

Abstract
The incidence of cutaneous malignant melanoma, tumours arising from melanocytes, has increased markedly over the past few years in many countries. Although early melanoma is curable through surgical excision, the prognosis of advanced melanoma is very poor, this tumour being resistant to the current therapies. Thus, there is a need for new therapies to improve the treatment of advanced melanoma. This review provides an overview of the recent discoveries in the genetics of melanoma which could offer new therapeutic opportunities.
Keywords: Animals; Antineoplastic Agents; therapeutic use; Early Diagnosis; Genes, ras; Humans; MAP Kinase Signaling System; genetics; Melanocytes; metabolism; Melanoma; epidemiology; genetics; metabolism; therapy; Mutation; Oncogenes; Prognosis; Proto-Oncogene Proteins c-kit; genetics; Signal Transduction; genetics; Skin Neoplasms; epidemiology; genetics; metabolism; therapy; raf Kinases; genetics
INTRODUCTION
Cutaneous melanoma, a tumour arising from melanocytes, has become a major public health problem in many countries. Episodic exposure of fair-skinned individuals to intense sunlight is thought to be responsible for the steadily increasing worldwide incidence of melanoma over recent decades. Since the mid 1960s, the incidence of melanoma has risen by 3–8% per year in most people of European background. Despite this increase, and an overall rise in mortality due to melanoma, the survival rate has improved substantially. Roughly 60% of those diagnosed with melanoma in the 1960s died of the disease, compared with just 11% more recently, an improvement attributed mainly to early detection [66]. However, although early melanoma is curable through surgical excision, the prognosis of advanced melanoma is very poor because it is resistant to most chemotherapeutic agents. Therefore, identifying molecular targets for the diagnosis and treatment of patients is of particular importance and has been the focus of recent melanoma research. At least three signal transduction pathways have been implicated in melanoma genesis: The Mitogen Activated Protein Kinase (MAPK) pathway, the Phosphatidylinositol 3-Kinase (PI3K) pathway and the cyclic AMP (cAMP) pathway (figure 1). Their involvement in melanoma development and their therapeutic implications are detailed below.
Figure 1.
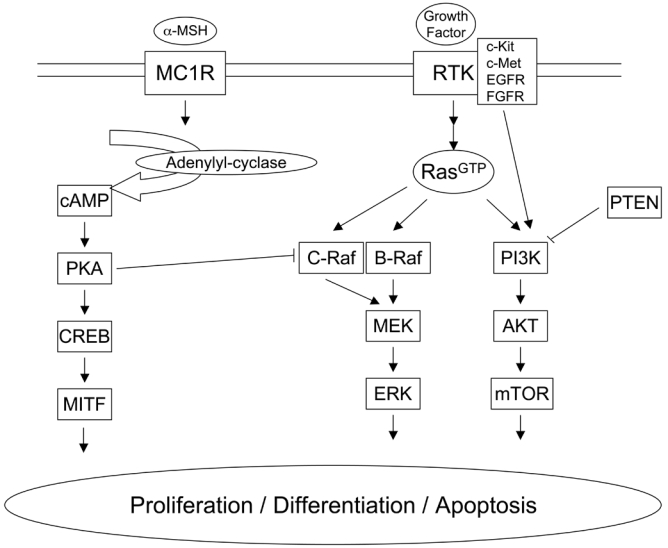
Description of the three signal transduction pathways which have been implicated in melanocyte proliferation, differentiation and apoptosis and in melanoma genesis. cAMP pathway (left), MAPK pathway (centre) and PI3K pathway (right).
THE MAPK PATHWAY
The MAPK pathway is activated downstream of most growth factor receptors and is an essential signal transduction cascade that controls cell survival, growth, differentiation and transformation [13]. Activation of the MAPK pathway is initiated by Receptor Tyrosine Kinases (RTK), which are trans-membrane cell surface proteins that contain an N-terminal extracellular ligand binding domain and a C-terminal intracellular tyrosine kinase domain. Most RTK dimerise following binding to their ligand which induces receptor activation and autophosphorylation of tyrosine residues in the RTK intracellular domain. These phosphorylated tyrosines serve as binding sites for adaptor proteins responsible for the activation of several molecules including the small GTPase of the Ras family [47]. Ras is the critical link that allows RTK to signal to the Extracellular Regulated Kinase (ERK). Once activated, Ras recruits the Raf kinase to the plasma membrane where it gets activated. Raf phosphorylates and activates a second protein kinase called MAPK/ERK kinase (MEK); this in turn activates the third protein kinase termed ERK. ERK regulates downstream signalling complexes of transcription factors that affect gene expression, rearrangements of the cytoskeleton and metabolism [67] (figure 1). The MAPK pathway regulates melanocyte proliferation downstream of agonists such as basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF) and stem cell factor (SCF) [26]. The recent discovery in melanoma of frequent mutations in the MAPK pathway represents a major breakthrough in the genetics of this cancer and brings great hope for its treatment as detailed above.
