

Abstract
Cells have intrinsic defenses against virus infection, acting before the innate or the adaptive immune response. Preexisting antiviral proteins such as PML, Daxx, and Sp100 are stored in specific nuclear domains (ND10). In herpes simplex virus type 1 (HSV-1), the immediate-early protein ICP0 serves as a counterdefense through degradation of the detrimental protein PML. We asked whether interferon (IFN)-upregulated Sp100 is similarly antagonized by ICP0 in normal human fibroblasts by using a selective-knockdown approach. We find that of the four Sp100 isoforms, the three containing a SAND domain block the transcription of HSV-1 proteins ICP0 and ICP4 at the promoter level and that IFN changes the differential splicing of the Sp100 transcript in favor of the inhibitor Sp100C. At the protein level, ICP0 activity does not lead to the hydrolysis of any of the Sp100 isoforms. The SAND domain-containing isoforms are not general inhibitors of viral promoters, as the activity of the major immediate-early cytomegalovirus promoter is not diminished, whereas the long terminal repeat of a retrovirus, like the ICP0 promoter, is strongly inhibited. Since we could not find a specific promoter region in the ICP0 gene that responds to the SAND domain-containing isoforms, we questioned whether Sp100 could act through other antiviral proteins such as PML. We find that all four Sp100 isoforms stabilize ND10 and protect PML from ICP0-based hydrolysis. Loss of either all PML isoforms or all Sp100 isoforms reduces the opposite constituent ND10 protein, suggesting that various interdependent mechanisms of ND10-based proteins inhibit virus infection at the immediate-early level.
Herpes simplex virus type 1 (HSV-1) is a common human pathogen that causes recurrent infections through its ability to establish a latent state in sensory ganglia after primary epithelial infection (for a general review, see reference 43). During lytic infection, HSV-1 tegument protein VP16 efficiently redirects the host's transcriptional machinery to express viral genes in a tightly regulated temporal cascade consisting of the sequential expression of three gene classes: the immediate-early (IE), delayed-early (DE), and late (L) genes. The five IE genes (ICP0, -4, -22, -27, and -47) are expressed shortly after entry into the host cell, and they are essential for efficient expression of DE genes, the majority of which encode proteins involved in viral DNA replication, as well as L genes, which encode predominantly structural proteins.
ICP0 is a RING finger E3 ubiquitin ligase (3) that is required for efficient entry into the lytic cycle and is essential for the reactivation of latent or quiescent genomes (reviewed in references 10, 11, 21-23, and 42). ICP0 influences many cellular pathways, and one of its most prominent activities is its ability to localize to and disrupt nuclear substructures known as ND10 (also known as PML nuclear bodies; reviewed in references 8, 12, and 31). This disruption occurs through ICP0-induced degradation of PML (14), the key component of ND10 that is required for the assembly of these structures (26, 53). During lytic infection, the RING finger of ICP0 is able to recruit UbcH5a and UbcH6 (3), which are required for efficient degradation of PML and Sp100 (20). HSV-1 mutants that do not express ICP0 or that express mutant ICP0 proteins that lack RING finger activity are unable to degrade PML and disrupt ND10 (3, 9, 14, 32, 33). Such HSV-1 mutants have a profound defect in gene expression, especially after infection of human fibroblasts (7, 17, 46, 47).
Although viral IE gene expression is decreased in cells pretreated with interferons (IFNs) (35, 40, 41), HSV-1 is relatively resistant to the effects of IFNs in cell culture, in part by counteracting an IFN-induced block to virus transcription (24, 36, 37). ICP34.5 and ICP0 are two HSV-1 protein components of IFN resistance (24, 37), and ICP0 is sufficient to inhibit the activation of IFN-stimulated genes (6). The major function of ICP34.5 is to counteract PKR phosphorylation of eIE2 in the cytoplasm, whereas the major function of ICP0 takes place in the nucleus. However, in the absence of ICP0, HSV-1 can still inhibit the expression of IFN-stimulated genes and can replicate, but only at a high multiplicity of infection (MOI). This suggests that more than one viral gene product inhibits the intrinsic cellular defense (37) and that ICP0 may enhance the expression of those viral genes.
Like PML, Sp100 is IFN upregulated and is part of an intrinsic defense mechanism (5, 29). Sp100 is a single-copy gene located on human chromosome 2q37 (50) that gives rise to a number of alternatively spliced Sp100 variants. Sp100B contains a SAND domain, Sp100HMG contains a SAND domain and an HMG box, and newly described Sp100C contains SAND, PHD, and Bromo domains (19, 45; Fig. 1A contains a schematic representation). All of these isoforms share the N-terminal 476 amino acids with the most abundant isoform, Sp100A, a protein of 480 amino acids, which aberrantly migrates at 100 kDa. Sp100A most likely does not bind to DNA alone but may be recruited to DNA via association with DNA-binding proteins such as hHMG2/DSP1 (30), the B-cell-specific transactivator Bright (54), or ETS-1 (49). While interaction of Sp100 with hHMG2/DSP1 or Bright mediates transcriptional repression, binding to ETS-1 stimulates the expression of ETS-1 target genes in one condition (49) and repression in the others (51, 52). Interestingly, two other proteins of the Sp family, Sp110 and Sp140, contain the same motifs and all of the genes of this family encode a SAND domain (named after Sp100, AIRE-1, NucP41/75, and DEAF-1). SAND domains of AIRE, NUDR, DEAF-1, and GMEB-1, all transcriptional regulators, bind to DNA. For the Sp100 SAND domain, DNA-binding properties have not been demonstrated but the structure of the SAND domain was recently resolved and allows speculation about such properties (1). A highly conserved tryptophan occurs at the DNA-binding interface of the SAND domain of each of these proteins. Unlike wild-type Sp100B, the Sp100B variant, constructed with a mutation of this tryptophan (W655Q), lost its ability to repress expression from transfected DNA or infecting virus (39). We have established that Sp100 isoforms with a SAND domain (Sp100B, Sp100C, and Sp100HMG) are required for the suppression of IE gene expression in HEp2 cells (39).
FIG. 1.

Characterization of human BJ fibroblasts with suppressed Sp100-SAND isoforms. (A) Diagram of the structural and functional domains of Sp100 isoforms. Each short line at the bottom, labeled shRNA, indicates the position of shRNA used to knock down all Sp100 transcripts (left line) or only the SAND domain-containing isoforms (right line). (B) Quantitative PCR was used to determine the relative levels of the different Sp100 isoform transcripts in vector-transduced BJ fibroblasts left untreated or treated with 1,000 U/ml IFN-β for 18 h before total cellular RNA extraction. (C) Levels of Sp100 isoforms were analyzed by quantitative PCR in stable BJ fibroblast lines that had been transduced with control vector or expressing shSp100BCH, which knocked down the Sp100 isoforms containing SAND domains. (D) Western blot assay of HSV-1 IE protein expression in BJ fibroblasts (BJV, vector-transduced control BJ fibroblasts; BJ-SAND, shRNA-transduced BJ minus SAND fibroblasts) at 3 h postinfection with a low number of HSV-1 PFU. The blot assayed with anti-ICP0 antibodies was stripped, and the membrane was reprobed with antibodies reactive with ICP4 and PML. The values on the left are molecular sizes in kilodaltons.
We set out to determine whether the IFN repression in normal human cells changes the ND10-associated repressive proteins and how these proteins suppress HSV-1 propagation. We find that the SAND domain-containing Sp100 isoforms suppress IE HSV-1 proteins at the promoter level and that IFN changes the splicing pattern of the Sp100 transcript to the suppressing Sp100C isoform. At the protein level, ICP0 activity does not lead to the hydrolysis of any of the Sp100 isoforms and that all four Sp100 isoforms stabilize ND10 and protect PML from ICP0-based hydrolysis. In this context, we hypothesize that the two constituent ND10-associated proteins interact at various levels in the intrinsic cellular defense against viruses.
