

Abstract
Gain-of-function mutations in the proto-oncogene c-kit that induce constitutive kinase activity of its product, KIT protein, are characteristic of human mast cell disease and are believed to play a central role in mast cell leukemia oncogenesis, proliferation and survival. Nuclear overexpression of the Wnt effector β-catenin and deregulated β-catenin nuclear signaling can promote malignant transformation in solid tumors and hematologic malignancies. However, a role for β-catenin in mast cell leukemia has not been described. Nuclear accumulation of β-catenin is upregulated by its tyrosine phosphorylation, a process that can be exacerbated by de-regulated expression of oncogenic tyrosine kinases. Here, we investigated the relationship between activated KIT and β-catenin signaling in mast cell leukemia. β-catenin was tyrosine-phosphorylated in cells with KIT activated by either gain-of-function mutation or incubation with the KIT ligand stem cell factor. β-catenin tyrosine-phosphorylation depended on KIT activity but not on PI3K-AKT activation. Tyrosine-phosphorylation of β-catenin was associated with its nuclear localization and enhanced transcription of target genes c-myc and cyclin D1. Endogenous KIT and β-catenin were found to associate in mast cell leukemia cells, and in vitro kinase assay demonstrated that active KIT phosphorylates tyrosine residues of β-catenin directly. Aberrant β-catenin-driven transcription caused by de-regulated KIT may represent a significant new target for treatment of mast cell leukemia.
Keywords: c-kit, mast cell, β-catenin, Tyrosine-phosphorylation
Introduction
The proto-oncogene c-kit encodes the transmembrane type III tyrosine kinase, KIT protein (1), which is the receptor for stem cell factor (SCF) (2, 3). This receptor kinase is characterized structurally by 5 extracellular immunoglobulin-like repeats and a split tyrosine kinase domain (4). Binding of SCF to KIT induces homodimerization of the receptor and autophosphorylation at the Y568 and Y570 tyrosine residues in the juxtamembrane domain (5). These residues act as docking sites for Src homology 2 (SH2) domain-containing signaling molecules, such as Janus kinase (JAK), signal transducer and activator of transcription (STAT), Src kinase, mitogen-activated protein (MAP) kinases, and phosphatidylinositol-3 (PI3) kinase (6). KIT is expressed on melanocytes, mast cells, hematopoietic stem cells, germ cells and interstitial cells of Cajal (7). Gain-of-function point mutations that result in ligand-independent constitutive phosphorylation of KIT protein have been described in various neoplastic diseases including mast cell leukemia (MCL), systemic mastocytosis and gastrointestinal stromal tumors (8–11). Downstream signaling pathways, including PI3 kinase (PI3K)/AKT, are inappropriately activated, and this is believed to contribute to the abnormal proliferation and survival of these neoplastic cells.
β-catenin is a multifunctional protein that plays an important role in both cell-cell interactions (12, 13) and transcriptional regulation (14–16). In epithelial cells, β-catenin is localized in the cytoplasm and at the inner surface of the plasma membrane, where in conjunction with E-cadherin it functions as part of the adherens junction, a specialized cytoskeletal complex that regulates cell-cell adhesion (17). As a transcriptional regulator, β-catenin is the critical effector of the canonical Wnt signaling pathway, in which nuclear β-catenin co-activates transcription in association with T-cell factor/lymphoid enhancer factor (LEF/TCF) family members. In the absence of secreted Wnts, the modular protein axin provides a scaffold for the binding of glycogen synthase kinase-3 β (GSK-3 β), adenomatous polyposis coli (APC) protein, and β-catenin. This facilitates serine/threonine-phosphorylation in the amino terminus of β-catenin by GSK3-β and subsequent rapid degradation of β-catenin by a proteasome-dependent process (18, 19). On the other hand, Wnt stimulation leads to β-catenin stabilization, nuclear accumulation and interaction with TCF/LEF proteins to regulate genes important for proliferation and survival (14, 20–22). While GSK3-β-mediated phosphorylation promotes degradation of β-catenin, tyrosine-phosphorylation is associated with the Wnt-independent nuclear localization of β-catenin and subsequent enhancement of its transcriptional activity(23). Recently, a number of oncogenic tyrosine kinases have been reported to directly promote tyrosine-phosphorylation of β-catenin in melanoma, breast and pancreatic cancer, and in chronic myelogenous leukemia (24–26).