Receptor Tyrosine Kinases
c-Kit
The c-Kit RTK is the receptor for the SCF and is centrally involved in the development of multiple cell lineages, including melanocytes. Insights into the roles of this receptor and its cognate ligand have been greatly facilitated by the large series of naturally occurring mutations in the murine genes that encode these molecules, the dominant white spotting (W) and steel (Sl) loci, respectively. Thus, mice with loss-of-function in either the W or Sl loci exhibit white spotting, highlighting the importance of c-Kit in melanocyte development [31]. c-Kit is a type III RTK characterised by the presence of five immunoglobulin-like domains in its extracellular portion. SCF binds to the second and third immunoglobulin domains while the fourth domain plays a role in receptor dimerisation [56]. Binding of SCF to c-Kit induces receptor dimerisation and autophosphorylation of specific tyrosine residues. These phosphorylation events create docking sites for specific Src homology 2 (SH2) domain-containing proteins, which in turn control various intracellular signalling pathways. Recruitment of particular targets is mediated by the ability of their SH2 domains to recognise specific phosphotyrosine (pTyr)-containing motifs on the activated receptor. Numerous signaling molecules have been identified as binding partners for specific pTyr residues on activated c-Kit, including the p85 subunit of PI3K (by means of tyrosine 721), phospholipase C (by means of tyrosine 730), and the Grb2 and Grb7 adapter proteins (by means of tyrosine residues 703 and 936). Additionally, signalling molecules, including Src family kinases and the protein tyrosine phosphatases Shp-1 and Shp-2, have been shown to associate with a dual tyrosine motif in the juxtamembrane region of c-Kit (tyrosine residues 568 and 570) [36]. Until recently, data suggested that c-Kit might act as a tumour suppressor gene in melanoma. Indeed, c-Kit is expressed in melanocytes, benign melanocytic naevi and in situ malignant melanoma, but tends to be reduced or lost with tumour progression to invasive and metastatic melanoma [49]. Furthermore, c-Kit expression is lost in most melanoma cell lines [35]. However, a subset of melanoma have been found to maintain c-Kit overexpression, even in metastatic lesion and activating c-Kit mutations have recently been identified in 2–3% of melanoma [9]. The majority of c-Kit mutations are present in exons 11, 13 17 and 18 of the c-c-Kit gene, K642E and L576P being the most frequent. The biological significance of these mutations in melanoma remains unclear as it was recently shown that constitutive signalling from an active c-Kit mutant found in gastrointestinal stromal tumours (D814Y) did not stimulate melanocyte proliferation or induce their transformation [2]. Whether K642E and L576P are oncogenic in melanocytes remains to be demonstrated. This is particulary important as a c-Kit inhibitor is available in the clinic and could benefit a subset of melanoma patients (table 1). Imatinib mesylate is an oral tyrosine kinase inhibitor that targets bcr-Abl, c-Kit and platelet-derived growth factor receptor (PDGFR) alpha and beta, leading to remarkable clinical responses in several cancers such as gastrointestinal stromal tumors [59] and myeloid leukaemia [70]. Unfortunately, clinical trials in melanoma patients did not show promising responses. Indeed, different phase II trials were undertaken but no clinical responses were noted among those patients. Tumor biopsies were obtained from patients who received Imatinib treatment and cell lines were established from these tumors. Ex vivo Imatinib treatment of the cell lines showed no anti-proliferation effect [68]. The lack of efficacy of Imatinib in those clinical trials can be explained by the fact that no mutations were found in any of the analysed c-Kit sequences. A clinical phase II trials with imatinib is currently in process in patients affected by acral or mucosal melanoma which often carry c-Kit mutation.
Table 1.
Targets and selected drugs for melanoma therapy
| Main targets | Inhibitors | Other known targets |
|---|---|---|
| c-Kit | Imatinib | Bcr-Abl, PDGFRα/β |
| SU11248(sunitinib) | VEGFR, PDGFRα/β | |
| c-Met | SU11274 | Unknown |
| PHA-665752 | Unknown | |
| Ras | Tipifarnib (Zanestra) | Farnesylated proteins |
| SCH66336 | Farnesylated proteins | |
| Raf | Sorafenib (BAY-439006) | VEGFR2, PDGFRβ |
| CHIR-265 | VEGFR, PDGFRα/β | |
| PLX720 | Unknown | |
| MEK | CI-1040 (PD184352) | Unknown |
| ARRY-142886 (AZD6244) | Unknown | |
| PD0325901 | Unknown | |
| Lethal Factor (LF) | Unknown | |
| mTOR | Rapamycin | Unknown |
| CCI779 (temsirolimus) | Unknown | |
| RAD-001 (everolimus) | Unknown | |
| AP-23573 (deforolimus) | Unknown | |
c-Met
c-Met is an RTK that has been found mutated in several human cancers, including sporadic papillary renal carcinoma, gastric and liver cancer, small cell and non–small cell lung cancers, and metastases of head and neck squamous cell carcinoma and is known to stimulate the invasive growth of cancer cells and increase their metastatic potential [6]. The natural ligand for c-Met is HGF, which is also known as scatter factor. HGF is a multifunctional cytokine acting as a mitogen and morphogen for many epithelial cells. Recent studies by Herlyn’s group have shown that most melanoma cells, but not normal melanocytes, produce HGF, which can induce sustained activation of its receptor, the MAPK and the PI3K pathways [38]. Hence, an autocrine HGF/c-Met signalling loop may be involved in the development of melanoma. In transgenic mice ubiquitously expressing HGF, hyperpigmentation of the skin is observed and melanocytes are located within the dermis and epidermis, whereas mouse melanocytes are normally located in the hair follicule [51]. Moreover, melanoma arose spontaneously but could also be UV induced in these transgenic animals [50]. It is therefore suggested that c-Met autocrine activation induces the development of malignant melanoma and the acquisition of the metastatic phenotype. Recently, a mutation in exon 13 in the juxtamembrane domain (N948S) of c-Met was found in three melanoma cell lines. Also, in 14 melanoma tumours a mutation of c-Met was discovered (R988C) in exon 14 in the juxtamembrane domain. This mutation has also been seen in non–small cell and small cell lung cancer tumors but this is the first report of a mutation in c-Met in human melanoma tumors [54]. Whether these c-Met mutants are oncogenic in melanocytes remain to be demonstrated but therapy targetting c-Met could be suitable for melanoma treatment. Several therapies targetting the HGF/c-Met signalling pathway are being developed. One therapeutic strategy is to develop antagonism of ligand-receptor interaction. NK4, composed of the N-terminal hairpin and subsequent four kringle domains of HGF, acts as the competitive antagonist for HGF and angiogenesis inhibitor. In experimental models of distinct types of cancers, NK4 protein administration or NK4 gene therapy inhibited tumour invasion, metastasis, and angiogenesis-dependent tumor growth [33]. Recently, five different humanised monoclonal antibodies that bound to and neutralized human HGF were generated and characterised. Antibodies with subnanomolar affinities for HGF blocked binding of human HGF to c-Met and inhibited HGF-mediated c-Met phosphorylation, cell proliferation, survival, and invasion [5]. Recent successes in the treatment of chronic myeloid leukemia, gastrointestinal stromal tumours and other cancers using RTK inhibitors strongly support the potential efficacy of this therapeutic strategy in targeting c-Met (table 1). SU11274 and PHA665752 are two selective synthetic inhibitors of c-Met ATP binding, which block HGF stimulated activities in cultured cells and tumourigenicity in well characterised c-Met driven xenograft models [53]. Clinical trials are now underway.