MATERIALS AND METHODS
Expression plasmids.
The monomeric red fluorescent protein (RFP)- or green fluorescent protein (GFP)-tagged Sp100 constructs were described previously (38, 39). P110 and pCMV-110 were obtained from Roger Everett (Glasgow, United Kingdom).
Cells and viruses.
HEp-2 carcinoma, human embryonic kidney 293 (HEK293), 293-S, and human BJ diploid foreskin fibroblast cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% antibiotics. All cells were grown at 37°C in a humidified 5% CO2 atmosphere. For immunohistochemical staining, cells were grown on round coverslips in 24-well plates (Corning Glass Inc., Corning, NY) until approximately 80% confluent before fixation. HSV-1 strain 17+ was obtained from R. D. Everett. Titers of all virus stocks were determined in Vero cells.
Cell infection.
In all experiments, replicate cell cultures grown in six-well plates were either mock infected or exposed to virus at the PFU/cell ratio indicated in Results. Virus was adsorbed for 1 h at room temperature in DMEM with 1% FCS. The inoculum was replaced with DMEM supplemented with 10% FCS and 1% antibiotics, and cultures were incubated for 3 h at 37°C.
Sp100 and PML knockdown.
For lentiviral vector-based knockdown, clones TRCN000019224 (targeting nucleotides 1759 to 1780, 5′-GCCAACACTAGACCTTTGAA-3′; these short hairpin RNAs [shRNAs] recognize the targeting sequence common to the mRNAs of Sp100 isoforms Sp100B, Sp100C, and Sp100HMG) and TRCN000019227 (targeting nucleotides 716 to 725, 5′-CGCTAGGAAGCCAACAAACAA-3′; these shRNAs recognize the targeting sequence common to the mRNAs of all Sp100 isoforms) were purchased from Open BioSystems (Huntsville, AL). These lentiviruses expressed The RNAi Consortium human shRNA from the pLKO.1 vector. pLKO vectors were cotransfected with packaging plasmids pCMV-VSVG and pCMV-deltaR8.2 into 293T cells on a 6-cm dish by standard calcium phosphate precipitation. Viral supernatants were collected from transfected 293T cells at 24, 36, and 48 h posttransfection, clarified by centrifugation, filtered, and used for the infection of 5 × 104 target BJ fibroblasts grown on six-well plates in the presence of 4 μg/ml Polybrene (Sigma, St. Louis, MO). Stably transduced cell populations were selected in DMEM with 0.5 μg/ml puromycin for 3 days. To knock down PML, we used previously characterized siPML2 based on retrovirus vector pSIREN-RetroQ (16). Retrovirus stocks were prepared by cotransfecting a pSIREN-RetroQ plasmid with pVSV-G (BD Biosciences) into GP-293 cells.
Indirect immunofluorescence assay.
Two days after plating on round glass coverslips, cells were fixed at room temperature for 15 min with freshly prepared 1% paraformaldehyde in phosphate-buffered saline (PBS) and treated as described previously (25). Cells were analyzed with a Leica confocal laser scanning microscope. Leica image enhancement software was used in balancing signal strength, and fourfold scanning was used to separate the signal from the noise. Because of the variability among cells in any culture, the most prevalent cells were photographed and are presented as small groups of cells at high magnification. ND10 proteins were visualized with the following antibodies: monoclonal antibody (MAb) PG-M3 (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), which recognizes PML; rabbit antibody AB1380 (1:1000 dilution; Chemicon International, Temecula, CA) against Sp100; anti-ICP0 MAb 11060 (1:5 dilution), which has been described previously (13), and anti-ICP4 MAb (1:2 dilution; American Type Culture Collection, Manassas, VA).
Mammalian cell transfection and reporter assays.
HEp-2 cells, HEK293 cells, or 293-S cells (cells that do not contain Sp100) were cultured in DMEM plus 10% FCS and transfected with plasmids by using the SuperFect (Qiagen) reagent according to the manufacture's protocol. For luciferase reporter gene assays, 1 × 105 cells were seeded into each well of a 12-well plate and transfected with a total of 2 μg of plasmid DNA, which routinely included 0.1 μg of pCMVmin-β-gal to monitor transfection efficiency. In transfections with increasing doses of effector plasmids, the total amount of plasmid containing the simian virus 40 or cytomegalovirus (CMV) enhancer was maintained by the addition of empty expression vector pET1. Cells were harvested at 24 to 48 h posttransfection, washed with PBS, and lysed in 100 μl of hypotonic lysis buffer. The cytoplasmic fraction was used to measure protein content (1 μl), β-galactosidase activity (2 μl), and luciferase activity (20 μl). The nuclear fraction was used to check the expression of effector proteins. Three independent transfections were carried out, and the mean n-fold repression was calculated relative to the basal luciferase activity obtained from cells transfected with pET1. Absolute luciferase values and n-fold activation or repression differed among similar experiments performed on different days. Thus, all of the data within a panel were derived from transfections performed on the same day and assayed as a set.
Western blotting.
Total cellular protein extract was prepared from cells by lysis as described above, except that radioimmunoprecipitation assay buffer contained 1% sodium dodecyl sulfate. Protein concentration was quantitated by the Bradford method (Bio-Rad Laboratories, Hercules, CA). Total cellular protein (30 μg) was fractionated by 4 to 12% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred to a nylon membrane by standard techniques. Immunoblotting was performed with 5% nonfat dry milk in PBS containing 0.1% Tween 20 as the blocking agent. Filters were blocked overnight at 4°C, incubated with primary antibody for 1 h at room temperature, washed three times for 10 min per wash in PBS containing 0.1% Tween 20, and incubated for 1 h with goat anti-mouse or anti-rabbit immunoglobulin G antibody conjugated to horseradish peroxidase (GIBCO/BRL, Rockville, MD) according to the manufacturer's recommendations. Filters were washed again as described above, and reactivity was detected by enhanced chemiluminescence (Amersham Bioscience Corp., Piscataway, NJ) according to the manufacturer's recommendations. The antibodies applied were rabbit anti-Sp100 antibody AB1380 (1:30,000 dilution), rabbit anti-Daxx antibody 2133 (1:300 dilution) (28), MAb anti-ICP0 (1: 50 dilution), MAb anti-ICP4 (1:25 dilution), and MAb anti-GFP B-2 (1: 2,000 dilution) from Santa Cruz Biotechnology (Santa Cruz, CA), and MAb anti-tubulin (1:10,000 dilution) and MAb anti-FLAG M2 (1:50,000 dilution) from Sigma (St. Louis, MO).
qRT-PCR.
