger, M. Duterque-Coquillaud, and F. Morle. 2003. Functional cross-antagonism between transcription factors FLI-1 and EKLF. Mol. Cell. Biol. 231390-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Starck, J., A. Doubeikovski, S. Sarrazin, C. Gonnet, G. Rao, A. Skoultchi, J. Godet, I. Dusanter-Fourt, and F. Morle. 1999. Spi-1/PU.1 is a positive regulator of the Fli-1 gene involved in inhibition of erythroid differentiation in Friend erythroleukemic cell lines. Mol. Cell. Biol. 19121-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stopka, T., D. F. Amanatullah, M. Papetti, and A. I. Skoultchi. 2005. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. 243712-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramanian, A., P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, and J. P. Mesirov. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 10215545-15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subramanian, A., H. E. Teal, P. H. Correll, and R. F. Paulson. 2005. Resistance to friend virus-induced erythroleukemia in W/W(v) mice is caused by a spleen-specific defect which results in a severe reduction in target cells and a lack of Sf-Stk expression. J. Virol. 7914586-14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun, C., A. Dobi, A. Mohamed, H. Li, R. L. Thangapazham, B. Furusato, S. Shaheduzzaman, S. H. Tan, G. Vaidyanathan, E. Whitman, D. J. Hawksworth, Y. Chen, M. Nau, V. Patel, M. Vahey, J. S. Gutkind, T. Sreenath, G. Petrovics, I. A. Sesterhenn, D. G. McLeod, and S. Srivastava. 2008. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 275348-5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun, X. X., M. S. Dai, and H. Lu. 2007. 5-Fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 2828052-8059. [DOI] [PubMed] [Google Scholar]
- 60.Tamir, A., J. Howard, R. R. Higgins, Y. J. Li, L. Berger, E. Zacksenhaus, M. Reis, and Y. Ben-David. 1999. Fli-1, an Ets-related transcription factor, regulates erythropoietin-induced erythroid proliferation and differentiation: evidence for direct transcriptional repression of the Rb gene during differentiation. Mol. Cell. Biol. 94452-4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Terada, M., E. Epner, U. Nudel, J. Salmon, E. Fibach, R. A. Rifkind, and P. A. Marks. 1978. Induction of murine erythroleukemia differentiation by actinomycin D. Proc. Natl. Acad. Sci. USA 752795-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Truong, A. H., and Y. Ben-David. 2000. The role of Fli-1 in normal cell function and malignant transformation. Oncogene 196482-6489. [DOI] [PubMed] [Google Scholar]
- 63.Xi, H., Y. Yu, Y. Fu, J. Foley, A. Halees, and Z. Weng. 2007. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res. 17798-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamada, T., N. Kondoh, M. Matsumoto, M. Yoshida, A. Maekawa, and T. Oikawa. 1997. Overexpression of PU.1 induces growth and differentiation inhibition and apoptotic cell death in murine erythroleukemia cells. Blood 891383-1393. [PubMed] [Google Scholar]
- 65.Yang, J. Y., C. S. Zong, W. Xia, H. Yamaguchi, Q. Ding, X. Xie, J. Y. Lang, C. C. Lai, C. J. Chang, W. C. Huang, H. Huang, H. P. Kuo, D. F. Lee, L. Y. Li, H. C. Lien, X. Cheng, K. J. Chang, C. D. Hsiao, F. J. Tsai, C. H. Tsai, A. A. Sahin, W. J. Muller, G. B. Mills, D. Yu, G. N. Hortobagyi, and M. C. Hung. 2008. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10138-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zeng, X., L. Chen, C. A. Jost, R. Maya, D. Keller, X. Wang, W. G. Kaelin, Jr., M. Oren, J. Chen, and H. Lu. 1999. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 193257-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang, P., G. Behre, J. Pan, A. Iwama, N. Wara-Aswapati, H. S. Radomska, P. E. Auron, D. G. Tenen, and Z. Sun. 1999. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc. Natl. Acad. Sci. USA 968705-8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zochodne, B., A. H. Truong, K. Stetler, R. R. Higgins, J. Howard, D. Dumont, S. A. Berger, and Y. Ben-David. 2000. Epo regulates erythroid proliferation and differentiation through distinct signaling pathways: implication for erythropoiesis and Friend virus-induced erythroleukemia. Oncogene 92296-2304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.