Ras GTPase
Ras proteins are small G-proteins embedded on the inner surface of the plasma membrane through post-translational processing. Ras acts as a molecular switch that is ‘on’ when bound to GTP and ‘off’ when bound to GDP. Following binding of cytokines, growth factors or mitogens to their appropriate receptors, the protein complex Shc/Grb2/SOS is recruited to the receptor. Upon stimulation by Shc/Grb2/SOS, the inactive Ras exchanges GDP for GTP, undergoes a conformational change and becomes active [57]. The signalling responses induced by Ras activation are highly varied, owing largely to the myriad effector molecules that bind Ras in its GTP-bound state, such as the Raf kinase family, PI3K, RalGDS and Phospho Lipase C ε (PLCε) [42]. There are three closely related Ras proteins H, K, and N-Ras. Mutations in codon 12, 13 or 61 of one of the three Ras genes convert these genes into active oncogenes. This results in the Ras proteins remaining predominantly bound to GTP compared with wild-type and losing the ability to be deregulated. Around 20% of melanomas harbour oncogenic mutations in the Ras genes, most mutations being located in codon 61 of N-Ras (Q61R or Q61K) [9]. Ras has several effectors that may contribute to the oncogenic property of mutated Ras. However, the existence in melanoma of frequent mutations in B-Raf, an effector of Ras, suggests that the Ras/Raf/MEK/ERK pathway is the predominant effector of Ras in melanoma [12]. The presence of mutations in Ras or upstream of Ras, in RTK, makes Ras a good therapeutic target for melanoma treatment. However, the only clinical agents used to date that affect Ras activity are the farnesyltransferase inhibitors (FTIs) (table 1). These agents impair the post-translational modification of the Ras proteins and prevent their membrane localisation, which is required for signalling activity [27]. Several strategies have been developed to inhibit the farnesylation of Ras, the most common being the design of compounds that mimic the carboxy-terminal CAAX motif of Ras and compete for binding to farnesyltransferase. Another strategy has been to make compounds that compete with the farnesyl pyrophosphate group. A large number of highly effective FTIs have been identified, mostly through high-throughput screening of compound libraries [29]. Clearly, farnesylation is not a process that is limited to the Ras proteins; thus, this class of agents is surely limited by lack of specificity. Nonetheless, in preclinical models and in limited correlative studies in clinical trials, there is evidence that Ras activity is impaired by farnesyltransferase inhibitors. Indeed, a phase II study was undertaken to evaluate the activity and tolerability of the FTI tipifarnib (Zarnestra) as well as its ability to inhibit protein farnesylation and oncogenic pathways in patients with relapsed multiple myeloma. Results show that tipifarnib can induce disease stabilisation, and can inhibit farnesylation [3]. Data from a limited number of patients (4/34) of this phase II study show that treatment with tipifarnib affected the main oncogenic and tumour survival pathways differently. The data show that tipifarnib inhibited phospho-signal transducer and activator of transcription (STAT)-3 levels in all four patients. However, this inhibition did not correlate with clinical activity, since two out of the four patients progressed. Similarly, the effects on phosphorylated ERK1/2 levels did not correlate either. In contrast, tipifarnib treatment inhibited the phosphorylated Akt levels in the two patients that stabilised but not in the patients which progressed [3]. In conclusion, the significance of this observation could not be determined due too limited number of samples. Another phase II study was undertaken in patients with metastatic pancreatic adenocarcinoma but no anti-tumour activity was measured [7]. Although, H-Ras is exclusively modified by farnesyltransferase, K-Ras and, to a lesser extent, N-Ras can also be modified by geranylgeranyltransferase [74]. Geranylgeranylation (GGT) of K-Ras and N-Ras becomes important only when farnesylation is blocked. As the vast majority of Ras mutations in human tumours are in K-Ras, followed by N-Ras, with very few in H-Ras, it is likely that inhibition of mutant Ras farnesylation is not responsible for any antitumour effects of FTIs. Attempting to inhibit the function of KRas and NRas by using FTIs and GGT inhibitors together has failed because of the very high toxicity that is associated with this combination. However, combination studies with FTIs and chemotherapy in N-Ras mutated melanoma cells seems justified by preclinical data. Indeed, preclinical studies showed that FTI (SCH66336) inhibits farnesyl transferase, leads to morphological changes in melanoma cells associated with actin stress fibre formation and inhibits the growth melanoma cells. They also showed that SCH66336 induced a concentration and time-dependent increase in apoptosis, cell cycle arrest and, at last, increased melanoma sensitivity to cisplatin [61]. Clinical trials associating FTI and other inhibitors in melanoma patients are now underway.
