

Abstract
Protein scaffold complexes are a key mechanism by which a common signaling pathway can serve many different functions. Sequestering a signaling enzyme to a specific subcellular environment not only ensures that the enzyme is near its relevant targets, but also segregates this activity to prevent indiscriminate phosphorylation of other substrates. One family of diverse, well-studied scaffolding proteins are the A-kinase anchoring proteins (AKAPs). These anchoring proteins form multi-protein complexes that integrate cAMP signaling with other pathways and signaling events. In this review we focus on recent advances in the elucidation of AKAP function.
Introduction
Protein phosphorylation regulated by protein kinases and protein phosphatases is a primary mechanism for the regulation of enzymatic activities (Krebs, 1985) and inducing protein-protein interactions (Pawson & Nash, 2000). This bidirectional process is a flexible means of influencing cellular activity. Not surprisingly, a breakdown in signal transduction may lead to pathophysiological outcomes and the development of disease (Cohen, 1999).
The molecular organization of these signaling pathways is intriguing as most transduction cascades are composed of common elements. The initial signal is transduced through a receptor at the plasma membrane (such as a G-protein coupled receptor, or a receptor tyrosine kinase or phosphatase), which results in activation of the receptor or the mobilization of receptor-associated proteins to generate some form of intracellular message.
There has been a concerted research effort focused to understand how the subcellular location of protein kinases and phosphatases contributes to the regulation of phosphorylation events. Sequestering a signaling enzyme to a specific subcellular environment not only ensures that the enzyme is near its relevant targets, but also segregates this activity to prevent indiscriminate phosphorylation of other substrates. Thus protein scaffold complexes are a key mechanism by which a common signaling pathway can serve many different functions.
One family of diverse, well-studied scaffolding proteins are the A-kinase anchoring proteins (AKAPs) (Colledge & Scott, 1999). To date over 50 AKAPs have been identified in mammals and lower organisms (Wong & Scott, 2004). AKAPs contribute to the precision of intracellular signaling events by directing anchored enzyme pools to a subset of their physiological substrates at specific subcellular locations (Figure 1). AKAPs have little primary sequence similarity and thus were originally classified based on their ability to bind the cAMP-dependent protein kinase (protein kinase A; PKA) (Carr et al, 1991). As a result, evidence to support their function has primarily been gleaned from analysis of PKA phosphorylation events. However, it is now well recognized that an equally important role of AKAPs is their capacity to form multi-protein complexes that integrate cAMP signaling with other pathways and signaling events (Smith et al, 2006) (Jarnaess & Tasken, 2007). In this review we focus on recent advances in the elucidation of AKAP function.
Figure 1. Properties of AKAPs.
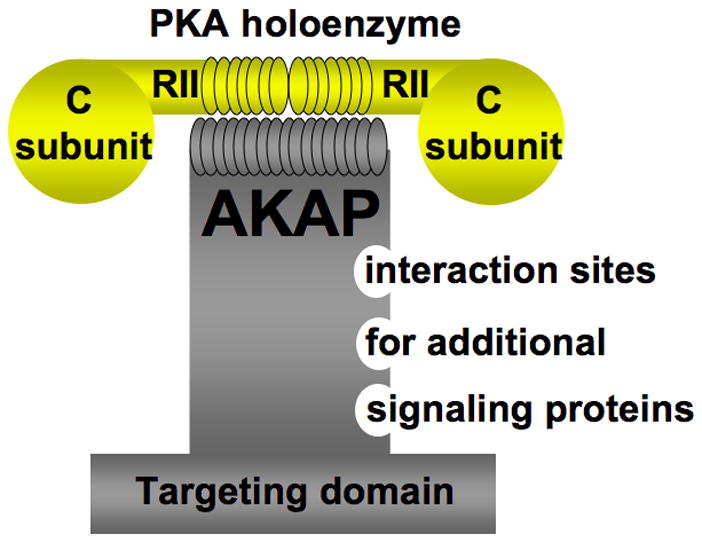
AKAPs bind to the regulatory subunit of PKA. AKAPs also bind additional signaling proteins (for example other protein kinases, protein phosphatases, phopshodiesterases, adenylyl cyclases and small G proteins). AKAPs function to target signaling complexes to discreet locations inside a cell.
PKA
PKA is a broad specificity Ser/Thr kinase that can phosphorylate a range of proteins. Many functions have been ascribed to this protein kinase, including metabolism (McKnight et al, 1998), learning and memory (Abel & Nguyen, 2008) and exocytosis (Szaszak et al, 2008). Several of these processes contribute to various disease states. For example, PKA is implicated in pancreatic b-cell function and may provide a novel means for combating diabetes (Nesher et al, 2002). PKA is also important in the molecular mechanisms of learning and memory, making it an attractive target for therapies seeking to inhibit cognitive decline resulting from neurodegenerative diseases (Bauman et al, 2004). The activity of PKA is regulated by its two regulatory subunits, which form a dimer that binds to the two catalytic subunits. Activation of PKA occurs through the binding of cAMP to the regulatory dimer, causing dissociation of the catalytic subunits (Corbin et al, 1973) (Kim et al, 2005) (Kim et al, 2007). The two main PKA subtypes are defined by the identity of their regulatory subunits, RI and RII (Scott, 1991). Anchoring of PKA by AKAPs confines PKA activity to a relevant subset of potential substrates. The majority of known AKAPs bind specifically to the RII holoenzyme. However, several dual specificity AKAPs, which bind to both PKA subtypes, have been identified. These include the dual-function anchoring proteins D-AKAP1 (Huang et al, 1997b) and D-AKAP2 (Huang et al, 1997a). While the intracellular regulation of PKA activity has been well described in terms of anchoring proteins, specific biological functions for AKAPs have been harder to identify. At present, proteomic and biochemical techniques have been exhaustively used to identify the majority of AKAPs and to characterize the components of their signaling complexes (Smith & Scott, 2006). Only recently, however, are we beginning to gain an understanding of the role that AKAPs play in cellular physiology.
PKA Anchoring
Most AKAPs contain a recognizable sequence that forms a binding site for the R subunits. This motif is a hallmark of all AKAPs and was first identified in the human thyroid anchoring protein, AKAP-Lbc. Biochemical studies showed that a 24-residue peptide (originally called Ht31) binds RII with nanomolar affinity (Carr et al, 1992; Carr et al, 1991). The Ht31 sequence is predicted to form an amphipathic helix, and structural studies indicate that the hydrophobic face of this region fits into a binding pocket formed by the N-terminal regions of the RII dimer of PKA (Newlon et al, 1997; Newlon et al, 2001) (Gold et al, 2006). Cellular delivery of this peptide or related derivatives (using cell-soluble stearated forms, or plasmid-based expression) has become a standard means to delineate a role for AKAPs in the coordination of cAMP-responsive events by disrupting PKA-RII anchoring inside cells (Figure 2).
Figure 2. Study of AKAP function by peptide-mediated disruption of PKA anchoring.
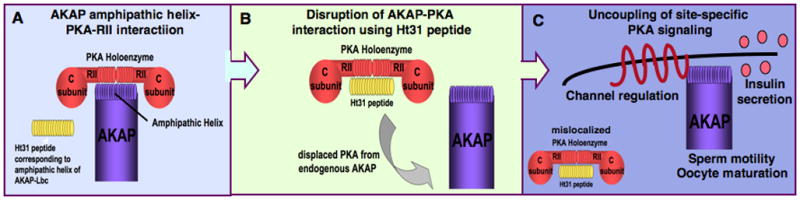
A) The AKAP-amphipathic helix-PKA-RII interaction can be disrupted in vivo by introduction of a competing peptide called Ht31.
B) Ht31 will bind to PKA-RII, thereby displacing PKA from an AKAP inside a cell.
C) Mislocalization of PKA by displacement from an AKAP may lead to uncoupling of site-specific PKA signaling. This has been demonstrated in the processes of channel regulation, insulin secretion, sperm motility and oocyte maturation.
The utility of this peptide as a disruptor of PKA anchoring was first demonstrated in studies showing that perfusion of Ht31 into cultured hippocampal neurons uncoupled PKA from α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors (Rosenmund et al, 1994). This attenuates post-synaptic AMPA receptor currents and synaptic events. Similarly, the Ht31 peptide has been used to demonstrate that disruption of PKA anchoring uncouples cAMP-dependent regulation of the L-type Ca2+ channel (Johnson et al, 1994). The importance of AKAPs in channel regulation relating to learning and memory and in cardiac physiology will be described later. The Ht31 peptide has also been used to demonstrate that sperm motility (Vijayaraghavan et al, 1997), oocyte maturation (Newhall et al, 2006) and GLP-1-mediated insulin secretion (Lester et al, 1997) are AKAP-dependent processes. The roles of relevant AKAPs in these processes will be described later.