In this study, we investigated the relationship between KIT and β-catenin in several cell lines derived from patients with MCL, in which a role for de-regulated β-catenin has not been described. β-catenin was tyrosine-phosphorylated in the presence of KIT activated by either gain-of-function mutation or SCF. β-catenin tyrosine-phosphorylation depended on KIT activation but not on signaling via PI3K/AKT. In cells with activated KIT kinase, β-catenin was localized mainly in the nucleus. In contrast, pharmacologic inhibition of KIT or its molecular knockdown with c-kit siRNA caused β-catenin to redistribute to the cytosol, coinciding with reduced transcription of β-catenin target genes. Finally, we observed the physical interaction between endogenous KIT and β-catenin in MCL, and in vitro kinase assay revealed that active KIT can directly phosphorylate tyrosine residues of β-catenin.
Materials and methods
Antibodies and reagents
The tyrosine-kinase inhibitor imatinib was kindly provided by Novartis (Basel, Switzerland). The PI3K inhibitor LY294002 was purchased from Calbiochem (San Diego, CA). KIT siRNA, β-catenin siRNA and control siRNA were purchased from Dharmacon (Lafayette, CO). PKC412 was purchased from LC laboratories (Woburn, MA). Anti-β-catenin monoclonal antibody (clone 14) was purchased from BD Biosciences (San Jose, CA). Anti-KIT-antibody PC34 was purchased from Oncogene Research (San Diego, CA) Anti-phospho-tyrosine monoclonal antibody 4G10 was purchased from Upstate (Charlottesville, VA). Anti-phospho KIT (Tyr719), anti-phospho AKT antibody, anti-AKT antibody and active KIT kinase were purchased from Cell Signaling Technology (Danvers, MA). Western blotting blocking reagent was purchased from Roche Applied Science (Indianapolis, IN). GammaBind Plus Sepharose beads and horseradish peroxidase (HRP)-labeled goat anti-mouse and goat anti-rabbit antibodies were purchased from Amersham Biosciences (Uppsala, Sweden). BCA Protein Assay reagent, western blot chemiluminescence reagents and Restore Western Blot Stripping Buffer were purchased from Pierce Chemical Co (Rockford, IL). PVDF membranes were purchased from Millipore (Billerica, MA).
Cells
HMC-1 cells (27), derived from a patient with MCL, were kindly provided by Dr. Joseph Butterfield (Mayo Clinic, Rochester, MN). HMC-1.2 cells contain the mutations D816V and V560G and are resistant to imatinib, while HMC-1.1 cells are sensitive to imatinib and contain a single mutation, V560G. These cell lines were maintained in Iscove’s medium with 25 mM HEPES and L-glutamine (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen). LAD 2,, an SCF-dependent mast cell line derived from a patient with mast cell sarcoma/leukemia (28), were kindly provided by Dr. Dean D. Metcalfe (National Institutes of Health, Bethesda, MD). LAD 2 was maintained in serum-free medium (StemPro-34; Life Technologies, Grand Island, NY) supplemented with 2 mM L-glutamine and 100 ng/ml rhSCF (R&D systems, Minneapolis, MN).
Cell growth assay
Cell growth was measured using the XTT assay kit (Roche). Briefly, cells grown in 96-well tissue culture plates were incubated with the treatment indicated for 24 to 72 h. XTT solution was added, cells were incubated for an additional 4 h, and the formation of formazan was spectrophotometrically quantified using an ELISA plate reader at an absorption wavelength of 450 nm.