Total RNA was purified from BJ fibroblasts with RNeasy minicolumns (Qiagen). For quantitative real-time PCR (qRT-PCR) analysis, 2.5 μg of total RNA was used to synthesize cDNA with the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer's protocol. Fifty nanograms of cDNA was added to quantitative 2 × PCR master mix, which also contained SYBR green (Applied Biosystems) and each gene-specific primer at 250 nM. PCR was carried out at 60°C (annealing temperature) for 40 cycles. The primers used were Sp100A sense (5′-TGGGAACTCCTTTTTGCATT) and antisense (5′-CAAACGACAATGATGTCAACC), Sp100B sense (5′-AGGAAGCGATTCAAACAAGGA) and antisense (5′-AGACGAGACATTGGCAGAAG), Sp100C sense (5′-AAGCCAATCAGGTCATCAGG) and antisense (5′-ATGTCCTGCACAAACCCTTC), Sp100HMG sense (5′-TAGCCCTGTCCTGGTGGTAT) and antisense (5′-TGTCAACAAAACAGCTGCAA), PML4 sense (5′-GCAGCTCGGAAGACTC) and antisense (5′-GTAGCCCCAGGAGAAC), and GAPDH sense (5′-CTGGGCTACACTGAGCACCAG) and antisense (5′-CCAGCGTCAAAGGTGGAG).
qRT-PCR was performed with triplicate samples and a 348-well plate on a 7900HT Fast Real Time PCR system (Applied Biosystems) according to the manufacturer's instructions. Melting curves were performed to document single-product formation, and agarose electrophoresis confirmed product size. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. Amplification of the product with the expected size was confirmed by analysis of the completed reaction product in 2% agarose gels stained with ethidium bromide. For quantitative analysis, semilogarithmic plots were constructed of delta fluorescence versus cycle number, and a threshold was set for the changes in fluorescence at a point in the linear PCR amplification phase (CT). The CT values for each gene were normalized to the CT values for GAPDH with the ΔCT equation. The level of target RNA, normalized to the endogenous reference and relative to the mock-infected and untreated cells, was calculated by the comparative CT method with the 2−ΔΔCT equation.
RESULTS
Effect of HSV-1 infection on PML in SAND domain-containing Sp100 isoform-depleted normal fibroblasts.
Since most viruses have developed mechanisms to counter the cell's defenses, we began to investigate the Sp100-based intrinsic nuclear defense in normal human cells and whether ICP0 can counter the repressive effects of the Sp100 isoforms containing SAND domains. Human BJ fibroblasts were first characterized as to their complement of Sp100 isoform transcripts with and without IFN exposure since we had previously noticed substantial differences between cell types. As shown in Fig. 1B, Sp100A and Sp100C transcripts are present in larger amounts than the Sp100B and Sp100HMG isoforms. This was unexpected, since in another fibroblast strain, FF2425, the Sp100HMG isoform was dominant (39). In addition, the Sp100 transcript mix changes dramatically upon IFN exposure. We find a substantial relative increase in Sp100C transcripts, suggesting that differential splicing is strongly dependent on IFN, at least for Sp100 transcripts and BJ fibroblasts. This extends our observation that different cell types have variable ratios of Sp100 isoforms to different strains of the same cell type (BJ versus FF2425 fibroblasts), at least at the transcript level.
Overexpression of Sp100 SAND domain-containing isoforms is detrimental to the long-term propagation of cells (39; our unpublished observations). We therefore decided to take the opposite approach and developed BJ fibroblast lines where only the suppressive SAND domain-containing isoforms were knocked down. We expected that this would result in a less repressed environment for IE HSV-1 protein expression. For this purpose, we used one of the two previously characterized lentiviruses expressing shRNA against Sp100 (39). The diagram in Fig. 1A shows the position of the shRNA sequence located outside of the splice isoform of Sp100A transcripts. Due to the lack of isoform-specific antibodies, we confirmed the downregulation of individual Sp100 isoforms containing SAND domains by qRT-PCR in two different subcultures, S1 and S2 (shown for S1 in Fig. 1C). We find no apparent diminishment of the dominant Sp100A transcripts but a substantial reduction of Sp100B, Sp100C, and Sp100HMG (Fig. 1C). In Western blot assays with rabbit antibodies that recognize all four isoforms, Sp100-immunoreactive bands appeared not much changed from controls and IFN exposure increases those bands. However, it should be mentioned that the immunoreactive species is mostly Sp100A and its SUMOylated forms (not shown). This isoform is also the dominant one at ND10, as previously reported (39). Over time, the BJ fibroblasts with downregulated Sp100 SAND domain-containing isoforms (BJ-SAND) did not show any abnormalities with respect to PML and Daxx expression, as assayed by Western blot analysis (data not shown). The only growth effect detected upon long-term propagation was that BJ-SAND fibroblasts senesced earlier than control BJV fibroblasts (passage 45 versus passage 55; data not shown). All experiments with the BJ-SAND fibroblasts were therefore conducted before passage 35 (about five passages posttransduction).
Since overexpression of the Sp100 SAND domain-containing isoforms had a highly detrimental effect on HSV-1 IE protein expression (39), knockdown of expression of these isoforms in normal cells should prevent repression by IFN. To test this proposition, we infected BJ-SAND fibroblasts (BJ fibroblasts minus SAND domain-containing Sp100 isoforms) and control BJV fibroblasts (with integrated empty vector, V) with wild-type HSV-1 at an MOI of 5 for 3 h with or without overnight IFN-β pretreatment. Western blot analysis of the infected cells shows that, at 3 h postinfection, ICP0 and ICP4 are expressed in the absence of IFN but barely recognizable in its presence (Fig. 1D, top, lanes 3 and 4). ICP0 levels are increased above the level of control cells and barely diminished by IFN (lanes 7 and 8). This demonstrates that most of the repressive effect of IFN can be eliminated by suppression of Sp100 SAND domain transcripts and presumably their translated products. The same can be seen for ICP4; however, some effect of IFN remains (Fig. 1D, middle, lanes 7 and 8). In addition, the normal ICP0-dependent hydrolysis of PML in infected control cells (compare Fig. 1D, bottom, lanes 1 and 3) is inhibited by IFN (lanes 2 and 4) but hydrolysis takes place in IFN-treated BJ-SAND fibroblasts (compare lanes 7 and 8), in contrast to control IFN-treated cells (lane 4). These results suggest that wild-type virus, i.e., one with competent ICP0, is repressed in normal fibroblasts by the Sp100 SAND domain-containing isoforms and that IFN-mediated repression of HSV is largely dependent on these IFN-upregulated proteins.
The Sp100 SAND domain-containing isoforms suppress ICP0 expression at the transcriptional level.
The counterdefense of the virus to the repressive effects of ND10-associated proteins, such as PML, is thought to be through ICP0-dependent ubiquitination that leads to protein degradation. The cellular Sp100-based defenses may be at various levels, and we tested first the possibility that Sp100 functions on the transcriptional level of viral IE gene expression. The experiments were conducted with cells that had no indigenous Sp100, i.e., 293-S cells (39). Plasmids that expressed ICP0 from its own promoter were cotransfected with the various plasmids expressing Sp100 isoforms or PML. Western blot assays developed with anti-GFP antibodies determined the effect on protein expression (Fig. 2, top panel; yellow fluorescent protein is recognized by anti-GFP antibodies). Expression control is shown for Sp100 isoforms and PML-IV in the middle and lower panels, respectively. ICP0 expression is strongly diminished, with the largest effects seen for the SAND domain-containing isoforms, the Sp100B isoform having the strongest effect. We also tested the addition of PML-IV since PML was supposedly inhibiting IE virus protein expression. Surprisingly, PML-IV had a strong enhancing effect on ICP0 expression (Fig. 2, top panel, lanes 11 and 12).
FIG. 2.