Whole animal models; flies, fish and mice
Surprisingly, there is still little detail described for the physiological roles of AKAPs through the use of genetically modified model organisms. For many years Drosophila have been the most widely described genetically tractable organism, however there have been few studies involving AKAP function. Collectively, Drosophila studies have reinforced experiments carried out in cultured neurons indicating that AKAPs do indeed play a role in the subcellular organization of signaling components involved in learning and memory (Lu et al, 2007; Schwaerzel et al, 2007; Terman & Kolodkin, 2004).
Zebrafish are another genetically tractable organism. The combination of protein knockdown using morpholino-oligonucleotides combined with relative ease of analysis of embryogenesis make the use of this organism attractive for developmental studies. In particular, a recent study points to a role for gravin in gastrulation (Weiser et al, 2007) (see Reproduction & Development section).
As for mice, there are relatively few reports of AKAP knockouts. This may be for several reasons including lethality or lack of obvious phenotype. Recent studies in our laboratory suggest subtle alterations in phenotype, which could in part be due to compensatory mechanisms. We also have experience with embryonic lethality of certain AKAP knockouts, suggesting a previously unappreciated role in development. There is also now a well recognized role for AKAPs in sperm and oocyte function that may also impinge upon successful generation of homozygous knockout mice.
Notable knockout or transgene studies include WAVE-1 (Soderling et al, 2003) and AKAP150 (Hall et al, 2007) (see Learning and Memory section), AKAP149 (AKAP1) (Newhall et al, 2006), AKAP82 (AKAP4) (Miki et al, 2002) (see Reproduction & Development section) and D-AKAP2 (Tingley et al, 2007) (see Genetic Studies section).
Reproduction & Development
Sperm function
AKAPs play important roles in sperm function, including regulation of motility, sperm capacitation and the acrosome reaction (figure 3). Several AKAPs have been identified in sperm, including AKAP84 (AKAP1), AKAP110 (AKAP3), AKAP82 (AKAP4), AKAP95 (AKAP8), AKAP220 (AKAP11), WAVE-1 and MAP2 (Carr & Newell, 2007). The sperm function of AKAPs involves targeting of other signaling molecules in addition to PKA, including PDE’s (eg. PDE4A in the case of AKAP110 in flagella (Bajpai et al, 2006)) and protein phosphatases, for example PP1 in the case of AKAP220 (Schillace & Scott, 1999). In addition to scaffolding PKA, AKAPs also bind to a group of four proteins that share homology to the RII dimerization/docking (R2D2) domain. R2D2 proteins are expressed at high levels in the testis. These proteins function in the regulation of flagella and cilia independent of PKA activity and, unlike RII do not bind cAMP (Fiedler et al, 2008). Recently it has been demonstrated that these proteins are expressed in a wide variety of tissues suggesting a more general function in both motile and primary cilia (Fiedler et al, 2008).
Figure 3. Development – sperm function, oocyte maturation and embryogenesis.
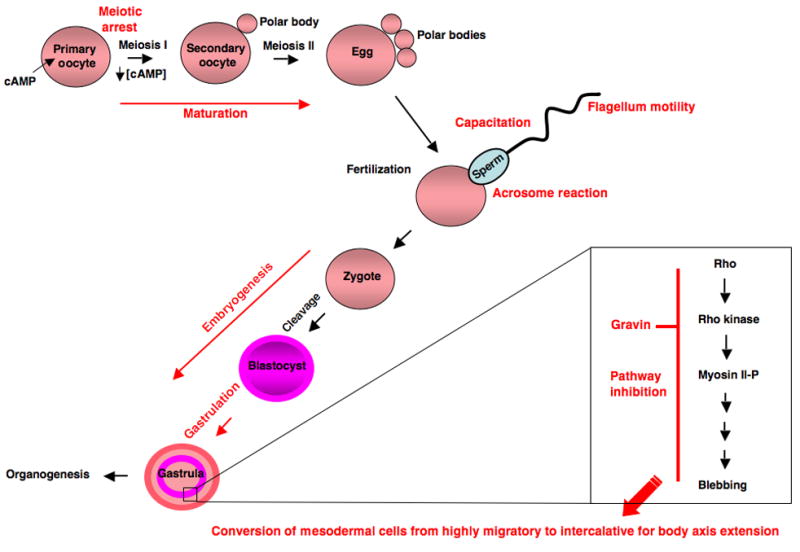
AKAPs are highly abundant in testes, where they play important roles in sperm function including regulation of motility, capacitation and the acrosome reaction. AKAPs also function in oogenesis where they function to regulate PKA localization maintaining initial meiotic arrest, followed by subsequent meiotic divisions to produce an egg ready for fertilization. Following fertilization, AKAPs play a role in embryogenesis. Specifically, gravin has been demonstrated to play a role in gastrulation regulating the movement of germ layer cells to form an embryonic body plan.
The Akap4 gene has been knocked out in mice. These (male) animals are infertile due to a lack of sperm motility. AKAP82 (AKAP4), also known as fibrous sheath component 1 is sperm specific and is the major fibrous sheath protein of the principle piece of sperm flagellum (Miki et al, 2002).
Oocyte function
Studies have implicated PKA in the regulation of oocyte maturation and several AKAPs have been identified in oocytes. PKA activity maintains the oocyte in meiotic arrest in response to cAMP generated by ovarian follicle cells that is transmitted to the oocyte through gap junctions. Meiosis is arrested at the first prophase, corresponding to the G2 phase of the cell cycle. Re-initiation of meiosis occurs following a decrease in cAMP in the oocyte, in response to the pituitary leutenizing hormone (LH) (Kovo et al, 2006). A change in PKA localization is also observed as the oocyte resumes meiosis (Rawe et al, 2004b). It is thought that AKAPs function in the initial maintenance of meiotic arrest and the subsequent irreversible progression to the polar body extruded stage by efficiently targeting PKA activity to specific sites and substrates in the oocyte (Dekel, 2005). The major AKAP expressed in mouse oocyte is 140 kDa in size and is a product of the Akap1 gene. This gene (encoding AKAP1 also known as AKAP140/149 and other splice variants AKAP121, sAKAP84 or D-AKAP-1) has recently been knocked out in mice (Newhall et al, 2006). Interestingly, while AKAP1 knockout males are fertile, the females exhibit infertility, or subfertility, suggesting a role in oocyte maturation. Further investigation demonstrates that PKA activity is disregulated in the knockout oocytes. Furthermore, these cells are more sensitive to cAMP-activated arrest because of a change in PKA localization. Results show that anchoring of RIIα-PKA on mitochondria through targeting by AKAP1 effectively prevents the holoenzyme from interfering with the maturation process once it is initiated. In addition, further studies using the Ht31 peptide in AKAP1 knockout oocytes suggest that PKA localization to an AKAP other than AKAP1 is essential at an early stage in order for cAMP to maintain the arrest of oocytes in meiosis I prophase. The identity of this AKAP is not known, however it is postulated that it may interact with the protein phosphatase cdc25 and/or the protein kinase wee1 to facilitate phosphorylation of these substrates, thereby regulating these enzymes to inhibit oocyte maturation in response to cAMP.
WAVE-1 is another AKAP demonstrated to play a role in oocytes. Specifically, immunofluorescence experiments show that WAVE-1 exhibits different cellular locations depending upon the phase of oocyte maturation. WAVE-1 redistributes from the cortex in germinal vesicle oocytes to the cytoplasm in oocytes arrested in meiosis II. Following fertilization, WAVE-1 relocalizes to developing male and female pronuclei. WAVE-1 then redistributes to the cytoplasm upon nuclear envelope breakdown at mitosis and concentrates at the cleavage furrow during embryonic cell division. Currently, although the localization of WAVE-1 in the process of oocyte maturation has been well documented it is unclear as to the role of the anchored signaling proteins in the WAVE-1 complex. Association of WAVE-1 with PKA is detected in both meiosis II stage oocytes and pronucleate zygotes, but interaction with Arp2/3 is observed only in meiosis II stage oocytes (Rawe et al, 2004a).