Immunoprecipitation and immunoblotting assays
Cells were washed twice with ice-cold PBS, and were lysed in lysis buffer: 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM phenylmethylsulfonyl fluoride (PMSF). After incubation for 1 hour at 4°C, lysates were spun at 12,000 ×g for 25 minutes, and pellets were discarded. Lysates were immunoprecipitated with each primary antibody overnight at 4°C. GammaBind Plus Sepharose beads were added, and the mixture was rocked for 1 hour at 4°C. The beads were subsequently washed 3 times with lysis buffer and mixed with sodium dodecyl sulfate (SDS) sample buffer. After boiling for 5 minutes, samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and electroblotted onto PVDF membranes. The membranes were incubated overnight with primary antibody in 10% blocking reagent in TNE washing buffer: 50 mM NaCl, 10 mM Tris-HCl, pH 7.5, 2.5 mM EDTA, 0.1% Tween 20. Primary antibodies were detected by HRP-labeled secondary antibody (1:2000), and were visualized using chemiluminescence reagents. Optical density of the band was measured by GS-800 densitometer with Quantity One software (Bio-Rad).
Immunocytochemistry
Cells that had been attached to glass slides by cytocentrifugation (Shandon, Pittsburgh, PA) were fixed with 3.7% formaldehyde in PBS for 10 minutes and permeabilized with 0.2% Triton X-100 for 10 minutes at room temperature. The cells were stained with monoclonal anti-β-catenin antibody, Alexa Fluor 488 goat anti-mouse immunoglobulin (Molecular Probes, Carlsbad, CA) and 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes). The cells were observed using a Leica DM IRB fluorescence microscope (100× objective) equipped with a Z-axis motor (Ludl Electronics, Hawthorne, NY). Stacks of images (13 to 19 optical sections at a step size of 0.3 μm) were taken with a digital camera (Hamamatsu) and processed using Openlab Volume Deconvolution software (Improvision, Lexington, MA). Nuclear β-catenin was determined using the folocalization feature in Openlab.
Quantitative RT-PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA), and cDNA was prepared using the TaqMan Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Analysis of mRNA expression was carried out using the ABI Prism 7700 Sequence Detection System (Applied Biosystems). All samples were normalized to the level of 18S ribosomal RNA. The primer and probe sequences for real-time RT-PCR were as follows, 18S rRNA forward 5′-AGTCCCTGCCCTTTGTACACA-3′, 18S rRNA reverse 5′-CGATCCGAGGGCCTCACTA-3′, c-myc forward 5′-CGTCTCCACACATCAGCACAA-3′, c-myc reverse 5′ TCTTGGCAGCAGGATAGTCCTT-3′, cyclin D1 forward 5′ GCATGTTCGTGGCCTCTAAGAT-3′, cyclin D1 reverse 5′-TCGGTGTAGATGCACAGCTTCT-3′. Where shown, p values were determined by t-test using the StatView software package (SAS, Cary, NC).
In vitro kinase assay
Recombinant β-catenin (Upstate) was incubated with or without recombinant KIT kinase in reaction buffer (60 mM HEPES pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 3 μM Na3VO4, 1.25 mM DTT, 200 μM ATP) for 30 minutes at 25°C. After the reaction, samples were mixed with SDS-sample buffer and were resolved by SDS-PAGE, electrotransferred and immunoblotted with anti-phospho-tyrosine antibody. The membrane was stripped and reprobed with anti-β-catenin antibody, anti-KIT antibody or anti-albumin antibody.
RNAi-mediated silencing of c-kit gene expression
HMC-1.2 cells (5 × 106 in 100 μl) were transfected with 1 μM c-kit siRNA, β-catenin siRNA or control siRNA by electroporation (Nucleofector; Amaxa Biosystems, Gaithersburg, MD) following the manufacturer’s instructions. Following transfection, the cells were incubated for 48 h and then were subjected to quantitative RT-PCR, immunoprecipitation and immunoblot assay. Optical density of the band was measured by GS-800 densitometer with Quantity One software.