Raf serine/threonine kinase
Raf proteins are serine/threonine kinases that play a pivot role in the MAPK pathway. The Raf kinase family is composed of three members: A-Raf, B-Raf and C-Raf (also termed Raf-1) that share a common architecture, with three conserved regions: two (CR1 and CR2) in the N terminus and the third (CR3), which encodes the kinase domain, in the C terminus [71]. Raf proteins are subject to complex regulation, which is reflected by the presence of numerous phosphorylation sites that are distributed throughout the proteins. Some of the sites are conserved in all three isoforms, which indicates common mechanisms of regulation, but others are not conserved, which shows that these proteins can be independently regulated [17]. Raf proteins are recruited to the plasma membrane by direct binding to activated Ras and are activated through many steps, including lipid binding, binding to other proteins, conformational changes and phosphorylation. Raf positively regulates MEK1 and 2 by phosphorylating serine residues in the catalytic domain. All three Raf family members are able to activate MEK but different biochemical potencies have been observed (B-Raf>C-Raf>A-Raf). Downstream MEK targets are ERK1 and 2 serine threonine kinases, which can directly regulate many transcription factors including Ets-1, c-Jun and c-Myc [48]. The most commonly mutated component of the MAPK pathway in melanoma is B-Raf which is mutated in 50% to 70% of cutaneous melanoma [12]. The most common mutation is found within the kinase domain of B-Raf, with a single substitution (T to A) of glutamate for valine at codon 600 (V600E) being responsible for 80% of observed mutations [23]. V600EB-Raf stimulates constitutive ERK signalling, driving proliferation and survival and providing essential tumour growth [28, 30]. V600EB-Raf also contributes to neo-angiogenesis by stimulating autocrine vascular endothelial growth factor (VEGF) secretion [58]. Because activating mutations in B-Raf allow the kinase to remain constitutively active irrespective of upstream Ras activity, Ras is an unattractive target in B-Raf mutant melanoma (table 1). Sorafenib (BAY 43-9006) is an agent with B-Raf inhibitory activity that has reached clinical trials [73]. This agent was selected for clinical development before B-Raf was identified as an oncogene relevant to melanoma and is most potent against C-Raf. Nonetheless, the homology in the ATP binding sites of the Raf kinases provides for potent cross-reactivity against B-Raf. Although sorafenib was found to be a potent inhibitor of VEGF receptors later in its clinical development, its activity against B-Raf remains the focus of interest in the context of melanoma. In melanoma cell lines, sorafenib induces cell cycle arrest and apoptosis and was able to elicit a significant retardation in the growth of human melanoma tumour xenografts even if the animals were not cured [30]. In clinical trials, as a single-agent sorafenib has been associated with few objective responses and only a modest degree of tumor stabilisation [18, 19]. Yet, phase I/II trials combining sorafenib with chemotherapy has been undertaken and combination with dacarbazine and carboplatin/paclitaxel showed promising efficacy and acceptable tolerability [21] It remains to be determined in appropriately controlled trials in patients with melanoma whether combination therapies with sorafenib show superior efficacy to chemotherapy alone and whether this effect is mediated through inhibition of the Raf kinase pathway, rather than inhibition of angiogenesis [52].
CHIR-265, a molecule with B-Raf and VEGF inhibitory activity, is in phase I clinical trial in melanoma patients [39]. Pre-clinical experiments show that CHIR-265 induces tumour regression in melanoma tumour xenografts that express V600EB-Raf. Significant and sustained inhibition of downstream pathway biomarkers (phospho-MEK, phospho-ERK and cyclin D1) was associated with this tumour response whereas sorafenib caused tumour stasis and no detectable inhibition of Ras/Raf/MEK/ERK pathway biomarkers.
PLX720 is a novel inhibitor of oncogenic B-Raf which shows specificity toward the V600EB-Raf mutant [20]. PLX720 induces an inhibition of proliferation in A375 melanoma cells ex vivo and an inhibition of tumour growth can be measured in an A375 tumour xenograft model. The inhibition of proliferation correlates with the inhibition of the MAPK pathway. Clinical trials in patients harbouring the V600EB-Raf mutant are underway.
MEK serine/threonine kinase
Although MEK has never been found mutated in melanoma, it is the immediate and sole downstream target of Raf and therefore an appealing therapeutic target in melanoma with mutations in the MAPK pathway. Due to the prevalence of mutations in the MAPK pathway in human cancers in general, many MEK inhibitors are in clinical development and could benefit melanoma therapy (table 1).
The highly specific MEK inhibitor CI-1040 inhibit ex vivo proliferation, soft agar colony formation and matrigel invasion of V600EB-Raf mutant human melanoma cells. Moreover, administration of CI-1040 to mice bearing A375MR-Luc xenograft prevents pulmonary metastases formation and cause established metastases regression [8]. CI-1040 reached phase II clinical trials but its development was stopped due to insufficient efficacy [55].
ARRY-142886 (AZD6244) is a second generation and ATP-uncompetitive inhibitor of MEK, which has shown better efficacy than CI-1040 in preclinical models and is currently in early clinical development. It has been recently shown that ARRY-142886 was able to inhibit cellular growth and induce apoptosis of several lines containing B-Raf and Ras mutations. Moreover, ARRY-142886 demonstrated its efficacy against pancreatic BxPC3 xenograft model and human colon carcinoma tumours [75]. This compound is currently being investigated in clinical studies.
PD0325901 is a derivative of CI 1040 that has improved oral bioavailability and induces a longer duration of target suppression. Preclinical studies show that PD0325901 completely suppressed the growth of V600EB-Raf mutant xenografts [62]. Phase I trials were performed in patients with breast, colon nonsmall cell lung cancer (NSCLC) or melanoma. According to the analysis performed on serial biopsies, PD0325901 treatment show greater than 80% of ERK inhibition. But, despite target inhibition, it appears that as a single agent PD0325901 is not sufficient to induce tumor regression and stabilisation [41]. The combination of MEK inhibition with chemotherapy seems justified but remains to be clinically investigated.