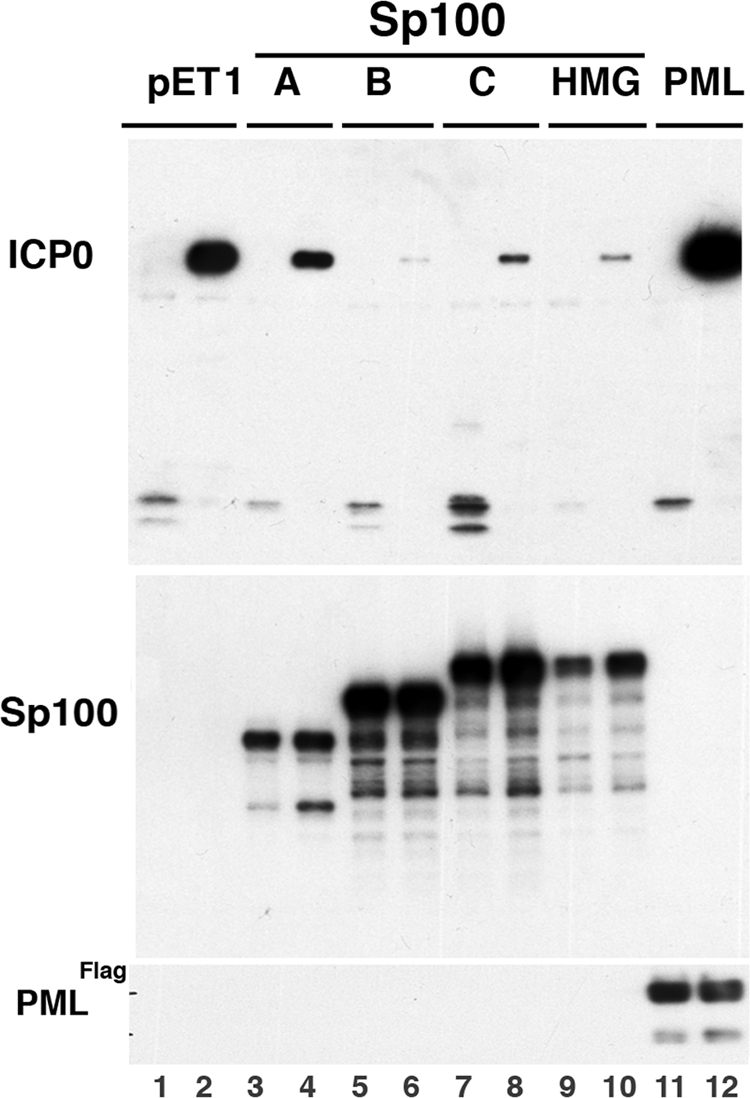
Test of Sp100 isoform suppression of ICP0 protein expression. An ICP0 expression vector driven by its own promoter was cotransfected into 293-S cells with a control empty vector (pET1) or p110 (expressing ICP0 under the control of the ICP0 promoter) and different Sp100 isoforms and probed with anti-Sp100 rabbit antibodies or with an expression vector for FLAG-tagged PML-IV. Western blots of the nuclear fractions (30 μg/lane) were probed with antibodies to GFP (top panel) or Sp100 and FLAG antibodies (middle and bottom panels).
A luciferase reporter assay with the ICP0 promoter was used to verify these results. We cotransfected 293-S cells with the reporter driven by the ICP0 promoter and plasmids expressing different Sp100 isoforms. Results are shown in Fig. 3; arbitrary units of luciferase were normalized to the level expressed in cells that had been cotransfected with the pET1 empty vector. In agreement with previously published results (39), the Sp100A isoform enhanced expression whereas the three isoforms containing the SAND domain suppressed luciferase expression (lane 2 versus lanes 3, 4, and 5). The single amino acid mutation W665Q in the SAND domain of Sp100B (Sp100BQ) eliminated the repressive effects (lane 6). The suppressive effect of these isoforms is therefore most likely at the promoter level. The discrepancy of a diminishment of ICP0 protein expression by Sp100A (as shown in Fig. 2, vector versus Sp100A) and the enhancement at the promoter level (as shown in Fig. 3, lanes 1 and 2) may be due to some influence of expressed Sp100A on ICP0 protein (see below).
FIG. 3.

Determination of different promoters relative to their repressibility by Sp100 isoforms. 293-S cells were cotransfected with luciferase reporter vectors driven by promoters of ICP0, CMV, and the long terminal repeat (LTR) of Rouse sarcoma virus. Cells were harvested 48 h later, and luciferase activity (relative light units [RLU]) from the cytoplasmic fraction was determined and normalized to the protein concentration. Results are presented as a percentage of the luciferase activity of the pET1 vector, which was considered 100%. Mean values and standard deviations from three independent experiments are shown.
Because of the known effect of ICP0 on PML degradation and the resulting diminishment of its antiviral effects, we wanted to know whether ICP0 can also reduce the suppressive effects of Sp100. Due to the strong effect of the Sp100 isoforms on the expression of ICP0, it was not feasible to test the effect of ICP0 on Sp100 as long as ICP0 was expressed from its own promoter. We therefore determined the effect of Sp100 on several commonly used promoters to find one unresponsive to Sp100 isoforms. In Fig. 3 are shown results for two of these promoters that had extremely different effects upon exposure to the different Sp100 isoforms. All Sp100 isoforms had no significant effects on the CMV promoter (Fig. 3, lanes 7 to 12), whereas they had a major effect, similar to that of the ICP0 promoter, on the long terminal repeat of a retrovirus: the Sp100A isoform strongly stimulated expression over that obtained with the vector control (lane 13 versus lane 14), whereas the SAND domain-containing isoforms strongly inhibited expression (lanes 15 to 17). The Sp100 isoforms are therefore likely to affect the individual promoters rather than a general transcription or translation function.
After obtaining these results, we used the CMV promoter from which to express ICP0. However, the interpretation of the intended experiments might also be problematic if ICP0 degrades Sp100. To test this possibility, we addressed two questions with an Sp100-unresponsive CMV promoter-driven ICP0 expression vector and 293-S cells, which contains only a small amount of the Sp100A isoform (39). We asked first whether ICP0 destroys Sp100 as it does PML in other cell lines and second whether ICP0 can reduce some of the repressive effects of the Sp100 isoforms.
To answer the first question, we titrated the CMV-driven ICP0 expression vector into 293-S cells together with fixed concentrations of the individual Sp100 isoform vectors predetermined to express Sp100 at low levels. As shown in Fig. 4A, there is no diminishment of any of the Sp100 isoforms (lower panel) with increasing ICP0 concentrations (middle panel). This result indicates that ICP0 does not affect Sp100 protein concentrations in this cell type.
FIG. 4.

Effects of Sp100 isoforms on luciferase expression from the ICP0 promoter. (A) 293-S cells were cotransfected with a luciferase reporter plasmid under the control of the ICP0 promoter or the minimum CMV promoter-β-galactosidase reporter; equal amounts of GFP-tagged isoforms Sp100A, Sp100B, Sp100C, Sp100HMG, and Sp100BQ; and increasing amounts (0.5, 1, and 2 μg) of effector plasmids expressing ICP0 from its own promoter. Cells were harvested 48 h later, and luciferase activity (relative light units [RLU]) was determined from the cytoplasmic fraction and normalized to the protein concentration (top). Results are presented as a percentage of the luciferase activity of the pET1 vector, which was considered 100%. Mean values and standard deviations from three independent experiments are shown. Nuclear fractions (30 μg/lane) were electrophoretically separated and probed and reprobed with antibodies to GFP to ensure similar levels of expression and with antibodies to ICP0 to show the increasing amount of ICP0 expressed. (B) Repeat of the experiment with HEp-2 cells except that no increasing ICP0 concentrations were used. pC3 is the CMV promoter control vector.