Results
β-catenin tyrosine-phosphorylation status in imatinib-sensitive and imatinib-resistant MCL
We first compared tyrosine-phosphorylation of β-catenin in imatinib-sensitive and imatinib-resistant cell lines. As previously described (27), the growth of the SCF-independent human MCL cell line, HMC-1.1, expressing an activating juxtamembrane mutation of c-kit at codon 560 (V560G), was inhibited by 200 nM imatinib, while that of HMC-1.2, a human MCL cell line which has an additional activating mutation (D816V) in the kinase domain, was insensitive to imatinib (Fig. 1A). Both cell lines expressed β-catenin protein and β-catenin was tyrosine phosphorylated in the absence of imatinib (Fig. 1B). While treatment with imatinib (500 nM for 3h) markedly suppressed the tyrosine-phosphorylation of β-catenin in imatinib-sensitive HMC-1.1 cells, no decrease was observed in imatinib-insensitive HMC-1.2 cells.
Figure 1.

Imatinib suppresses tyrosine-phosphorylation of β-catenin in imanitib-sensitive mast cell leukemia cells.
A: Effect of imatinib on cell growth of HMC-1.1 and HMC-1.2 cells. Cell growth was monitored by XTT assay as described in “Methods”. Cells were incubated with or without indicated concentration of imatinib for 24 h. Mean (+/− standard deviation) of three independent experiments are shown.
B: Effect of imatinib on expression and tyrosine phosphorylation of β-catenin. To detect tyrosine phosphorylation of β-catenin, 1 mg of total protein from cell lysates was precipitated using 2 μg of anti-β-catenin antibody. Proteins were resolved by reducing 10% SDS-PAGE, electrotransferred to PVDF membrane, and western blotted using corresponding antibodies. Optical density ratio of β-catenin/tubulin and tyrosine phosphorylated/total β-catenin (phospho/total β-catenin) was measured using a GS-800 densitometer with Quantity One software.
Unlike imatinib, the kinase inhibitor PKC412 has been reported to suppress activation of the D816V KIT mutant (29, 30). Treatment with PKC412 (500 nM for 3h), inhibited KIT in HMC-1.2 and effectively abrogated tyrosine-phosphorylation of β-catenin in these cells (Fig. 2A). LAD 2 is a recently described SCF-dependent mast cell line lacking mutation at codon 816 of KIT. Activation of KIT in LAD 2 cells was observed in the presence of SCF, while SCF-starvation suppressed KIT phosphorylation, as previously reported (28). Tyrosine-phosphorylation of β-catenin in these cells was also SCF-dependent (Fig. 2A).
Figure 2.

KIT activation is related to β-catenin tyrosine phosphorylation status, but does not depend on PI3K/AKT activation.
A: HMC-1.1 and HMC-1.2 cells were treated with or without the KIT inhibitors imatinib (500 nM for 3h) or PKC412 (250 nM for 3h). LAD 2 was starved of SCF for 48 h, and then treated with or without rhSCF (100 ng/ml for 3h). After the treatments, cells were lysed and lysates were subjected to western blot and immunoprecipitation. The HMC1.1 β-catenin and phosphotyrosine blots are reproduced from Fig. 1B for comparison.