Lethal factor (LF), one of the three proteins of the anthrax toxin of Bacillus anthracis, inhibits MAPK signalling by proteolitically cleaving MEK 1 and 2. Preclinical studies show that LF induces regression of human melanoma xenografts [1, 32]. These studies should pave the way for melanoma clinical trials using LF.
It is important to note that B-Raf mutated melanoma cell lines were recently shown to be more sensitive to MEK inhibition than Ras mutated cell lines ex vivo. Moreover, pharmacological MEK inhibition completely inhibited tumour growth in B-Raf mutant xenografts whereas Ras mutant tumours were only partially inhibited [62]. These data suggest that MEK inhibitors may provide a therapeutic strategy for B-Raf but not Ras mutant melanoma. We have recently shown that Ras mutated melanoma cell lines switch their signalling from B-Raf to C-Raf [16]. This finding has important therapeutic implications because its suggests that both B-Raf and C-Raf are valid targets in melanoma, but this depends whether B-Raf or Ras are mutated in the tumour.
PI3K PATHWAY
Type I PI3K is another effector of Ras. Activated PI3K converts phosphatidylinositol 4,5 phosphate (PIP2) into phosphatidylinositol 3,4,5 phosphate (PIP3) which regulates the activity and localisation of a number of target proteins including those that contain pleckstrin homology (PH) domains [22] (figure 1). One such PH domain-containing protein regulated by lipids is the serine/threonine kinase Akt (also called protein kinase B). Targets for Akt include kinases, glycogen-synthase kinase 3, p70S6 kinase, transcription factors, FKHR and cAMP-response element-binding protein, as well as proteins associated with apoptosis, caspase 9 and BAD [43]. This pathway is negatively regulated by the phosphatase and tensin homolog deleted on chromosome ten (PTEN) and Src homology 2 domain-containing inositol phosphatase (SHIP)-1 and 2 which remove the phosphates from PIP3 [11]. PTEN is a protein and lipid phosphatase that inhibits Akt activation by PI3K [60]. Loss of heterozygosity in the region of chromosome 10q, which harbors the PTEN locus, was demonstrated in 30–50% of melanomas and somatic mutations of the PTEN gene were detected in 10% of melanomas [4, 25 ]. Furthermore, constitutive activation of Akt3, the predominant Akt isoform in melanoma, was recently reported in more than 60% of melanomas [63], emphasising the importance of the PI3K pathway activation in melanoma development. The expression of phosphorylated and therefore active Akt protein increases dramatically with melanoma invasion and metastasis and is inversely correlated with patient survival [10]. Increased Akt activity prolongs cell survival through the inactivation of the Bcl-2 antagonist of cell death (BAD) protein. In model systems, suppression of Akt3, a member of the Akt family, reduces the survival of melanoma cell and the growth of human melanoma implanted in immunodeficient nude mice [63]. Therefore, the PI3K is a good therapeutic target for melanoma treatment. Given that restoration of PTEN is more technically challenging than pharmacologic inhibition of components of the PI3K pathway, current efforts are aimed at identifying inhibitory compounds for PI3K, Akt and mammalian target of rapamycin (mTOR), a downstream target of Akt. The lack of PI3K and Akt inhibitors for clinical use has turned attention to mTOR, for which numerous inhibitors are in clinical development (table 1). mTOR is a member of the family of protein kinases termed phosphatidylinositol 3-kinase-related kinase (PIKKs), which are involved in many regulatory cellular functions such as protein translation, cell cycle progression and cellular proliferation [45]. Aberrations in the upstream regulators of the PI3K pathway, leads to alterations in mTOR regulation activity.
Rapamycin is an inhibitor of mTOR with poor aqueous solubility and chemical stability which precluded its clinical development as an anti-cancer agent. Recently, a series of rapamycin analogs with improved aqueous solubility and stability have been synthesised and evaluated such as CCI-779 (temsirolimus), RAD-001 (everolimus) and AP-23573. CCI-779, a soluble ester analog of rapamycin, was selected for development as an anti-cancer agent based on its prominent anti-tumour profile and pharmaceutical and toxicological characteristics in preclinical studies [15]. Preclinical studies suggest that sensitivity to mTOR inhibitors may correlate with activation of the PI3K pathway or with aberrant expression of cell cycle regulatory or anti-apoptotic proteins. CCI-779 has demonstrated activity against melanoma in pre-clinical models and shown clinical benefit in patient with breast and renal carcinoma. However, a phase II clinical trial with CCI-779 resulted in only one objective response among 33 melanoma patients and early closure of the study [44]. The lack of single-agent anti-tumour efficacy does not preclude a chemotherapy-enhancing effect, which is suggested by pre-clinical studies. Indeed, preclinical studies show that CCI-779 has the potential to increase the chemotherapeutic efficacy of dacarbazine in human melanoma xenografts [65]. The combination of CCI-779 with cisplatin has shown promising results in a human melanoma xenograph model in severe combined immunodeficient (SCID) mice injected with three melanoma cell lines (518A2, Mel-JUSO or 607B). Four of six tumours of the 518A2 cell line were completely eradicated and the two others were reduced by 97% by CCI-779. Combination of both treatments also had a significant anti-tumour effect in the tumours of Mel-JUSO and 607B [64].