In the same experiment, we asked whether ICP0 could stimulate expression from its own promoter. To answer this question, we cotransfected the luciferase reporter driven by the ICP0 promoter with plasmids expressing the respective Sp100 isoforms and the ICP0 expression vector driven by the CMV promoter. The top part of Fig. 4A shows that increasing ICP0 concentrations improve expression from the ICP0 promoter in the absence of Sp100 (lanes 1 to 4; vector control) and more so when Sp100A is present (lanes 5 to 8). The Sp100B isoform suppression is substantially relieved by small amounts of ICP0, but relief was apparently not very ICP0 concentration dependent (lanes 9 to 12). The Sp100C isoform inhibition can be reduced in an ICP0 concentration-dependent manner (lanes 13 to 16), whereas the Sp100HMG suppression relief is quite minor (lanes 17 to 20). The mutant Sp100BQ isoform mimics the Sp100A isoform apparent ICP0 promoter activation with increasing ICP0 protein expression. These results indicate that, contrary to PML, ICP0 cannot efficiently counter the Sp100 repressive effect.
Since 293-S cells have very little PML and thus the degradation of PML as a control for the activity of ICP0 cannot be ascertained, we repeated the experiment with HEp2 cells, which have a normal set of Sp100 isoforms and in which ICP0 can efficiently degrade PML (Fig. 5A). By transfection into HEp2 cells, we shifted the normal balance of these four Sp100 isoforms strongly to one of them. We find the same trend as for the 293-S cell line; i.e., ICP0 can moderately increase its own promoter activity with the Sp100A isoform and marginally with the Sp100C isoform. No effect is found in the presence of Sp100B and Sp100HMG (Fig. 4B). The effect of ICP0 in HEp2 cells with endogenous Sp100 is much weaker than in 293-S cells, which have no endogenous Sp100. Surprising was an observation in the Western blot assay control for the appropriate expression of the transfected proteins. We find no effect of ICP0 on the quantity of the Sp100 SAND domain-containing isoforms, except that an apparent secondary band is eliminated in the presence of ICP0 (compare lanes 5 and 6, 7 and 8, and 9 and 10). This elimination does not take place on the Sp100A isoform (lanes 3 and 4) and on the Sp100BQ isoform (lane 11), which has the same effects as the Sp100A isoform on IE promoter stimulation. From these observations, we have to conclude that the inhibitory effect of Sp100 isoforms is not affected by ICP0-induced degradation.
FIG. 5.
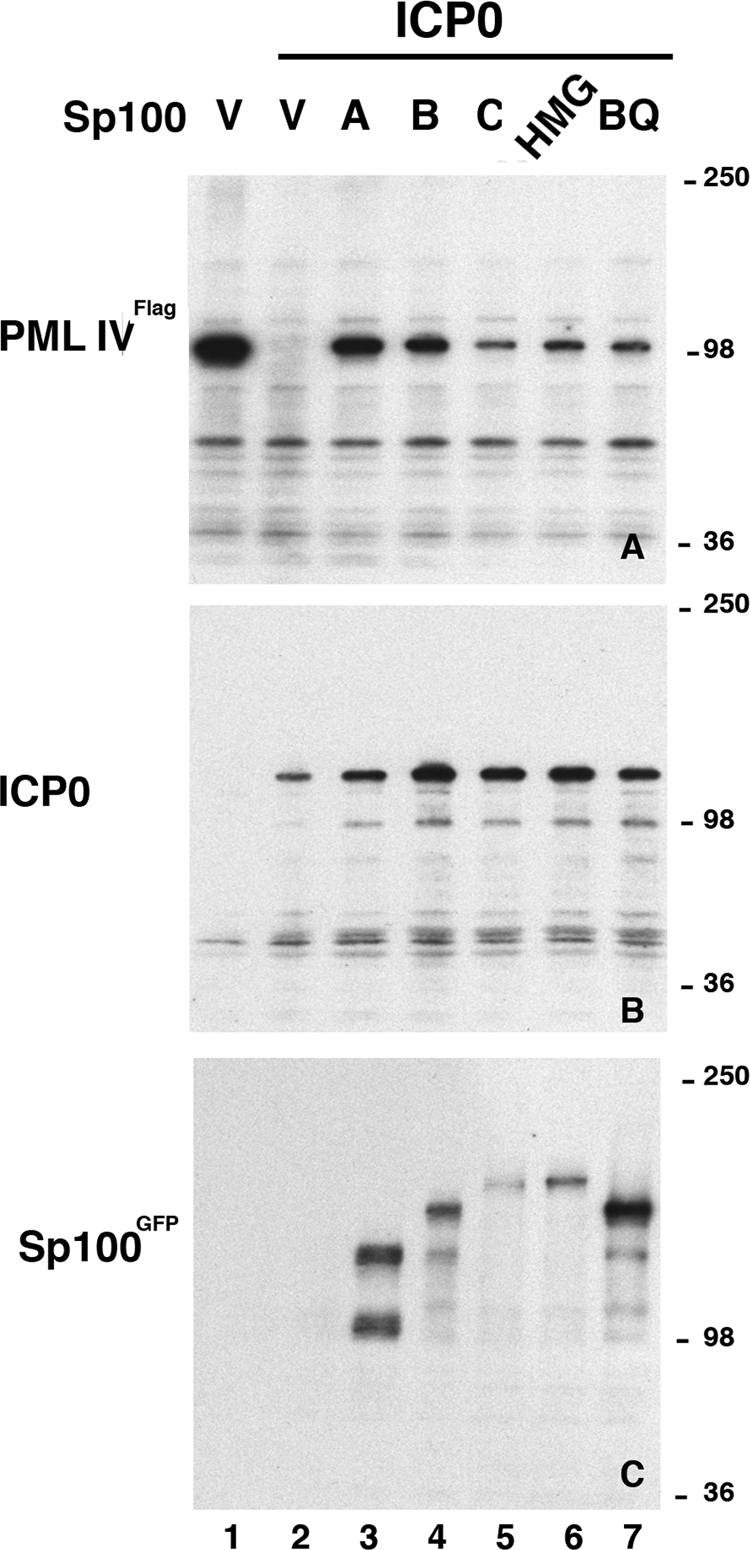
Effects of Sp100 isoforms on the degradation of PML by ICP0. HEp-2 cells were cotransfected with a plasmid expressing FLAG-PML-IV, ICP0 driven by the CMV promoter, and the different GFP-tagged Sp100 isoforms. Lanes V contained the empty-vector (pET1) control. Western blot assay of nuclear fractions probed with antibodies to FLAG (top), ICP0 (middle), or GFP (bottom). The values on the right are molecular sizes in kilodaltons.
All Sp100 isoforms protect PML from ICP0-mediated degradation.
We had previously determined that ICP0 did not equally lead to the degradation of PML in different cell types. In fibroblasts, this degradation was quite evident and was most strongly present in HEp2 cells, whereas less degradation of PML was recorded for 293-S cells (data not shown). Wild-type HSV-1 seems not to be affected by PML; however, after knockdown of PML, the absence of ICP0 improved virus production, although total reconstitution of wild-type viral replicative success was not achieved (15, 16). We asked whether the cellular antiviral defense of Sp100 isoforms functions by protecting PML from hydrolysis, thus retaining PML's antiviral properties. For the test of potential PML degradation protection, FLAG-tagged expression vectors for PML-IV, the CMV-driven ICP0 expression vector, and the different GFP-tagged Sp100 isoforms were cotransfected into HEp2 cells and nuclear extracts obtained from cultures at 24 h posttransfection were analyzed by Western blot assay. The blots were stripped and reprobed to ensure adequate protein expression. As shown in Fig. 5A, lane 1, PML-IV is well expressed in the absence of ICP0 but hydrolyzed at 24 h posttransfection when ICP0 is coexpressed (lane 2). When the different Sp100 isoforms are added to the mixture, we find that isoforms Sp100A and Sp100B substantially suppress ICP0-dependent PML degradation (lanes 3 and 4). Isoforms Sp100C, Sp100HMG, and Sp100BQ (SAND domain mutant protein) also repress the function of ICP0, although marginally less well. In the cases of Sp100C and Sp100HMG, the lower protection is possible because of their lower expression levels (Fig. 5C, lanes 5 and 6).