B: HMC-1.1 and 1.2 cells were treated with or without imatinib (500 nM for 3h) or the PI3K inhibitor LY294002 (5 μM for 3h). After the treatment, cells were lysed and subjected to western blot and immunoprecipitation assay. C: HMC-1.1 and 1.2 cells were treated with or without imatinib (500 nM), LY294002 (5 μM), or PKC412 (250 nM) for 24 h. LAD 2 cells were incubated with or without rhSCF (100 ng/ml) for 72 h, following an initial starvation period of 48 h. Cell proliferation was determined by XTT assay. Mean (+/− standard deviation) of three experiments are shown. D: HMC-1.2 cells were transfected with either 1 μM c-kit siRNA or control siRNA. Following transfection, cells were incubated for 48 h. After 48 h, cells were lysed and protein expression of KIT, β-catenin and tubulin was analyzed by western blot, and tyrosine phosphorylation of β-catenin was analyzed by immunoprecipitation assay. Optical density ratios of KIT/tubulin, β-catnein/tubulin and phospho/total β-catenin were determined using a GS-800 densitometer and Quantity One software.
To clarify further the relationship between KIT and β-catenin tyrosine-phosphorylation, we knocked down KIT expression in HMC-1.2 cells with c-kit siRNA. As shown in Figure 2D, β-catenin tyrosine-phosphorylation was suppressed by silencing the c-kit gene. These results support the hypothesis that tyrosine-phosphorylation of β-catenin depends on activated KIT in MCL cell lines.
β-catenin tyrosine-phosphorylation is not mediated by KIT-induced PI3K/AKT signaling
AKT has been shown to be a downstream target of KIT via KIT-dependent PI3K activation (6). Since AKT directly phosphorylates and inhibits the activity of GSK3-β thereby stabilizing β-catenin levels (31), we wished to determine whether it played a role in KIT-dependent tyrosine-phosphorylation of β-catenin. Although imatinib treatment suppressed AKT phosphorylation in HMC-1.1, little change was observed in HMC-1.2. However, PKC412 (500 nM for 3h) effectively reduced AKT activation in HMC-1.2 (Fig. 2A). In LAD 2 cells, AKT phosphorylation was strongly dependent on SCF (Fig. 2A). To investigate the possible role of AKT signaling in mediating KIT-dependent β-catenin tyrosine-phosphorylation, we used the PI3K inhibitor LY294002. As shown in Figure 2B, treatment with LY294002 (5 μM for 3h) suppressed AKT phosphorylation in both HMC-1.1 and HMC-1.2 cells without altering the tyrosine-phosphorylation status of KIT. Although the total protein level of β-catenin was decreased slightly in LY294002-treated cells, presumably as a result of reversing AKT-mediated inhibition of GSK3-β, β-catenin tyrosine-phosphorylation was relatively preserved. As shown in Figure 2C, at the concentration used LY294002 did not affect the growth of these cells, while KIT inhibition in all 3 cell lines reduced growth. These data suggest that in MCL neither KIT-stimulated tyrosine-phosphorylation of β-catenin nor KIT-dependent cell growth are mediated via KIT activation of the PI3K/AKT pathway.
Suppression of KIT activation decreased nuclear β-catenin
Because tyrosine-phosphorylation of β-catenin has been reported to be associated with its increased nuclear localization (21–23, 32, 33), we examined the possible KIT dependence of the subcellular distribution of β-catenin in these MCL lines. β-catenin was located mainly in the nucleus in the KIT-activated cell lines HMC-1.1 and 1.2 (Fig. 3A & B, upper panel). Nuclear localization of β-catenin was also observed in SCF-stimulated LAD 2 (Fig. 3C, upper panel). In contrast, nuclear localization of β-catenin was markedly decreased after treatment of HMC-1.1 with imatinib (Fig. 3A, lower panel). Although imatinib was unable to alter the nuclear localization of β-catenin in HMC-1.2 (Fig. 3B, middle panel), exposure of these cells to PKC412 caused a marked redistribution of β-catenin to the cytoplasm (Fig. 3B, two lower panels). Similarly, removal of SCF from LAD 2 cells caused a dramatic relocalization of β-catenin from nucleus to cytoplasm (Fig. 3C, lower panel). Thus, KIT activation status in 3 independent MCL lines correlates with the subcellular localization of β-catenin.