cAMP PATHWAY AND MITF
The microphthalmia-associated transcription factor (MITF) is a member of the basic helix-loop-helix leucine zipper (bHLH-Zip) transcription factors conserved in essentially all vertebrate species [40]. The M-MITF isoform is considered as a master regulator of differentiation of the melanocyte lineage and is induced through the cAMP pathway. M-MITF transcription is stimulated by hormones such as the α Melanocyte Stimulating Hormone (α-MSH) which binds to the melanocortin 1 receptor (MC1R), and stimulates cAMP production. cAMP activates the serine/threonine kinase protein kinase A (PKA) which phosphorylates the cAMP responsive element binding protein (CREB) transcription factors, which in turn, stimulate the M-MITF promoter (figure 1) [37]. MITF is located in the center of multiple signalling pathways which control differentiation, morphology, proliferation and survival of the various cells of the melanocyte lineage: melanoblasts, melanocytes and melanoma. MITF plays a major role in melanoblast differentiation, by inducing the key enzyme of melanogenesis, tyrosinase, and its secondary enzymes, Tyrp1 and Dct. MITF regulates morphology and migration of melanocytes, particularly by regulating cytoskeleton organisation and cell-cell adhesion [69]. MITF also regulates the transcription of the melanocyte-specific genes silver homologue (SILV) and melan-A, whose immunohistochemical detection points to the diagnosis of melanoma [14]. Recently, amplification of MITF was identified in 10% of primary cutaneous melanoma, 21% of metastatic tumours but not in naevi [24]. MITF amplification is more prevalent in metastatic disease and correlated with decreased overall patient survival. The cell lines that harboured MITF amplifications also contained B-Raf mutations and p16 inactivation. Ectopic MITF expression in conjunction with the V600EB-Raf mutant transformed primary human melanocytes showing that MITF can function as a melanoma oncogene [24]. How MITF acts as an oncogene is not clear but it may be linked to the transcriptional regulation of the c-MET proto-oncogene. It has been shown that MITF directly regulates the c-MET promoter and is essential for the homeostatic upregulation of c-MET expression following activation of the receptor by its ligand HGF [46]. c-MET is highly expressed in human melanoma and has been linked to the metastatic potential of melanomas (see above). It was also observed that MITF regulates c-MET expression in primary melanocytes and that the ability of HGF to stimulate invasive growth of melanocytes and melanoma cells in culture was abolished upon suppression of endogenous MITF [46]. MITF may also contribute to melanocyte transformation through its ability to regulate apoptosis [34]. It was recently shown that reduction of MITF activity sensitizes melanoma cells to chemotherapeutic agents suggesting that targeting MITF in combination with B-Raf or cyclin-dependent kinase inhibitors may offer a rational therapeutic avenue into melanoma [24]. However, others have also demonstrated that MITF is an antiproliferative factor in melanoma that is downregulated by B-Raf signalling and that this is a crucial event for the progression of melanomas harboring oncogenic B-Raf [72]. The therapeutic potential of MITF awaits further studies.
CONCLUSION
The future of melanoma therapy relies on tailored targeted therapies that are likely to result from the significant progress in understanding the molecular mechanisms leading to melanoma. It becomes evident that the response to targeted therapies will depend on which type of genetic alteration is responsible for the dysregulation. Recent melanoma genetics studies allow the classification of melanoma into four subtypes: Melanoma occurring on skin without signs of chronic sun-induced damage (non-CSD melanoma), on sun-protected skin such as the palms, soles or subungual sites (acral melanoma), on mucosal membranes (mucosal melanoma) and finally on skin showing evidence of chronic sun-induced sun damage (CSD melanoma). Signalling pathways are activated differently according to melanoma subtypes. Indeed, although B-Raf mutations are highly prevalent in non-CSD melanoma (59%), they are significantly less frequent in acral and mucosal melanoma and are uncommon in CSD melanoma. However, N-Ras mutations hit approximately 20% of melanomas in all four subtypes. c-Kit mutations are more frequent in acral (36%), mucosal (39%) and CSD melanoma (28%) but absent in non-SCD melanomas [9]. Recent discoveries in melanoma genetics raises high expectations for the rational design of more effective anti-melanoma therapies (table 1). However, it is evident that it will be necessary to vary the single pharmacological approaches to inhibit one pathway and to combine inhibitors of different pathways with classical as well as new chemotherapeutic agents.
Acknowledgments
We thank Dr Kirsten Dumaz for proofreading the manuscript. Work in our laboratory is funded by the Institut National de la Santé et de la Recherche Médicale (INSERM), Paris XII university, Société Française de Dermatologie, Société de Recherche Dermatologique and the Association pour la Recherche sur le Cancer (ARC).