Gastrulation
Gravin was one of the first AKAPs to be identified as a multi-functional kinase scaffold protein. In addition to PKA, gravin also binds PKC (Nauert et al, 1997) and Src (Tao et al, 2007). Studies using cultured mammalian cells have implicated gravin in the regulation of cell morphology (Gelman et al, 1998). Gravin is thought to function as a tumor suppressor (Gelman, 2002). Orthologs of gravin (also known as SSeCKS in rat and mouse) have been identified in xenopus and zebrafish and studies with these organisms demonstrate a developmental role in embryo morphogenesis. Xenopus-gravin is zygotically expressed in a highly dynamic manner, beginning at the onset of gastrulation (stage 10). Gastrulation involves the coordinated movement of germ layers, resulting in the development of an embryonic body plan. Convergent extension of the mesoderm is a major process in this event (Klingbeil et al, 2001). Recent studies in zebrafish show that gravin is required for the conversion of mesodermal cells from a highly migratory behaviour to a medio-lateral intercalative behaviour required for body axis extension. In particular, gravin functions to inhibit the blebbing movement of the mesodermal cells through inhibition of a Rho/ROCK/myosin II pathway (Weiser et al, 2007). The precise mechanism of this inhibition relating to gravin remains unknown however. This observed role of gravin in embryogenesis has lead to speculation as to the tumor supressor function of this AKAP. It is conceivable that gravin may act to inhibit the blebbing movements observed in some tumors that may cause cell invasion and metastasis.
Learning and Memory (Channel Regulation)
One of the first physiological roles identified for AKAPs was in learning and memory.
AKAP79 (or the mouse ortholog AKAP150) can regulate synaptic plasticity through regulation of neuron channel response. This is accomplished by AKAP79 through scaffolding, targeting and regulation of the signaling molecules PKA, protein kinase C (PKC) and protein phosphatase 2B (PP2B, calcineurin) at the post-synaptic membrane (Dell’Acqua et al, 2006). Phosphorylation of specific channel subunits (including AMPA- and NMDA-glutamate receptors (Lee et al, 1998; Roche et al, 1996), the inwardly rectifying potassium channel Kir2.1, (Dart & Leyland, 2001) the β-adrenergic receptor (Tao et al, 2003), mGluR1/5 metabotropic glutamate receptor (Francesconi & Duvoisin, 2000) and GABA receptors (Moss et al, 1992) at inhibitory synapses) may modulate synaptic efficiency either directly or indirectly by regulating receptor surface expression, (which has been shown for AMPA receptors) (Colledge et al, 2003; Gardner et al, 2006). Recently there have been a couple of studies published characterizing the knockout mouse ortholog of AKAP79, called AKAP150 (Navedo et al, 2008) (Tunquist et al, 2008). A knock-in mouse has been generated where AKAP150 has been replaced by a mutant form of this scaffold protein that is unable to bind PKA. Expression of PKA is highly reduced within postsynaptic densities (PSDs) of neurons cultured from these mice indicating that AKAP150 is the predominant AKAP responsible for targeting of PKA to the PSD in neurons. Further studies suggest that AKAP150-targeted PKA contributes to LTP in the adult hippocampus (Hall et al, 2007). In addition, AKAP150 null mice also exhibit deficits in motor coordination and strength, consistent with a role for AKAP150 in the cerebellum (Tunquist et al, 2008). Interestingly, AKAP79/150 can also tether adenylyl cyclase V/VI, thereby forming a protein configuration to regulate cAMP production. The anchoring of both an adenylyl cyclase (to generate cAMP) followed by the subsequent activation of PKA (which can inhibit the cyclase) leads to a negative feedback loop which may be important in generating specific pools of localized cAMP (Bauman et al, 2006). The physiological significance of this is currently unknown, but may be important in the context of characterizing the knockout mouse.
WAVE-1 (Wiskott-Aldrich syndrome, verprolin-homology domain containing protein) is another AKAP with important neuronal functions. Expression of this scaffolding protein is restricted to the central nervous system where it functions to organize protein networks involved in the regulation of the cytoskeleton (Soderling et al, 2003). In particular this AKAP belongs to a family of proteins that link Rho GTPases to actin assembly (Takenawa & Suetsugu, 2007). Interestingly, WAVE-1 is likely to exist in many different combinatorial complexes relating to its spatiotemporal function. For example, the RII-binding region of this protein overlaps with the actin-binding domain and in vitro binding experiments demonstrate that actin competes for the RII-binding site (Westphal et al, 2000). Synaptic remodeling, through actin polymerization, is believed to play a role in synaptic plasticity, learning, and memory. Insight into the role of WAVE-1 in this process comes from the characterization of WAVE-1 knockout mice which display defects in hippocampal learning and memory (Soderling et al, 2003). WAVE-1 knockout mice displayed defects in two tests (novel open arena and elevated zero-maze) used to determine anxiety levels associated with neural networks in the amygdala. Additionally, these knockout mice also displayed poor performance in tests for balance and coordination (rotarod, inclined screen and balance beam tests). The precise molecular mechanisms governing the observed phenotype of this mouse are not known. Interestingly, mutations in WRP (MEGAP/srGAP3) (a Rho GTPase) have been linked to 3p-syndrome mental retardation in humans, which also results in impaired learning and memory, poor balance, and reduced coordination (Endris et al, 2002). Further studies carried out in mice generated to express a form of WAVE-1 without the WRP binding sites indicate that the anchored pool of WRP associated with WAVE-1 plays a role in the control of cytoskeletal events that underlie normal neuronal development and synaptic plasticity (Soderling et al, 2007). Thus, WAVE1 localization of WRP is important for regulation of the actin cytoskeleton relating to processes of learning and memory. Currently, it is unclear if ablation of the WAVE-1 complex may also affect actin bundle formation through disruption of structural proteins involved in this process, such as the Arp2/3 complex and/or, if anchored PKA and/or other anchored signaling molecules such as Abl (a tyrosine kinase) may play a role in the aetiology of this phenotype. It is believed that dendritic morphology may be regulated by direct phosphorylation of WAVE-1 itself (Kim et al, 2006).
Cardiac Functions
To date, perhaps the most evidence for the physiological roles of a number of different AKAPs lies in the study of cardiac function (see summary figure 4). PKA phosphorylates numerous substrates that may influence contractility in cardiac myocytes, including the L-type Ca2+ channel, the ryanodine receptor, phospholamban and troponin I (Olson, 2004). RIα is likely to play a role in cardiac physiology because it is the predominant isoform associated with the sarcolemma in cardiac myocytes (Robinson et al, 1996). Mutations in the RIa gene are associated with both familial cardiac myxomas and Carney complex, implicating this isoform in cardiac growth and differentiation (Wilkes et al, 2006). However, in addition to RI, many AKAPs targeting type II PKA have been characterized in the heart. AKAPs play a critical role in modulating phosphorylation of numerous PKA dependent substrates that regulate cardiac function. Thus, altered expression of AKAP’s would seem likely to result in drastic consequences. In particular, mAKAP (Dodge-Kafka et al, 2005), yotiao (Kurokawa et al, 2004), AKAP18 (Lygren et al, 2007), AKAP-Lbc (Appert-Collin et al, 2007), synemin (Russell et al, 2006), and myospryn (Reynolds et al, 2007) are all reported to have physiological functions in the heart. It should also be noted that the dual specific AKAP2 (D-AKAP2) (which targets both type I and type II PKA) plays an important role in the heart (Tingley et al, 2007) and will be discussed later (see Genetic Studies).
Figure 4. Cardiac AKAPs.
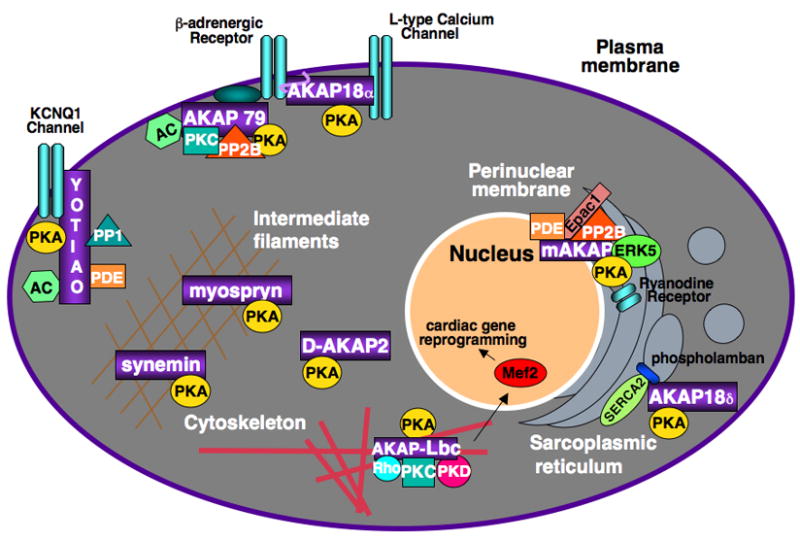
AKAPs play a prominent role in the regulation of cardiac function.