Figure 3.

Nuclear localization of β-catenin in activated c-kit cell lines.
A, B: HMC-1.1 (A) and HMC-1.2 (B) cells were treated with or without imatinib (500 nM for 3 h) or PKC412 (250 ng/ml for 3 h), and then were cytocentrifuged onto glass slides. Cells were fixed in 3.7% formaldehyde in PBS and permeabilized with 0.2% Triton X-100. β-catenin was visualized by immunofluorescence (green, left panel). The DNA-intercalating dye DAPI was used to identify cell nuclei (blue, center panel). The right panel presents a merged image to highlight the nuclear pool of β-catenin.
C: Localization of β-catenin in LAD 2 cells. After incubation with or without rhSCF, LAD 2 cells were cytocentrifuged onto glass slides. Cells were fixed and stained as above. Images were collected using a 100× objective.
Inactivation and silencing of KIT down-regulates β-catenin target genes in MCL
Because enhanced nuclear localization of β-catenin correlated with the activation status of KIT, we wished to determine whether β-catenin-dependent transcription in MCL was dependent on KIT activity. To examine this question, we measured the mRNA levels of two β-catenin target genes, cyclin D1 (14, 20) and c-myc (20) using real-time RT-PCR. After imatinib treatment, expression of both cyclin D1 and c-myc was markedly decreased in HMC-1.1, while little change was observed in HMC-1.2 (Fig. 4A). In contrast, PKC412 decreased expression of both cyclin D1 and c-myc in the imatinib-resistant cells (Fig. 4A). Further, c-kit- and β-catenin-specific siRNAs each decreased expression of both target genes in HMC-1.2 (Fig. 4B), and the degree of target gene downregulation was similar to the degree of downregulation of KIT and β-catenin proteins, respectively (Fig. 4B). Moreover, SCF-induced activation of KIT in LAD 2 cells coincided with increased expression of both cyclin D1 and c-myc genes (Fig. 4C).
Figure 4.

Effect of KIT activation on β-catenin target gene expression in MCL cell lines.
A: HMC-1.1 and 1.2 were treated with or without imatinib (500 nM) or PKC412 (250 nM) for indicated time periods. Total RNA was isolated from the cells and quantitative RT-PCR was performed in duplicate. All samples were normalized to the level of 18S ribosomal RNA. The mean of two individual experiments (+/− standard deviation) are shown.
B: HMC-1.2 cells were transfected with either c-kit siRNA, β-catenin siRNA or control siRNA. Following incubation for 48 h, total RNA was isolated and quantitative RT-PCR was performed.
C: LAD 2 cells were starved of SCF for 48 h and then were treated with or without rhSCF (100 ng/ml) for an additional 4 h. Total RNA was isolated from the cells and quantitative RT-PCR was performed. Where shown, p values were determined by t-test using the StatView software package (SAS, Cary, NC).
Active KIT binds to β-catenin and catalyzes its tyrosine phosphorylation
We examined the possible physical interaction between KIT and β-catenin by co-immunoprecipitation. In HMC-1.1, a large amount of endogenous KIT was co-immunoprecipitated with endogenous β-catenin. This association was significantly reduced in cells treated with imatinib (Fig. 5A, left panel). Similarly, in the reciprocal experiment, endogenous β-catenin was co-immunoprecipitated by anti-KIT antibody in untreated cells, but this association was inhibited by imatinib (Fig. 5A, right panel). These results demonstrate that β-catenin preferentially interacts with active KIT. To determine whether active KIT can directly phosphorylate tyrosine residues of β-catenin, we performed an in vitro kinase assay using purified recombinant active KIT kinase as enzyme source, and purified recombinant β-catenin as substrate. As shown in Figure 5B, no tyrosine phosphorylation of β-catenin was detected in the absence of KIT protein (upper panel, lane 2). Addition of active KIT kinase induced tyrosine-phosphorylation of β-catenin, while inclusion of imatinib decreased tyrosine-phosphorylation of both KIT and β-catenin (upper panel, lanes 3 and 4). These results suggest that active KIT can directly phosphorylate tyrosine residues of β-catenin.