Abbreviations
- α-MSH
α melanocyte stimulating hormone
- BAD-Bcl
2 antagonist of cell death
- bFGF
basic fibroblast growth factor
- bHLH-Zip
basic helix-loop-helix leucine zipper
- cAMP
cyclic AMP
- CREB
cAMP responsive element binding protein
- CSD
chronic sun-induced damage
- EGF
epidermal growth factor
- ERK
extracellular regulated kinase
- FTI
farnesyltransferase inhibitor
- MAPK
mitogen activated protein kinase
- GGT
geranylgeranyltransferase
- HGF
hepatocyte growth factor
- LF
lethal factor
- NSCLC
non small cell lung cancer
- MEK
MAPK/ERK kinase
- MC1R
melanocortin 1 receptor
- MITF
microphthalmia-associated transcription factor
- mTOR
mammalian target of rapamycin
- PDGFR
platelet-derived growth factor receptor
- PH
pleckstrin homology
- PI3K
phosphatidylinositol 3-kinase
- PIKK
phosphatidylinositol 3-kinase-related kinase
- PIP2
phosphatidylinositol 4,5 phosphate
- PIP3
phosphatidylinositol 3,4,5 phosphate
- PKA
protein kinase A
- PLC
phospholipase-C
- pTyr
phosphotyrosine
- PTEN
phosphatase and tensin homolog deleted on chromosome ten
- RTK
receptor tyrosine kinase
- SCID
severe combined immunodeficient
- SCF
stem cell factor
- SH2
src homology 2
- SHIP
SH2 domain-containing inositol phosphatase
- SILV
silver homologue
- STAT
signal transducer and activator of transcription
- VEGF
vascular endothelial growth factor
References
- 1.Abi-Habib RJ, Urieto JO, Liu S, Leppla SH, Duesbery NS, Frankel AE. BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol Cancer Ther. 2005;4:1303–1310. doi: 10.1158/1535-7163.MCT-05-0145. [DOI] [PubMed] [Google Scholar]
- 2.Alexeev V, Yoon K. Distinctive role of the cKit receptor tyrosine kinase signaling in mammalian melanocytes. J Invest Dermatol. 2006;126:1102–1110. doi: 10.1038/sj.jid.5700125. [DOI] [PubMed] [Google Scholar]
- 3.Alsina M, Fonseca R, Wilson EF, Belle AN, Gerbino E, Price-Troska T, Overton RM, Ahmann G, Bruzek LM, Adjei AA, et al. Farnesyltransferase inhibitor tipifarnib is well tolerated, induces stabilization of disease, and inhibits farnesylation and oncogenic/tumor survival pathways in patients with advanced multiple myeloma. Blood. 2004;103:3271–3277. doi: 10.1182/blood-2003-08-2764. [DOI] [PubMed] [Google Scholar]
- 4.Birck A, Ahrenkiel V, Zeuthen J, Hou-Jensen K, Guldberg P. Mutation and allelic loss of the PTEN/MMAC1 gene in primary and metastatic melanoma biopsies. J Invest Dermatol. 2000;114:277–280. doi: 10.1046/j.1523-1747.2000.00877.x. [DOI] [PubMed] [Google Scholar]
- 5.Burgess T, Coxon A, Meyer S, Sun J, Rex K, Tsuruda T, Chen Q, Ho SY, Li L, Kaufman S, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006;66:1721–1729. doi: 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 6.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 7.Cohen SJ, Ho L, Ranganathan S, Abbruzzese JL, Alpaugh RK, Beard M, Lewis NL, McLaughlin S, Rogatko A, Perez-Ruixo JJ, et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J Clin Oncol. 2003;21:1301–1306. doi: 10.1200/JCO.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Collisson EA, De A, Suzuki H, Gambhir SS, Kolodney MS. Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res. 2003;63:5669–5673. [PubMed] [Google Scholar]
- 9.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 10.Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 11.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 13.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 14.Du J, Miller AJ, Widlund HR, Horstmann MA, Ramaswamy S, Fisher DE. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol. 2003;163:333–343. doi: 10.1016/S0002-9440(10)63657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudkin L, Dilling MB, Cheshire PJ, Harwood FC, Hollingshead M, Arbuck SG, Travis R, Sausville EA, Houghton PJ. Biochemical correlates of mTOR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin Cancer Res. 2001;7:1758–1764. [PubMed] [Google Scholar]
- 16.Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R. In Melanoma, RAS Mutations Are Accompanied by Switching Signaling from BRAF to CRAF and Disrupted Cyclic AMP Signaling. Cancer Res. 2006;66:9483–9491. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- 17.Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. Febs J. 2005;272:3491–3504. doi: 10.1111/j.1742-4658.2005.04763.x. [DOI] [PubMed] [Google Scholar]
- 18.Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, Gibbens I, Hackett S, James M, Schuchter LM, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–586. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escudier B, Lassau N, Angevin E, Soria JC, Chami L, Lamuraglia M, Zafarana E, Landreau V, Schwartz B, Brendel E, et al. Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res. 2007;13:1801–1809. doi: 10.1158/1078-0432.CCR-06-1432. [DOI] [PubMed] [Google Scholar]
- 20.Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606–1620. doi: 10.1200/JCO.2006.06.0442. [DOI] [PubMed] [Google Scholar]
- 21.Flaherty KT. Chemotherapy and targeted therapy combinations in advanced melanoma. Clin Cancer Res. 2006;12:2366s–2370s. doi: 10.1158/1078-0432.CCR-05-2505. [DOI] [PubMed] [Google Scholar]
- 22.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Garnett MJ, Marais R. Guilty as charged; B-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 25.Guldberg P, thor Straten P, Birck A, Ahrenkiel V, Kirkin AF, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. 1997;57:3660–3663. [PubMed] [Google Scholar]
- 26.Halaban R. The regulation of normal melanocyte proliferation. Pigment Cell Res. 2000;13:4–14. doi: 10.1034/j.1600-0749.2000.130103.x. [DOI] [PubMed] [Google Scholar]
- 27.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 28.Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- 29.Johnston SR. Farnesyl transferase inhibitors: a novel targeted tnerapy for cancer. Lancet Oncol. 2001;2:18–26. doi: 10.1016/s1470-2045(00)00191-1. [DOI] [PubMed] [Google Scholar]
- 30.Karasarides M, Chiloeches A, Hayward R, Niculescu-Duvaz D, Scanlon I, Friedlos F, Ogilvie L, Hedley D, Martin J, Marshall CJ, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 31.Kimura Y, Jones N, Kluppel M, Hirashima M, Tachibana K, Cohn JB, Wrana JL, Pawson T, Bernstein A. Targeted mutations of the juxtamembrane tyrosines in the Kit receptor tyrosine kinase selectively affect multiple cell lineages. Proc Natl Acad Sci U S A. 2004;101:6015–6020. doi: 10.1073/pnas.0305363101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koo HM, VanBrocklin M, McWilliams MJ, Leppla SH, Duesbery NS, Woude GF. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc Natl Acad Sci U S A. 2002;99:3052–3057. doi: 10.1073/pnas.052707699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuba K, Matsumoto K, Date K, Shimura H, Tanaka M, Nakamura T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res. 2000;60:6737–6743. [PubMed] [Google Scholar]