To date AKAP18, AKAP79 and yotiao fuction in channel regulation, while myospryn and synemin act to target PKA to intermediate filaments. mAKAP and AKAP-Lbc appear to play a role in the disease process of cardiac hypertrophy.
The muscle specific A kinase anchoring protein (mAKAP) is highly expressed in cardiac tissue and localized to the perinuclear membrane and junctional sarcoplasmic reticulum where it is in close proximity to a variety of substrates such as L-type Ca2+ channels and the ryanodine receptor (RyR) (Kapiloff et al, 2001). By serving as a scaffold for a range of molecules such as PKA, PDE4D3, Epac1 and ERK5, mAKAP is able to assemble a highly structured network in which signals can be regulated and focused upon nearby substrates (Dodge et al, 2001). It is well known that PKA phosphorylation of the RyR receptor results in opening of this receptor and increased cardiac function and contractility and this is in large part due to the mAKAP-mediated localization of PKA to the RyR (Marx et al, 2001). Tight regulation of cardiac contractility is critical for efficient function of the heart. mAKAP plays a role in this by scaffolding phosphodiesterases which are able to rapidly hydrolyze localized cAMP and restore PKA to an inactive state. In addition to a role in mediating cardiac contractility, mAKAP is also implicated in cardiac hypertrophy. Two components of the mAKAP complex that are involved in the hypertrophic response are the mitogen activated kinase, ERK5 (Dodge-Kafka et al, 2005), and the phosphatase PP2B (calcineurin) (Pare et al, 2005). Activation of ERK5 results in cardiac hypertrophy although ERK5 is readily dephosphorylated and inactivated by PP2B, thus once again demonstrating another feedback mechanism whereby mAKAP tightly regulates cardiac function.
AKAP15/18 is expressed in the heart and localizes to the plasma membrane (shorter isoforms α and β) due to an N-terminal lipid-anchoring domain (Fraser et al, 1998) (Gray et al, 1998).
Recently the long splice variant of the AKAP15/18 gene; AKAP15/18δ, (which displays a cytoplasmic localization) was found to target PKA to phospholamban, which is a critical regulator of the sarcoplasmic reticulum Ca2+-ATPase (SERCA) (Lygren et al, 2007). The interaction of AKAP15/18δ with phospholamban is necessary for PKA-mediated phosphorylation of phospholamban at Ser16, leading to the effects of adrenergic stimulation on calcium reuptake. Decreased SERCA expression/activity and phospholamban hypo-phosphorylation are intricately involved in heart failure, thus AKAP15/18δ plays a crucial role in maintaining cardiac function. Previous studies have demonstrated that expression of a pseudo-phosphorylated phospholamban (Hoshijima et al, 2002) or enhanced expression/activity of SERCA (Miyamoto et al, 2000) is sufficient not only to halt the progression of cardiomyopathy but can also restore cardiac function. Since AKAP15/18δ mediates phospholamban phosporylation and a subsequent increase in SERCA activity, it is intriguing to postulate that modulation of AKAP15/18δ may represent a novel pharmacologic target for restoring heart function.
Fractionation experiments suggest that this protein may serve as a scaffold for the β2-adrenergic receptor (Sacchetto et al, 2001) and the L-type Ca2+ channel (Gray et al, 1997). AKAP15/18 expression potentiates cAMP responsive Ca2+ currents while expression of the Ht31 peptide to disrupt PKA-AKAP interactions results in inhibition of PKA induced potentiation of the L-type Ca2+ channel (Hulme et al, 2003).
In addition to AKAP15/18, AKAP79 also plays a role in the regulation of Ca2+, specifically by targeting PKCα to the L-type Ca2+ channel in arterial myocytes. Recent studies demonstrate that AKAP150 knockout mice were found to lack persistant Ca2+ sparklets and have lower arterial wall intracellular calcium and decreased myogenic tone. These knockout mice were hypotensive and did not develop angiotensin II-induced hypertension (Navedo et al, 2008).
AKAP-Lbc serves as a scaffold for PKA, PKC, PKD (Carnegie et al, 2004), and also contains a DH (Dbl-homology)-PH (pleckstrin-homology) domain that acts as a guanine nucleotide exchange factor (GEF) for the low molecular weight GTPase Rho (Diviani & Scott, 2001) which is a known mediator of cardiac hypertrophy. As with many signaling pathways regulated by AKAP scaffolding, the Rho GEF activity of AKAP-Lbc is also tightly regulated. The GEF activity of AKAP-Lbc is activated via the G12/13 family of heterotrimeric G-proteins, while it is inactivated by phosphorylation from the AKAP-Lbc bound PKA. It appears that phosphorylation of AKAP-Lbc by bound PKA results in oligomerization and recruitment of 14-3-3 proteins which prevent AKAP-Lbc from being able to activate Rho (Diviani et al, 2004) (Jin et al, 2004). In addition to its involvement in hypertrophy via the Dbl domain, AKAP-Lbc also serves to activate PKD through recruitment of its upstream activator, PKC (Carnegie et al., 2004). PKD has received much attention recently as a result of its ability to promote the nuclear export of certain class II histone deacetylases (HDAC’s) thereby driving MEF2-dependent transcription of hypertrophic genes (Vega et al, 2004). Therefore AKAP-Lbc plays central role in coordinating multiple pathways involved with the disease process of cardiac hypertrophy (Carnegie et al, 2008). Synemin is a rather unique AKAP in that it is the first intermediate filament protein shown to bind RII and localize PKA. Synemin colocalizes in the heart at the Z line with RII and desmin. By localizing PKA near intermediate filaments, synemin may enhance PKA phosphorylation of substrates at the Z line such as desmin, vimentin, tubulin, and αB-crystallin. This anchoring protein may also serve to localize PKA near myofibril filaments for potential phosphorylation of substrates such as troponin I or myosin binding protein C. Localization of synemin at the Z line and the plasma membrane would make it ideally suited to be involved in cardiac remodeling following cardiac hypertrophy or even cardiomyopathies, as altered phosphorylation of PKA substrates is seen in these conditions (Russell et al, 2006).
In the mouse model of Duchenne Muscular Dystrophy (mdx) where dystrophin is knocked out, the AKAP myospryn (an alpha-actinin-interacting, costamere-localized protein expressed in striated muscle) and its anchored PKA is mislocalized in these mice. It is believed that this is because myospryn is unable to interact with dystrophin (Reynolds et al, 2008). Thus PKA activity in the muscle may not be properly targeted and regulated in muscle tissue of patients exhibiting DMD. Furthermore, expression of mAKAP, RyR, and SERCA2A is decreased in these mice, resulting in disrupted Ca2+ homeostasis and cardiac hypertrophy. Prolonged cardiac hypertrophy can transition to cardiac failure in which there is downregulation of β-adrenergic receptors, decreased phosphorylation of RII, and as a result, there may be a shift in the localization of RII due to the different affinities of AKAP for phosphorylated and unphosphorylated RII (Zakhary et al, 2000).
Yotiao serves as a scaffold in the heart for localizing PP1, PDE4D3, and RII to the IKS channel subunit hKCNQ1 by means of a C-terminal leucine zipper motif on hKCNQ1 (Marx et al, 2002). Mutations in this leucine zipper motif have been found in individuals with long QT syndrome and in particular the S1570L mutation has been found in 2% (1/50) individuals with long QT syndrome while being absent in over 1300 individuals not exhibiting this syndrome. Computational analysis of this mutation reveals that it reduces cAMP dependent phosphorylation of the IKS channel resulting in a prolonged action potential (Chen et al, 2007). (See Genetic Studies section for more detail).
Genetic Studies
In the age of functional genomics mapping disease loci has become a very important method to identify potential proteins that may play a role in pathology, or define the aetiology of genetic disorders. In this regard there is now significant genetic information coupled with suitable technology to identify single nucleotide polymorphisms (SNPs) in the DNA of individuals that may result in the coding of a mutant form of protein. A single nucleotide base change in a region of DNA coding for a protein may lead to the mutation of a single amino acid, or a completely novel protein sequence, or a prematurely truncated protein. All of these outcomes may have serious effects upon cell signaling pathways.