How the Sp100 isoforms might protect PML from degradation is not known. Sp100A normally aggregates with PML in ND10, an aggregation of several inhibitory proteins dispersed by ICP0. Increased Sp100 might protect PML by aggregation or segregation. We therefore tested by immunofluorescence assay whether Sp100 in the presence of ICP0 changes the distribution of PML. The same transfection experiment with HEp2 cells as described for the Western blot assay in Fig. 5 was conducted with HEp2 cells grown on coverslips and then prepared for immunofluorescence analysis. Figure 6A to D show the distribution of ICP0 (green) in HEp2 cells as diffuse and with aggregates throughout the nucleus. A substantial amount of ICP0 is present in the nucleolus in these transfection experiments. However, when the nucleus is filled with ICP0, PML is mostly dispersed (nucleus at the lower left of panel A). When there is very little ICP0, as in the nucleus at the right of panel A (arrow), ICP0 associates with PML even after 24 h posttransfection (shown in blue, Fig. 6D), suggesting that ICP0 can be held at these sites when at low abundance without hydrolysis of PML, i.e., in a segregated and inactive state.
FIG. 6.

Immunofluorescence analysis of HEp-2 cells cotransfected with ICP0 and different RFP-tagged Sp100 isoforms. HEp-2 cells grown on coverslips were transfected with plasmid constructs expressing the monomeric RFP (mRFP)-tagged nuclear localization signal (A to D) or the RFP-tagged Sp100A (E to H), Sp100B (I to L), Sp100C (M to P), or Sp100HMG (Q to T) isoform and analyzed by immunofluorescence assay with a rabbit antibody against ICP0 (green) and MAb PG-M3 against endogenous PML (blue). The left column shows the merged image, and the other columns show the respective colored labels. Colocalization of all three proteins appears as a white signal. Small groups have been selected to show the typical and variable distribution of PML, Sp100, and ICP0.
Cotransfection of ICP0 with Sp100A results in dispersion of PML, Sp100A, and ICP0. At lower ICP0 concentrations, small dots of ICP0 are found and not all of the PML or Sp100A is dispersed (lower left of panels E to H). During transfection, some cells do not receive the same ratio of expression vectors. Some express only one of the two. If it is Sp100A, the resulting protein closely associates with PML (bottom of panels E to H). Cotransfection of ICP0 and Sp100B results in strong segregation of ICP0, but PML is dispersed (left upper nucleus in panels I to L). Surprisingly, PML is also dispersed by Sp100B alone (nucleus at the upper right containing only Sp100B, red), the same as if it had only ICP0 (nucleus at the top of panel I, green), although this could be a reflection of cell cycle-based dispersion of ND10. Expression of Sp100C and ICP0 results in very close colocalization over a wide range of ICP0 expression levels, and PML is present in these aggregates (upper two nuclei in panels M to P). This is surprising, as this isoform, when overexpressed, forms aggregates separate from PML-labeled ND10 (see reference 38). ICP0 must modify or expose the molecular motif at the N terminus that determines ND10 association. The Sp100HMG isoform also colocalizes with ICP0 when overexpressed (top left two nuclei in panels Q to T). Aggregates of ICP0 and Sp100HMG at higher expression levels are devoid of PML (three nuclei at the lower right of panels Q to T).
The variable association of the different Sp100 isoforms with ICP0 and the presence or absence of PML in these aggregates make it unlikely that the Sp100 isoforms function in a similar way in blocking ICP0-mediated degradation of PML, i.e., through the segregation and inactivation of ICP0. We therefore asked whether the Sp100 isoforms alone stabilize PML. If Sp100 stabilizes PML, then Sp100 knockdown should lower the amount of PML in the cells.
To test this possibility, we transduced BJ fibroblasts with a lentivirus expressing shRNAs that resulted in the knockdown of all of the Sp100 isoforms. Immunofluorescence analysis showed the known strong expression and colocalization of Sp100 and PML in control cells (Fig. 7A to C). No apparent reduction in PML was obvious in BJ fibroblasts where the Sp100 SAND domain-containing Sp100 isoforms were knocked down (BJ-SAND; Fig. 7D to F). However, when cells were analyzed after all of the Sp100 isoforms were knocked down, few contained Sp100 in ND10 and the size and intensity of PML aggregates and diffuse PML were reduced. More than 80% of the cells showed this reduced Sp100 staining (blue rim showing nuclear boundary). In some additional cells, a low level of Sp100 was found in association with PML (upper cell in Fig. 7G to I). A few cells had the same intensity and distribution of PML and Sp100 as control cells and are thought to be cells that had not been transduced with the shRNA vector. When these cell populations were tested for the respective proteins by Western blot assays in the presence and absence of prior IFN exposure, we found the expected IFN-induced increase in Sp100 in the vector control cells (BJV) but an apparent absence of all Sp100 bands in BJ-Sp100 fibroblasts (Fig. 7J). Immunofluorescence is apparently more sensitive to small amounts of protein, as we reported before (39). The BJ-Sp100 cells have a substantially reduced amount of PML, both with and without IFN induction (Fig. 7J and L).
FIG. 7.

Comparison of vector control BJ fibroblasts (BJV), BJ fibroblasts minus SAND domain-containing Sp100 isoforms (BJ-SAND), and BJ fibroblasts minus all Sp100 isoforms (BJ-Sp100). (A to C) Normal distribution of PML and Sp100 in BJV fibroblasts. (D to F) BJ-SAND fibroblasts after knockdown of the SAND domain-containing Sp100 isoforms. (G to I) BJ-Sp100 fibroblasts in which all Sp100 isoforms are knocked down. The added blue outline indicates a nucleus with little PML and little Sp100. Cells were grown on coverslips and analyzed by immunofluorescence assay with rabbit antibody against endogenous Sp100 (green) and MAb PG-M3 against endogenous PML (red). The left column shows the merged image, and the other columns show the respective labels, as indicated in the top row. Colocalization appears as a yellow signal. (J to M) Western blot analysis of whole-cell extract (30 μg/lane) of replicate BJV and BJ-Sp100 cell cultures stimulated with 1,000 U/ml IFN-β for 18 h or left untreated as a control. They were probed with antibodies to Sp100 (J and K) or PML (L and M). (N) BJ minus PML fibroblasts (BJ-PML) double labeled for Sp100 (green) and PML (red) showing various redistributions of Sp100 in cells after two passages posttransduction.
We tested whether the maintenance of the two constituent ND10 proteins PML and Sp100 is reciprocal. BJ fibroblasts were transduced with a previously characterized shRNA retroviral vector against PML (16) and tested by Western blot assay. The loss of all PML isoforms, even after IFN exposure, is documented in Fig. 7M. In Fig. 7K, the increase in Sp100 after IFN exposure is evident in vector-transduced BJ fibroblasts. A substantial decrease in Sp100 is evident in BJ minus PML fibroblasts (BJ-PML). In addition, the comparison of the IFN-exposed cells shows that the upper bands, presumably Sp100C and Sp100HMG, are absent and there appears to be no IFN-induced increase in the Sp100A isoform. Also the SUMOylated Sp100A isoform is not completely eliminated, as has been previously shown (15, 16). These differences may be due to the use of different cell types. Also, our observations are valid for cultures 2 doubling times after transduction. In later passages, Sp100 may be lost completely (unpublished data). By immunofluorescence assay, we recognize a redistribution of Sp100 after PML depletion either as finely granulated with one or two irregular aggregates (bottom two cells) or a larger number of aggregates reminiscent of ND10 and a fine sandy distribution. The normal distribution of PML and Sp100 is evident in the very few cells that may not have been transduced (top left of panel N) and represents a control for the antibody specificity of PML, validating the absence of PML in the other cells.