- 34.Larribere L, Hilmi C, Khaled M, Gaggioli C, Bille K, Auberger P, Ortonne JP, Ballotti R, Bertolotto C. The cleavage of microphthalmia-associated transcription factor, MITF, by caspases plays an essential role in melanocyte and melanoma cell apoptosis. Genes Dev. 2005;19:1980–1985. doi: 10.1101/gad.335905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lassam N, Bickford S. Loss of c-kit expression in cultured melanoma cells. Oncogene. 1992;7:51–56. [PubMed] [Google Scholar]
- 36.Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- 37.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, Herlyn M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene. 2001;20:8125–8135. doi: 10.1038/sj.onc.1205034. [DOI] [PubMed] [Google Scholar]
- 39.Li N, Batt D, Warmuth M. B-Raf kinase inhibitors for cancer treatment. Curr Opin Investig Drugs. 2007;8:452–456. [PubMed] [Google Scholar]
- 40.Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445:843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- 41.Lorusso PKS, Rinehart JR, Nabell L, Croghan G, Varterasian M, Sadis SS, Menon SS, Leopold J, Meyer MB. Phase 1–2 clinical study of a second generation oral MEK inhibitor, PD 0325901 in patients with advanced cancer. Journal of Clinical Oncology. 2005;23:194S–194S. [Google Scholar]
- 42.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 43.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 45.McCormick F. Cancer: survival pathways meet their end. Nature. 2004;428:267–269. doi: 10.1038/428267a. [DOI] [PubMed] [Google Scholar]
- 46.McGill GG, Haq R, Nishimura EK, Fisher DE. c-Met expression is regulated by Mitf in the melanocyte lineage. J Biol Chem. 2006;281:10365–10373. doi: 10.1074/jbc.M513094200. [DOI] [PubMed] [Google Scholar]
- 47.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 48.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1-to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 49.Natali PG, Nicotra MR, Sures I, Santoro E, Bigotti A, Ullrich A. Expression of c-kit receptor in normal and transformed human nonlymphoid tissues. Cancer Res. 1992;52:6139–6143. [PubMed] [Google Scholar]
- 50.Noonan FP, Otsuka T, Bang S, Anver MR, Merlino G. Accelerated ultraviolet radiation-induced carcinogenesis in hepatocyte growth factor/scatter factor transgenic mice. Cancer Res. 2000;60:3738–3743. [PubMed] [Google Scholar]
- 51.Otsuka T, Takayama H, Sharp R, Celli G, LaRochelle WJ, Bottaro DP, Ellmore N, Vieira W, Owens JW, Anver M, et al. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998;58:5157–5167. [PubMed] [Google Scholar]
- 52.Panka DJ, Atkins MB, Mier JW. Targeting the mitogen-activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res. 2006;12:2371s–2375s. doi: 10.1158/1078-0432.CCR-05-2539. [DOI] [PubMed] [Google Scholar]
- 53.Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006;12:3657–3660. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 54.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R, Salgia R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–2253. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 55.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 56.Roskoski R., Jr Structure and regulation of Kit protein-tyrosine kinase--the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338:1307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 57.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 58.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 59.Siddiqui MA, Scott LJ. Imatinib: a review of its use in the management of gastrointestinal stromal tumours. Drugs. 2007;67:805–820. doi: 10.2165/00003495-200767050-00012. [DOI] [PubMed] [Google Scholar]
- 60.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 61.Smalley KS, Eisen TG. Farnesyl transferase inhibitor SCH66336 is cytostatic, pro-apoptotic and enhances chemosensitivity to cisplatin in melanoma cells. Int J Cancer. 2003;105:165–175. doi: 10.1002/ijc.11064. [DOI] [PubMed] [Google Scholar]
- 62.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L, Robertson GP. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 64.Thallinger C, Poeppl W, Pratscher B, Mayerhofer M, Valent P, Tappeiner G, Joukhadar C. CCI-779 plus cisplatin is highly effective against human melanoma in a SCID mouse xenotranplantation model. Pharmacology. 2007;79:207–213. doi: 10.1159/000101008. [DOI] [PubMed] [Google Scholar]
- 65.Thallinger C, Werzowa J, Poeppl W, Kovar FM, Pratscher B, Valent P, Quehenberger P, Joukhadar C. Comparison of a Treatment Strategy Combining CCI-779 Plus DTIC Versus DTIC Monotreatment in Human Melanoma in SCID Mice. J Invest Dermatol. 2007 doi: 10.1038/sj.jid.5700872. [DOI] [PubMed] [Google Scholar]
- 66.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005;365:687–701. doi: 10.1016/S0140-6736(05)17951-3. [DOI] [PubMed] [Google Scholar]
- 67.Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5:350–356. doi: 10.1016/j.coph.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Ugurel S, Hildenbrand R, Zimpfer A, La Rosee P, Paschka P, Sucker A, Keikavoussi P, Becker JC, Rittgen W, Hochhaus A, et al. Lack of clinical efficacy of imatinib in metastatic melanoma. Br J Cancer. 2005;92:1398–1405. doi: 10.1038/sj.bjc.6602529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vance KW, Goding CR. The transcription network regulating melanocyte development and melanoma. Pigment Cell Res. 2004;17:318–325. doi: 10.1111/j.1600-0749.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- 70.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- 71.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 72.Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J Cell Biol. 2005;170:703–708. doi: 10.1083/jcb.200505059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 74.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5:405–412. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 75.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]