A mutation in the AKAP Yotiao has been identified in patients with familial long-QT syndrome (LQTS) (Chen et al, 2007). Long QT syndrome (LQTS) is a congenital disorder characterized by a prolongation of the QT interval on ECG and a propensity to ventricular tachyarrhythmias, which may lead to syncope, cardiac arrest, or sudden death. (The QT interval represents the duration of activation and recovery of the ventricular myocardium). LQTS is caused by mutations in the genes coding for cardiac potassium and sodium or calcium ion channels, such as KCNQ1, LQT1, KCNE1 and LQT5 (Abbott & Goldstein, 2002). Yotiao forms a macromolecular complex with the slowly activating cardiac potassium channel IKs, which is critical for repolarization of the ventricular action potential in the heart (Marx et al, 2002). The S1570L form of yotiao disrupts this interaction and reduces the cAMP-induced phosphorylation of the channel. This eliminates the functional response of the I(Ks) channel to cAMP, thereby promoting a prolonged ventricular action potential in individuals inheriting this mutation (Chen et al, 2007).
A deleterious SNP that results in an amino acid change from Ile to Val in the dual-specific AKAP2 (D-AKAP2) gene was identified in an aging population of European-America individuals that may be predisposed to cardiac dysfunction (Kammerer et al, 2003). Further evidence for an important cardiac function of D-AKAP2 comes from a more recent study of cardiac myocytes that were derived from mouse embryonic stem (ES) cells in which the D-AKAP2 gene was disrupted, thereby affecting the last 51 amino acids of D-AKAP2 (ablating the PKA binding site and a C-terminal PDZ domain). An increased cardiac response to cholinergic signals was observed and mutant mice exhibited cardiac arrythmias and died prematurely (Tingley et al, 2007). To date, specific PKA signaling pathways associated with D-AKAP are not known. However, the Val D-AKAP2 variant binds to the RIα isoform of the PKA holoenzyme with a three-fold stronger affinity than the wild-type Ile D-AKAP2, thereby affecting compartmentalization of type I PKA. One function of D-AKAP2 is to target PKA to the mitochondria, thus an increase in the ability of the Val D-AKAP2 variant to anchor PKA at the mitochondria has been observed (Kammerer et al, 2003). Importantly, these studies suggest that factors affecting the compartmentalization of type I PKA may have significant pathological effects, possibly by affecting the phosphorylation state of PKA substrates.
Interestingly, this same IleVal SNP was also identified to be associated with familial breast cancer in an additional independent study (Wirtenberger et al, 2007). A SNP causing a Lys526Gln mutation in AKAP-Lbc was also identified in this study (Wirtenberger et al, 2007). This mutation is not in the PKA anchoring domain, or any other recognized domain of this protein and is of unknown function. AKAP-Lbc was also recently identified in a pharmacogenomic study designed to highlight gene expression markers predictive for a favorable response in acute myelogenous leukemia (AML) patients treated with the farnesyltransferase inhibitor tipifarnib (Zarnestra, R115777). Raponi et al. used microarray technology to monitor gene expression from bone marrow samples of relapsed and refractory AML patients undergoing treatment with tipifarnib. Several marker genes were identified that may predict a positive response to tipifarnib treatment, including AKAP-Lbc. Thus, when AKAP-Lbc is under-expressed, farneslytransferase inhibitors may exhibit greater antitumorigenic effects in AML patients. It was also demonstrated in this study that overexpression of proto and onco-Lbc increases resistance to tipifarnib, but not doxorubicin (a non-farneslytransferase inhibitor chemotherapeutic). This indicates that AKAP-Lbc plays a role in the progression of this disease, where increased AKAP-Lbc activity may lead to an increased cellular profile of transformation through a Rho-mediated pathway. The authors speculate that AKAP-Lbc may be a downstream target of the farneslytransferase inhibitor, however to date there is no primary evidence to date to support this.
Recently, mutations identified in the gene encoding the AKAP pericentrin (PCNT) were demonstrated to cause Seckel syndrome (Rauch et al, 2008). Seckel syndrome is a disorder associated with defective ATR-dependent DNA damage signaling, resulting in a marked reduction of brain and body size. While the mechanism underlying this disorder is not fully understood, the authors demonstrated a loss of expression of pericentrin. This was due to a homozygous single-base pair deletion in exon 12 leading to a frameshift, which was predicted to result in premature protein truncation after an additional 65 amino acids. An additional SNP was also characterized; an insertion in exon 18 resulting in a frameshift at codon 1190. Collectively, these mutations disrupt all mammalian isoforms of PCNT. Pericentrin (also known as kendrin) is a 360 kDa coiled-coil protein with a C-terminal PACT domain that targets it to the centrosome. This scaffolding protein localizes to the pericentriolar matrix, where it interacts with several structural centrosomal proteins including γ-tubulin as well as PKA and PKCβII. Pericentrin plays an important role in microtubule nucleation and spindle organization. Presumably, the loss of pericentrin from the centrosome results in the loss of regulated protein phosphorylation through PKA and PKC, functioning downstream of the ATR kinase in the DNA damage response pathway, including G2-M checkpoint activation.
Cancer
Pericentrin functions as an essential centrosomal protein that is necessary for directing centrosomal assembly (through interaction with dynein (Purohit et al, 1999)) and microtubule nucleation (through interaction with γ-tubulin (Dictenberg et al, 1998)). This protein plays a critical role in cell division, therefore disruption of this gene can have drastic consequences and irregularities in this AKAP are often seen in tumors in which there are defects in regulated cell division. Cells overproducing pericentrin develop extra centrosomes and spindle defects, display genomic instability and enhanced growth in soft agar. Pericentrin, is able to bind RII through a domain unlike the conventional domain found in other AKAPs (Diviani et al, 2000). Disruption of PKA binding to pericentrin results in similar spindle abnormalities (Pihan et al, 1998). Finally as dynein is a substrate for PKA, and inhibition of dynein function results in spindle abnormalities, it is possible that pericentrin anchored PKA regulates dynein function.
The AKAP gravin is localized in the cytosol as well as at the plasma membrane, where it is associated with β-adrenergic receptors and serves as a scaffold for PKA, PKC (Shih et al, 1999). The AKAP domain of gravin appears to be required for association at the plasma membrane with β-adrenergic receptors and this association is involved in resensitization of the receptor (Lin et al, 2000a). Gravin has recently been recognized as a tumor suppressor due to its ability to induce cell cycle arrest (via decreases in cyclin D1) and even apoptosis (via caspase 3 activation) in numerous cell types (Lin et al, 2000b; Yoon et al, 2007). Gravin is down-regulated in a number of tumor types including prostate, ovarian, and breast cancer and is associated with metastatic progression of these tumors, thereby providing further evidence supporting the role of gravin as a tumor suppressor (Gelman, 2002). It is thought that the hypermethylation of the gravin gene and its promoter may account for the down-regulation of gravin seen in tumor cells (Jin et al, 2008).
AKAP95 is localized to the nucleus and serves to recruit PKA to the nucleus during mitosis where it plays an integral role in DNA replication (Coghlan et al, 1994). Upon initiation of mitosis, RII is phosphorylated by the CDK1-cyclin-B complex which drives it to the nucleus to associate with AKAP95 (Collas et al, 1999) (Arsenijevic et al, 2004). Once in the nucleus, RII bound AKAP95 plays a critical role in initiation and elongation of DNA by providing a scaffold for p68 RNA helicase and the minichromosome maintenance protein 2, and is furthermore involved in chromatin condensation by recruitment of the condensing complex (Eide et al, 2002). In addition, AKAP95 binds caspase-3 where it mediates apoptotic nuclear morphological changes (Kamada et al, 2005). At the termination of mitosis, RII is dephosphorylated which causes it to dissociate from nuclear AKAP95 and translocate back to the centrosome-golgi area where it resides until the next mitotic cycle. The release of RII from AKAP95 releases inhibition of chromatin condensation and allows for chromosome decondensation as the nuclear envelope reforms. This phosphorylation dependent localization of RII to AKAP95 serves as a molecular switch for ensuring the proper timing of cell division (Landsverk et al, 2001).
A truncated form of AKAP-Lbc (AKAP13) missing both N- and C-terminal regulatory sequences, called Onco-Lbc (amino acids 1923-2347), was originally identified as an oncogene from myeloid leukemia patients (Toksoz & Williams, 1994). This oncogenic protein displays constitutive Rho-GEF activity and is able to induce cell transformation in a Rho-dependent manner. Another splice variant of the Lbc gene, called Brx (amino acids 1384-2817) has been identified that is specifically expressed in testis and estrogen-sensitive tissues (Rubino et al, 1998). Only the full-length version of AKAP-Lbc contains the PKA anchoring domain (amino acids 1236–1257).