In summary, we concluded that fibroblasts require the presence of at least Sp100A for PML stabilization and maintenance of ND10 and that in the absence of either PML or Sp100, the other constitutive ND10 protein is complementarily diminished.
DISCUSSION
Evidence from several lines of investigation suggests that wild-type virus infection of permissive normal cells can be suppressed by intrinsic cellular factors. The transcription cascade to lytic virus replication may be interrupted at several stages, which the virus may overcome through inhibitors of such intrinsic factors by using tegument proteins essential for starting transcription, such as VP16, and the IE transactivators ICP4 and ICP0, which promote the next step in the transcription cascade, leading to replication. The relative success of the cell at virus suppression or the virus in replication most likely depends on the level of infection of the individual cell, i.e., on a shift of balance between the intrinsic repressor molecules and the viral input of tegument repressors and accumulation speed of newly expressed viral IE proteins.
A higher MOI of wild-type HSV-1 can overcome IFN-induced repression in human fibroblasts. These cells provide high resistance to infection with the ICP0 null mutant (15, 16, 18). Human fibroblasts also normally have a high level of expression of PML and Sp100, both IFN-inducible proteins. Recently, it has been shown that downregulation of PML increases the success of human CMV (HCMV) (48) and nearly rescues the ICP0 null mutant of HSV-1 (15, 16). For HCMV, the repressive effect could be restored by the reintroduction of one of the PML isoforms (PML-VI). For HSV-1, the IFN-dependent suppression was not PML dependent and not dependent on the Stat-1and IRF-3 pathways (18); however, we can show that the introduction of PML-IV into cells with a very low PML level had a stimulatory effect on the ICP0 promoter. Thus, the different isoforms of PML may, like the different isoforms of Sp100, have opposing functions where the readout when all are suppressed reflects the respective differential of activating and repressing functions. For the PML isoforms, an analysis of their different functions relative to virus promotion or repression is outstanding.
Only the HSV-1 ICP0 null mutant has a phenotype that can be partly alleviated by the removal of PML and Sp100. Only a small effect was seen upon plaque formation in wild-type HSV-1, even when both PML and Sp100 were knocked down (15). Under those conditions, the activating Sp100A isoform is also knocked down, blunting any normal effect. The Sp100 SAND domain-containing isoforms are the likely repressors, as evidenced by the results of our investigation. The Sp100A isoform has a robust activation effect on the IE HSV-1 promoters. Evidence that Sp100 SAND domain-containing isoforms suppress wild-type HSV-1 infection in normal permissive cells such as fibroblasts comes from the increase in IE protein synthesis when these isoforms are downregulated by shRNA. This suppression of IE proteins such as ICP4 and ICP0 takes place at the promoter level, as shown by ICP0 promoter-based reporter assays. Cotransfection assays demonstrate that the Sp100 isoforms strongly affect the amount of ICP0 expressed. In the context of viral infection, such suppression would also reduce the ICP0 function of PML degradation. The finding that small amounts of ICP0 can be present over long periods of time without dispersing ND10 (Fig. 6A) demonstrates that small amounts of this protein might effectively be removed from functioning as an agent of PML degradation. This raises the possibility that the transcription cascade could be interrupted even after IE protein synthesis has started in low-level infections. For MCMV, such a shutoff of lytic progression toward replication has been demonstrated at the single-cell level by time-lapse imaging (34). For HSV-1, such a possibility needs to be investigated.
We contemplated that ICP0 might counter the suppressive effects of the Sp100 SAND domain-containing isoforms similar to the way PML is disabled. When we tested this possibility in 293-S cells and used PML as a control for ICP0-induced degradation, we found that this cell line has a defect in that PML is not degraded as in other human cell lines (not shown). Other currently unknown cellular factors need to be present for ICP0 activity to function. This may be the reason that in vitro attempts to degrade PML directly by ICP0 failed (2). However, in HEp2 cells, where PML can be degraded by ICP0 without any other viral components present, ICP0 also could not degrade any of the Sp100 isoforms. This unexpected result requires the assumption of an additional counterdefense by the virus, perhaps one analogous to the tegument-based pp71 counterdefense of Daxx repression in HCMV infection (4, 27, 44) or more simply just an MOI-based swamping of the cell's defenses where enough ICP4 and ICP0 is produced to overcome these repressive Sp100 isoforms.
PML and Sp100 are normally aggregated in ND10. If aggregation of proteins protects them from degradation, then Sp100 may protect PML from ICP0-based degradation and keep PML available for PML-based HSV-1 inhibition. We found that all of the Sp100 isoforms protect PML from ICP0 degradation. Control experiments established that the Sp100 isoforms not only protect PML from ICP0-induced degradation but also that, in the absence of all of the isoforms, normal-size ND10 disappears and the cell's PML content diminishes substantially, even in the absence of ICP0. This result differs from published accounts that show retention of PML and ND10 after Sp100 ablation (15). We have no good explanation for these differences, except possibly that even human fibroblasts differ in their physiology, as is evident when we compare the IFN response of FF2425 fibroblasts (39), where the Sp100HMG isoform is dominant, with that of BJ fibroblasts, where Sp100 transcripts are preferentially spliced to produce the Sp100C isoform, as shown here.
Our observed loss of PML after Sp100 ablation is mirrored by the loss of Sp100 after PML ablation. In the absence of PML, Sp100 isoforms are diminished, with an apparent loss of the SP100C and Sp100HMG isoforms and a strong reduction of Sp100A and its SUMOylated isoform. This differs, in part, form published accounts where only the un-SUMOylated isoform remains (16). Loss of all Sp100 isoforms has only been noticed after repeated passages where the cell may have changed its phenotype. Thus, the reported PML minus phenotype (15, 16, 18) may, in part, depend on the diminishment of the repressive action of Sp100 isoforms and a change in the cell phenotype.
Our results also suggest that the Sp100A isoform is required for ND10 integrity. In BJ fibroblasts, where only Sp100 isoforms with a SAND domain were suppressed, no effect on PML stability or ND10 structure was observed, but in cells where all of the Sp100 isoforms were downregulated, PML proteins diminished in subsequent passages. This suggests that a domain that is present in all of the Sp100 isoforms is involved in PML protection. The elimination of Sp100 isoform functions by viruses might affect PML and ND10 in infected cells. In the case of HSV-1, it is possible that ICP0 performs this function, but because expression of ICP0 itself is under strong suppression by SAND domain-containing Sp100 isoforms, additional and different viral proteins might be required to eliminate this suppression at the beginning of viral infection. As IFN increases the expression of all Sp100 isoforms and shifts splicing to more efficient production of Sp100 isoforms with SAND domains, specifically Sp100C, we speculate that this might benefit not the initially infected cell in vivo but neighboring cells through heterologous activation of IFN-activated ND10-associated proteins such as Sp100 and PML, making it more difficult for the virus to overcome these intrinsic defenses.
Acknowledgments
We thank Roger Everett for supplying ICP0-expressing plasmids and Thomas Stamminger for the siPML2 retroviral vector.
This study was supported by funds from NIH (AI 41136) and the Mathers Foundation. NIH core grant CA-10815 is acknowledged for the support of the microscopy and sequencing facility. This project was funded in part under a grant from the Pennsylvania Department of Health.
Footnotes
Published ahead of print on 11 March 2009.
REFERENCES
- 1.Bottomley, M. J., M. W. Collard, J. I. Huggenvik, Z. Liu, T. J. Gibson, and M. Sattler. 2001. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 8626-633. [DOI] [PubMed] [Google Scholar]
- 2.Boutell, C., A. Orr, and R. D. Everett. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 778686-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 806188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chelbi-Alix, M. K., L. Pelicano, F. Quignon, M. H. Koken, L. Venturini, M. Stadler, J. Pavlovic, L. Degos, and H. de The. 1995. Induction of the PML protein by interferons in normal and APL cells. Leukemia 92027-2033. [PubMed] [Google Scholar]
- 6.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 762180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Everett, R. D. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 70(Pt. 5)1185-1202. [DOI] [PubMed] [Google Scholar]
- 8.Everett, R. D. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 207266-7273. [DOI] [PubMed] [Google Scholar]
- 9.Everett, R. D. 2000. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 749994-10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everett, R. D. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22761-770. [DOI] [PubMed] [Google Scholar]
- 11.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 781763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D., and M. K. Chelbi-Alix. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89819-830. [DOI] [PubMed] [Google Scholar]
- 13.Everett, R. D., A. Cross, and A. Orr. 1993. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology 197751-756. [DOI] [PubMed] [Google Scholar]
- 14.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 726581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett, R. D., C. Parada, P. Gripon, H. Sirma, and A. Orr. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 822661-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 807995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett, R. D., G. Sourvinos, C. Leiper, J. B. Clements, and A. Orr. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 781903-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everett, R. D., D. F. Young, R. E. Randall, and A. Orr. 2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 828871-8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grötzinger, T., K. Jensen, H. H. Guldner, T. Sternsdorf, C. Szostecki, M. Schwab, L. Savelyeva, B. Reich, and H. Will. 1996. A highly amplified mouse gene is homologous to the human interferon-responsive Sp100 gene encoding an autoantigen associated with nuclear dots. Mol. Cell. Biol. 161150-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu, H., and B. Roizman. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 1008963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 782169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halford, W. P., C. D. Kemp, J. A. Isler, D. J. Davido, and P. A. Schaffer. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 756143-6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 753240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Härle, P., B. Sainz, Jr., D. J. Carr, and W. P. Halford. 2002. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-α/β. Virology 293295-304. [DOI] [PubMed] [Google Scholar]
- 25.Ishov, A. M., and G. G. Maul. 1996. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 134815-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishov, A. M., A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss III, and G. G. Maul. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishov, A. M., O. V. Vladimirova, and G. G. Maul. 2002. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 767705-7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishov, A. M., O. V. Vladimirova, and G. G. Maul. 2004. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J. Cell Sci. 1173807-3820. [DOI] [PubMed] [Google Scholar]
- 29.Lavau, C., A. Marchio, M. Fagioli, J. Jansen, B. Falini, P. Lebon, F. Grosveld, P. P. Pandolfi, P. G. Pelicci, and A. Dejean. 1995. The acute promyelocytic leukaemia-associated PML gene is induced by IFN. Oncogene 11871-876. [PubMed] [Google Scholar]
- 30.Lehming, N., A. Le Saux, J. Schuller, and M. Ptashne. 1998. Chromatin components as part of a putative transcriptional repressing complex. Proc. Natl. Acad. Sci. USA 957322-7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maul, G. G. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20660-667. [DOI] [PubMed] [Google Scholar]
- 32.Maul, G. G., and R. D. Everett. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 751223-1233. [DOI] [PubMed] [Google Scholar]
- 33.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 742679-2690. [DOI] [PubMed] [Google Scholar]
- 34.Maul, G. G., and D. Negorev. 2008. Differences between mouse and human cytomegalovirus interactions with their respective hosts at immediate early times of the replication cycle. Med. Microbiol. Immunol. 197241-249. [DOI] [PubMed] [Google Scholar]
- 35.Mittnacht, S., P. Straub, H. Kirchner, and H. Jacobsen. 1988. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology 164201-210. [DOI] [PubMed] [Google Scholar]
- 36.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 742052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mossman, K. L., and J. R. Smiley. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 761995-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Negorev, D., A. M. Ishov, and G. G. Maul. 2001. Evidence for separate ND10-binding and homo-oligomerization domains of Sp100. J. Cell Sci. 11459-68. [DOI] [PubMed] [Google Scholar]
- 39.Negorev, D. G., O. V. Vladimirova, A. Ivanov, F. Rauscher III, and G. G. Maul. 2006. Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 808019-8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oberman, F., and A. Panet. 1989. Characterization of the early steps of herpes simplex virus replication in interferon-treated human cells. J. Interferon Res. 9563-571. [DOI] [PubMed] [Google Scholar]
- 41.Oberman, F., and A. Panet. 1988. Inhibition of transcription of herpes simplex virus immediate early genes in interferon-treated human cells. J. Gen. Virol. 69(Pt. 6)1167-1177. [DOI] [PubMed] [Google Scholar]
- 42.Preston, C. M. 2000. Repression of viral transcription during herpes simplex virus latency. J. Gen. Virol. 811-19. [DOI] [PubMed] [Google Scholar]
- 43.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 44.Saffert, R. T., and R. F. Kalejta. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 819109-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seeler, J.-S., A. Marchio, R. Losson, J. M. P. Desterro, R. T. Hay, P. Chambon, and A. Dejean. 2001. Common properties of nuclear body protein SP100 and TIF1α chromatin factor: role of SUMO modification. Mol. Cell. Biol. 213314-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stow, E. C., and N. D. Stow. 1989. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J. Gen. Virol. 70(Pt. 3)695-704. [DOI] [PubMed] [Google Scholar]
- 47.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 672571-2585. [DOI] [PubMed] [Google Scholar]
- 48.Tavalai, N., P. Papior, S. Rechter, M. Leis, and T. Stamminger. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 808006-8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wasylyk, C., S. E. Schlumberger, P. Criqui-Filipe, and B. Wasylyk. 2002. Sp100 interacts with ETS-1 and stimulates its transcriptional activity. Mol. Cell. Biol. 222687-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weichenhan, D., B. Kunze, W. Traut, and H. Winking. 1998. Evolution by fusion and amplification: the murine Sp100-rs gene cluster. Cytogenet. Cell Genet. 80226-231. [DOI] [PubMed] [Google Scholar]
- 51.Yordy, J. S., R. Li, V. I. Sementchenko, H. Pei, R. C. Muise-Helmericks, and D. K. Watson. 2004. SP100 expression modulates ETS1 transcriptional activity and inhibits cell invasion. Oncogene 236654-6665. [DOI] [PubMed] [Google Scholar]
- 52.Yordy, J. S., O. Moussa, H. Pei, D. Chaussabel, R. Li, and D. K. Watson. 2005. SP100 inhibits ETS1 activity in primary endothelial cells. Oncogene 24916-931. [DOI] [PubMed] [Google Scholar]
- 53.Zhong, S., S. Muller, S. Ronchetti, P. S. Freemont, A. Dejean, and P. P. Pandolfi. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 952748-2752. [PubMed] [Google Scholar]
- 54.Zong, R. T., C. Das, and P. W. Tucker. 2000. Regulation of matrix attachment region-dependent, lymphocyte-restricted transcription through differential localization within promyelocytic leukemia nuclear bodies. EMBO J. 194123-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]