Diabetes
Impaired insulin secretion is a characteristic of non-insulin-dependent diabetes mellitus (NIDDM). Insulin secretion can be stimulated by the hormone glucagon-like peptide 1 (GLP-1). One mechanism of GLP-1 action is through activation of PKA. A role for AKAPs in the regulation of hormone (GLP-1)-mediated insulin secretion was first identified in studies using the PKA-AKAP anchoring inhibitor peptide Ht31 (Lester et al, 1997). Results demonstrate that insulin secretion can be regulated by the reversible phosphorylation of β-cell proteins through the AKAP79 targeted effects of PKA and PP2B. However, the exact mechanism of this regulation is still not understood. It is believed that somehow AKAP79 facilitates a feedback loop whereby PKA activity promotes activation of PP2B. The reciprocal function of these enzymes in turn regulates the phosphorylation of substrates involved in insulin secretion pathways, for example ion channels and proteins involved in endocytosis, such as synapsin (Lester et al, 2001). Clearly more work is required in this area if specific cellular mechanisms are to be elucidated. This is one area where trangenic models would be very useful to facilitate in vivo studies.
Exocytosis
More recently the Rab27a GTPase effector protein MyRIP has been identified as an AKAP through a yeast two-hybrid screen for RII interacting proteins (Goehring et al, 2007). MyRIP interacts with components of the exocyst complex which is involved in protein trafficking and exocytosis. Specifically, the exocyst complex may function to facilitate the expression of the glucose transporter Glut4 at the plasma membrane (and therefore cellular glucose uptake) in response to insulin. MyRIP may play a role in this process by directing PKA to the exocyst complex.
Melanosome transport
Most recently it has been demonstrated that the AKAP Rab32 is associated with melanosomes and links PKA to these organelles (Park et al, 2007) as well as mitochondria (Alto et al, 2002). Melanosomes are found in melanocytes where the pigment melanin is synthesised and stored. Melanocytes are present in the skin and eyes of mammals whose primary function relates to photoprotection. Therefore exposure to potentially damaging UV radiation results in an increase in melatonin production. Melanosome transport is regulated by PKA and in particular PKA activation appears to stimulate melanosome aggregation in response to melanin production. Currently however, the exact molecular mechanisms in this process are unknown. Interestingly Rab32 and another highly homologous small GTPase Rab38 (67% sequence identity) can functionally compensate for each other in the regulation of pigment biogenesis. A recent study showed that Rab32/38 regulate a critical step in the trafficking of melanogenic enzymes, in particular tyrosinase from the trans golgi network to melanosomes (Wasmeier et al, 2006). The authors do not comment on whether PKA activity is required in this process or whether Rab38 also interacts with PKA. Given the sequence similarity between Rab38 and the mapped PKA anchoring region (amino acids 178–192) on Rab32 it seems likely that PKA will interact with Rab38, performing an important function in this pathway.
In summary, we hope this review provides the reader with an insight into some of the physiological/pathophysiological processes facilitated by this interesting and well characterized family of scaffold proteins. With the combination of large functional genomic screens and the study of tissue or whole animal models it is likely that we will gain further understanding into the physiological roles of these scaffold proteins. Hopefully these studies may lead to improved aspects of human health through the integration of protein function with precise molecular mechanisms of action.
Acknowledgments
The authors wish to acknowledge support from the Fondation Leducq and the NIH.
References
- Abbott GW, Goldstein SA. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 2002;16(3):390–400. doi: 10.1096/fj.01-0520hyp. [DOI] [PubMed] [Google Scholar]
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto NM, Soderling J, Scott JD. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J Cell Biol. 2002;158(4):659–668. doi: 10.1083/jcb.200204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appert-Collin A, Cotecchia S, Nenniger-Tosato M, Pedrazzini T, Diviani D. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007;104(24):10140–10145. doi: 10.1073/pnas.0701099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic T, Degraef C, Dumont JE, Roger PP, Pirson I. A novel partner for D-type cyclins: protein kinase A-anchoring protein AKAP95. Biochem J. 2004;378(Pt 2):673–679. doi: 10.1042/BJ20031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai M, Fiedler SE, Huang Z, Vijayaraghavan S, Olson GE, Livera G, Conti M, Carr DW. AKAP3 Selectively Binds PDE4A Isoforms in Bovine Spermatozoa. Biol Reprod. 2006;74(1):109–118. doi: 10.1095/biolreprod.105.043588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD. Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology. 2004;46(3):299–310. doi: 10.1016/j.neuropharm.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23(6):925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol Cell. 2004;15:889–899. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol Cell. 2008;32(2):169–179. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem. 1992;267(19):13376–13382. [PubMed] [Google Scholar]
- Carr DW, Newell AE. The role of A-kinase anchoring proteins (AKaps) in regulating sperm function. Soc Reprod Fertil Suppl. 2007;63:135–141. [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser IDC, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104(52):20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Langeberg LK, Fernandez A, Lamb NJC, Scott JD. Cloning and characterization of AKAP95, a nuclear protein that associates with the regulatory subunit of type II cAMP-dependent protein kinase. J Biol Chem. 1994;269:7658–7665. [PubMed] [Google Scholar]
- Cohen P. The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol. 1999;3(4):459–465. doi: 10.1016/S1367-5931(99)80067-2. [DOI] [PubMed] [Google Scholar]
- Collas P, Le Guellec K, Tasken K. The A-kinase-anchoring protein AKAP95 is a multivalent protein with a key role in chromatin condensation at mitosis. J Cell Biol. 1999;147(6):1167–1180. doi: 10.1083/jcb.147.6.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9(6):216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40(3):595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin JD, Soderling TR, Park CR. Regulation of adenosine 3′,5′-monophosphate-dependent protein kinase. I. Preliminary characterization of the adipose tissue enzyme in crude extracts. J Biol Chem. 1973;248(5):1813–1821. [PubMed] [Google Scholar]
- Dart C, Leyland ML. Targeting of an A kinase-anchoring protein, AKAP79, to an inwardly rectifying potassium channel, Kir2.1. J Biol Chem. 2001;276(23):20499–20505. doi: 10.1074/jbc.M101425200. [DOI] [PubMed] [Google Scholar]
- Dekel N. Cellular, biochemical and molecular mechanisms regulating oocyte maturation. Mol Cell Endocrinol. 2005;234(1–2):19–25. doi: 10.1016/j.mce.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Dell’Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006;85(7):627–633. doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Zimmerman W, Sparks CA, Young A, Vidair C, Zheng Y, Carrington W, Fay FS, Doxsey SJ. Pericentrin and gamma-tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol. 1998;141(1):163–174. doi: 10.1083/jcb.141.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D, Abuin L, Cotecchia S, Pansier L. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004;23(14):2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D, Langeberg LK, Doxsey SJ, Scott JD. Pericentrin anchors protein kinase A at the centrosome through a newly identified RII-binding domain. Curr Biol. 2000;10(7):417–420. doi: 10.1016/s0960-9822(00)00422-x. [DOI] [PubMed] [Google Scholar]
- Diviani D, Scott JD. AKAP signaling complexes at the cytoskeleton. J Cell Sci. 2001;114(Pt 8):1431–1437. doi: 10.1242/jcs.114.8.1431. [DOI] [PubMed] [Google Scholar]
- Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20(8):1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437(7058):574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide T, Carlson C, Tasken KA, Hirano T, Tasken K, Collas P. Distinct but overlapping domains of AKAP95 are implicated in chromosome condensation and condensin targeting. EMBO Rep. 2002;3(5):426–432. doi: 10.1093/embo-reports/kvf089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endris V, Wogatzky B, Leimer U, Bartsch D, Zatyka M, Latif F, Maher ER, Tariverdian G, Kirsch S, Karch D, Rappold GA. The novel Rho-GTPase activating gene MEGAP/srGAP3 has a putative role in severe mental retardation. Proc Natl Acad Sci U S A. 2002;99(18):11754–11759. doi: 10.1073/pnas.162241099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler SE, Bajpai M, Carr DW. Identification and characterization of RHOA-interacting proteins in bovine spermatozoa. Biol Reprod. 2008;78(1):184–192. doi: 10.1095/biolreprod.107.062943. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM. Opposing effects of protein kinase C and protein kinase A on metabotropic glutamate receptor signaling: selective desensitization of the inositol trisphosphate/Ca2+ pathway by phosphorylation of the receptor-G protein-coupling domain. Proc Natl Acad Sci U S A. 2000;97(11):6185–6190. doi: 10.1073/pnas.97.11.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP- responsive membrane events. Embo J. 1998;17(8):2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the beta1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem. 2006;281(44):33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- Gelman IH. The role of SSeCKS/gravin/AKAP12 scaffolding proteins in the spaciotemporal control of signaling pathways in oncogenesis and development. Front Biosci. 2002;7:d1782–1797. doi: 10.2741/A879. [DOI] [PubMed] [Google Scholar]
- Gelman IH, Lee K, Tombler E, Gordon R, Lin X. Control of cytoskeletal architecture by the src-suppressed C kinase substrate, SSeCKS. Cell Motil Cytoskeleton. 1998;41(1):1–17. doi: 10.1002/(SICI)1097-0169(1998)41:1<1::AID-CM1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Goehring AS, Pedroja BS, Hinke SA, Langeberg LK, Scott JD. MyRIP anchors protein kinase A to the exocyst complex. J Biol Chem. 2007;282(45):33155–33167. doi: 10.1074/jbc.M705167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24(3):383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- Gray PC, Tibbs VC, Catterall WA, Murphy BJ. Identification of a 15-kDa cAMP-dependent protein kinase-anchoring protein associated with skeletal muscle L-type calcium channels. J Biol Chem. 1997;272:6297–6302. doi: 10.1074/jbc.272.10.6297. [DOI] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46(6):1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, Iwatate M, Li M, Wang L, Wilson JM, Wang Y, Ross J, Jr, Chien KR. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med. 2002;8(8):864–871. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci USA. 1997a;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel dual specificity protein kinase A anchoring protein, D-AKAP1. J Biol Chem. 1997b;272(12):8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100(22):13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarnaess E, Tasken K. Spatiotemporal control of cAMP signalling processes by anchored signalling complexes. Biochem Soc Trans. 2007;35(Pt 5):931–937. doi: 10.1042/BST0350931. [DOI] [PubMed] [Google Scholar]
- Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O’Donnell P, Taylor P, Taylor L, Zougman A, Woodgett JR, Langeberg LK, Scott JD, Pawson T. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr Biol. 2004;14(16):1436–1450. doi: 10.1016/j.cub.2004.07.051. [DOI] [PubMed] [Google Scholar]
- Jin Z, Hamilton JP, Yang J, Mori Y, Olaru A, Sato F, Ito T, Kan T, Cheng Y, Paun B, David S, Beer DG, Agarwal R, Abraham JM, Meltzer SJ. Hypermethylation of the AKAP12 promoter is a biomarker of Barrett’s-associated esophageal neoplastic progression. Cancer Epidemiol Biomarkers Prev. 2008;17(1):111–117. doi: 10.1158/1055-9965.EPI-07-0407. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels in skeletal muscle cells requires anchored cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1994;91:11492–11496. doi: 10.1073/pnas.91.24.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada S, Kikkawa U, Tsujimoto Y, Hunter T. A-kinase-anchoring protein 95 functions as a potential carrier for the nuclear translocation of active caspase 3 through an enzyme-substrate-like association. Mol Cell Biol. 2005;25(21):9469–9477. doi: 10.1128/MCB.25.21.9469-9477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer S, Burns-Hamuro LL, Ma Y, Hamon SC, Canaves JM, Shi MM, Nelson MR, Sing CF, Cantor CR, Taylor SS, Braun A. Amino acid variant in the kinase binding domain of dual-specific A kinase-anchoring protein 2: a disease susceptibility polymorphism. Proc Natl Acad Sci U S A. 2003;100(7):4066–4071. doi: 10.1073/pnas.2628028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapiloff MS, Jackson N, Airhart N. mAKAP and the ryanodine receptor are part of a multi-component signaling complex on the cardiomyocyte nuclear envelope. J Cell Sci. 2001;114(Pt 17):3167–3176. doi: 10.1242/jcs.114.17.3167. [DOI] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130(6):1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307(5710):690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442(7104):814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- Klingbeil P, Frazzetto G, Bouwmeester T. Xgravin-like (Xgl), a novel putative a-kinase anchoring protein (AKAP) expressed during embryonic development in Xenopus. Mech Dev. 2001;100(2):323–326. doi: 10.1016/s0925-4773(00)00527-x. [DOI] [PubMed] [Google Scholar]
- Kovo M, Kandli-Cohen M, Ben-Haim M, Galiani D, Carr DW, Dekel N. An active protein kinase A (PKA) is involved in meiotic arrest of rat growing oocytes. Reproduction. 2006;132(1):33–43. doi: 10.1530/rep.1.00824. [DOI] [PubMed] [Google Scholar]
- Krebs EG. The phosphorylation of proteins: a major mechanism for biological regulation. Biochemical Society Transactions. 1985;13:813–820. doi: 10.1042/bst0130813. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Motoike HK, Rao J, Kass RS. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci U S A. 2004;101(46):16374–16378. doi: 10.1073/pnas.0405583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsverk HB, Carlson CR, Steen RL, Vossebein L, Herberg FW, Tasken K, Collas P. Regulation of anchoring of the RIIalpha regulatory subunit of PKA to AKAP95 by threonine phosphorylation of RIIalpha: implications for chromosome dynamics at mitosis. J Cell Sci. 2001;114(Pt 18):3255–3264. doi: 10.1242/jcs.114.18.3255. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lester LB, Faux MC, Nauert JB, Scott JD. Targeted protein kinase A and PP-2B regulate insulin secretion through reversible phosphorylation. Endocrinology. 2001;142(3):1218–1227. doi: 10.1210/endo.142.3.8023. [DOI] [PubMed] [Google Scholar]
- Lester LB, Langeberg LK, Scott JD. Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc Natl Acad Sci USA. 1997;94:14942–14947. doi: 10.1073/pnas.94.26.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Wang H, Malbon CC. Gravin-mediated formation of signaling complexes in beta 2-adrenergic receptor desensitization and resensitization. J Biol Chem. 2000a;275(25):19025–19034. doi: 10.1074/jbc.275.25.19025. [DOI] [PubMed] [Google Scholar]
- Lin X, Nelson P, Gelman IH. SSeCKS, a major protein kinase C substrate with tumor suppressor activity, regulates G(1)-->S progression by controlling the expression and cellular compartmentalization of cyclin D. Mol Cell Biol. 2000b;20(19):7259–7272. doi: 10.1128/mcb.20.19.7259-7272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Lu YS, Shuai Y, Feng C, Tully T, Xie Z, Zhong Y, Zhou HM. The AKAP Yu is required for olfactory long-term memory formation in Drosophila. Proc Natl Acad Sci U S A. 2007;104(34):13792–13797. doi: 10.1073/pnas.0700439104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, Lorenz D, Wiesner B, Rosenthal W, Zaccolo M, Tasken K, Klussmann E. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007;8(11):1061–1067. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153(4):699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight GS, Cummings DE, Amieux PS, Sikorski MA, Brandon EP, Planas JV, Motamed K, Idzerda RL. Cyclic AMP, PKA, and the physiological regulation of adiposity. Recent Prog Horm Res. 1998;53:139–159. discussion 160–131. [PubMed] [Google Scholar]
- Miki K, Willis WD, Brown PR, Goulding EH, Fulcher KD, Eddy EM. Targeted disruption of the Akap4 gene causes defects in sperm flagellum and motility. Dev Biol. 2002;248(2):331–342. doi: 10.1006/dbio.2002.0728. [DOI] [PubMed] [Google Scholar]
- Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97(2):793–798. doi: 10.1073/pnas.97.2.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science. 1992;257(5070):661–665. doi: 10.1126/science.1323140. [DOI] [PubMed] [Google Scholar]
- Nauert JB, Klauck TM, Langeberg LK, Scott JD. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr Biol. 1997;7:52–62. doi: 10.1016/s0960-9822(06)00027-3. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008;102(2):e1–e11. doi: 10.1161/CIRCRESAHA.107.167809. [DOI] [PubMed] [Google Scholar]
- Nesher R, Anteby E, Yedovizky M, Warwar N, Kaiser N, Cerasi E. Beta-cell protein kinases and the dynamics of the insulin response to glucose. Diabetes. 2002;51(Suppl 1):S68–73. doi: 10.2337/diabetes.51.2007.s68. [DOI] [PubMed] [Google Scholar]
- Newhall KJ, Criniti AR, Cheah CS, Smith KC, Kafer KE, Burkart AD, McKnight GS. Dynamic anchoring of PKA is essential during oocyte maturation. Curr Biol. 2006;16(3):321–327. doi: 10.1016/j.cub.2005.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Hausken ZE, Scott JD, Jennings PA. The A-kinase anchoring domain of Type IIα cAMP-dependent protein kinase is highly helical. J Biol Chem. 1997;272:23637–23644. doi: 10.1074/jbc.272.38.23637. [DOI] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. Embo J. 2001;20(7):1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10(5):467–474. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118(Pt 23):5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- Park M, Serpinskaya AS, Papalopulu N, Gelfand VI. Rab32 regulates melanosome transport in Xenopus melanophores by protein kinase a recruitment. Curr Biol. 2007;17(23):2030–2034. doi: 10.1016/j.cub.2007.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14(9):1027–1047. [PubMed] [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, Doxsey SJ. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58(17):3974–3985. [PubMed] [Google Scholar]
- Purohit A, Tynan SH, Vallee R, Doxsey SJ. Direct interaction of pericentrin with cytoplasmic dynein light intermediate chain contributes to mitotic spindle organization. J Cell Biol. 1999;147(3):481–492. doi: 10.1083/jcb.147.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dorfler A, Kinning E, Megarbane A, Meinecke P, Semple RK, Spranger S, Toutain A, Trembath RC, Voss E, Wilson L, Hennekam R, de Zegher F, Dorr HG, Reis A. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- Rawe VY, Payne C, Navara C, Schatten G. WAVE1 intranuclear trafficking is essential for genomic and cytoskeletal dynamics during fertilization: cell-cycle-dependent shuttling between M-phase and interphase nuclei. Dev Biol. 2004a;276(2):253–267. doi: 10.1016/j.ydbio.2004.07.043. [DOI] [PubMed] [Google Scholar]
- Rawe VY, Ramalho-Santos J, Payne C, Chemes HE, Schatten G. WAVE1, an A-kinase anchoring protein, during mammalian spermatogenesis. Hum Reprod. 2004b;19(11):2594–2604. doi: 10.1093/humrep/deh513. [DOI] [PubMed] [Google Scholar]
- Reynolds JG, McCalmon SA, Donaghey JA, Naya FJ. Deregulated PKA signaling and myospryn expression in muscular dystrophy. J Biol Chem. 2008 doi: 10.1074/jbc.C700221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JG, McCalmon SA, Tomczyk T, Naya FJ. Identification and mapping of protein kinase A binding sites in the costameric protein myospryn. Biochim Biophys Acta. 2007;1773(6):891–902. doi: 10.1016/j.bbamcr.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson ML, Wallert MA, Reinitz CA, Shabb JB. Association of the type I regulatory subunit of cAMP-dependent protein kinase with cardiac myocyte sarcolemma. Arch Biochem Biophys. 1996;330(1):181–187. doi: 10.1006/abbi.1996.0240. [DOI] [PubMed] [Google Scholar]
- Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- Rubino D, Driggers P, Arbit D, Kemp L, Miller B, Coso O, Pagliai K, Gray K, Gutkind S, Segars J. Characterization of Brx, a novel Dbl family member that modulates estrogen receptor action. Oncogene. 1998;16(19):2513–2526. doi: 10.1038/sj.onc.1201783. [DOI] [PubMed] [Google Scholar]
- Russell MA, Lund LM, Haber R, McKeegan K, Cianciola N, Bond M. The intermediate filament protein, synemin, is an AKAP in the heart. Arch Biochem Biophys. 2006;456(2):204–215. doi: 10.1016/j.abb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Sacchetto R, Damiani E, Margreth A. Clues to calcineurin function in mammalian fast-twitch muscle. J Muscle Res Cell Motil. 2001;22(6):545–559. doi: 10.1023/a:1015010914328. [DOI] [PubMed] [Google Scholar]
- Schillace RV, Scott JD. Association of the type 1 protein phosphatase PP1 with the A-kinase anchoring protein AKAP220. Curr Biol. 1999;9(6):321–324. doi: 10.1016/s0960-9822(99)80141-9. [DOI] [PubMed] [Google Scholar]
- Schwaerzel M, Jaeckel A, Mueller U. Signaling at A-kinase anchoring proteins organizes anesthesia-sensitive memory in Drosophila. J Neurosci. 2007;27(5):1229–1233. doi: 10.1523/JNEUROSCI.4622-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD. Cyclic nucleotide-dependent protein kinases. Pharmacol Ther. 1991;50:123–145. doi: 10.1016/0163-7258(91)90075-w. [DOI] [PubMed] [Google Scholar]
- Shih M, Lin F, Scott JD, Wang HY, Malbon CC. Dynamic complexes of beta2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem. 1999;274(3):1588–1595. doi: 10.1074/jbc.274.3.1588. [DOI] [PubMed] [Google Scholar]
- Smith FD, Langeberg LK, Scott JD. The where’s and when’s of kinase anchoring. Trends Biochem Sci. 2006;31(6):316–323. doi: 10.1016/j.tibs.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Smith FD, Scott JD. Anchored cAMP signaling: onward and upward - a short history of compartmentalized cAMP signal transduction. Eur J Cell Biol. 2006;85(7):585–592. doi: 10.1016/j.ejcb.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Guire ES, Kaech S, White J, Zhang F, Schutz K, Langeberg LK, Banker G, Raber J, Scott JD. A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J Neurosci. 2007;27(2):355–365. doi: 10.1523/JNEUROSCI.3209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling SH, Langeberg LK, Soderling JA, Davee SM, Simerly R, Raber J, Scott JD. Loss of WAVE-1 causes sensorimotor retardation and reduced learning and memory in mice. Proc Natl Acad Sci USA. 2003;100(4):1723–1728. doi: 10.1073/pnas.0438033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaszak M, Christian F, Rosenthal W, Klussmann E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008;20(4):590–601. doi: 10.1016/j.cellsig.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8(1):37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC. Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J. 2003;22(24):6419–6429. doi: 10.1093/emboj/cdg628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Wang HY, Malbon CC. Src docks to A-kinase anchoring protein gravin, regulating beta2-adrenergic receptor resensitization and recycling. J Biol Chem. 2007;282(9):6597–6608. doi: 10.1074/jbc.M608927200. [DOI] [PubMed] [Google Scholar]
- Terman JR, Kolodkin AL. Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science. 2004;303(5661):1204–1207. doi: 10.1126/science.1092121. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Pawlikowska L, Zaroff JG, Kim T, Nguyen T, Young SG, Vranizan K, Kwok PY, Whooley MA, Conklin BR. Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc Natl Acad Sci U S A. 2007;104(20):8461–8466. doi: 10.1073/pnas.0610393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toksoz D, Williams DA. Novel human oncogene lbc detected by transfection with distinct homology regions to signal transduction products. Oncogene. 1994;9(2):621–628. [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD. Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A. 2008;105(34):12557–12562. doi: 10.1073/pnas.0805922105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24(19):8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan S, Goueli SA, Davey MP, Carr DW. Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem. 1997;272:4747–4752. doi: 10.1074/jbc.272.8.4747. [DOI] [PubMed] [Google Scholar]
- Wasmeier C, Romao M, Plowright L, Bennett DC, Raposo G, Seabra MC. Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J Cell Biol. 2006;175(2):271–281. doi: 10.1083/jcb.200606050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser DC, Pyati UJ, Kimelman D. Gravin regulates mesodermal cell behavior changes required for axis elongation during zebrafish gastrulation. Genes Dev. 2007;21(12):1559–1571. doi: 10.1101/gad.1535007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Soderling SH, Alto NM, Langeberg LK, Scott JD. Scar/WAVE-1, a Wiskott-Aldrich syndrome protein, assembles an actin- associated multi-kinase scaffold. EMBO J. 2000;19(17):4589–4600. doi: 10.1093/emboj/19.17.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes D, Charitakis K, Basson CT. Inherited disposition to cardiac myxoma development. Nat Rev Cancer. 2006;6(2):157–165. doi: 10.1038/nrc1798. [DOI] [PubMed] [Google Scholar]
- Wirtenberger M, Schmutzhard J, Hemminki K, Meindl A, Sutter C, Schmutzler RK, Wappenschmidt B, Kiechle M, Arnold N, Weber BH, Niederacher D, Bartram CR, Burwinkel B. The functional genetic variant Ile646Val located in the kinase binding domain of the A-kinase anchoring protein 10 is associated with familial breast cancer. Carcinogenesis. 2007;28(2):423–426. doi: 10.1093/carcin/bgl164. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP Signalling complexes: Focal points in space and time. Nature Reviews Molecular Cell biology. 2004;5(12):959–971. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Yoon DK, Jeong CH, Jun HO, Chun KH, Cha JH, Seo JH, Lee HY, Choi YK, Ahn BJ, Lee SK, Kim KW. AKAP12 induces apoptotic cell death in human fibrosarcoma cells by regulating CDKI-cyclin D1 and caspase-3 activity. Cancer Lett. 2007;254(1):111–118. doi: 10.1016/j.canlet.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Zakhary DR, Moravec CS, Bond M. Regulation of PKA binding to AKAPs in the heart: alterations in human heart failure. Circulation. 2000;101(12):1459–1464. doi: 10.1161/01.cir.101.12.1459. [DOI] [PubMed] [Google Scholar]