Figure 5.
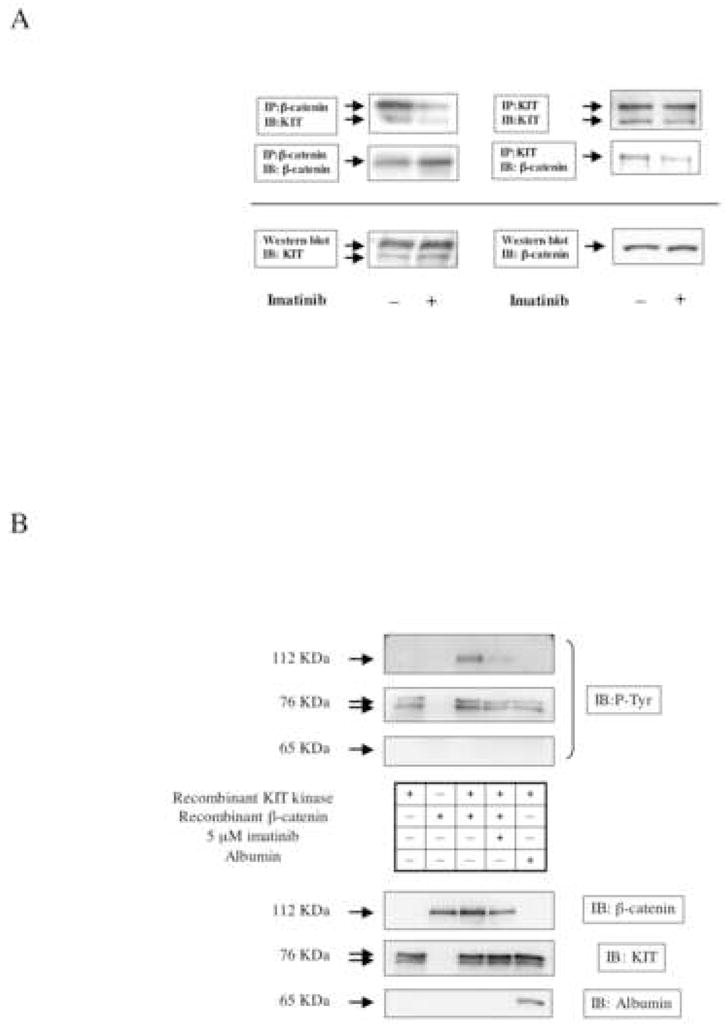
Active KIT binds to β-catenin and catalyzes β-catenin tyrosine phosphorylation.
A: HMC-1.1 cells were treated with or without imanitib (500 nM for 3 h). After cell lysis, 1 mg of total protein was immunoprecipitated with an antibody recognizing either β-catenin (left upper panel) or KIT (right upper panel). After SDS-PAGE and electrotransfer, blots were probed with antibodies to both β-catenin and KIT. The level of β-catenin and KIT protein in whole cell lysates is shown in the lower panel.
B: Recombinant β-catenin was incubated with recombinant active KIT in an in vitro kinase reaction buffer, as described in “Materials and Methods”. To demonstrate specificity of the reaction, albumin was substituted for β-catenin. After the reaction, proteins were resolved by SDS-PAGE and immunoblotted for phospho-tyrosine (upper panels). The membrane was stripped and re-probed with anti-β-catenin, anti-KIT and anti-albumin antibody to verify amounts of proteins present in the assay.
Discussion
Tyrosine kinase deregulation is commonly observed in both solid tumors and hematologic malignancies (34). Deregulated kinases increase cell proliferation and promote anti-apoptotic signaling, and as a class, tyrosine kinases are one of the most important targets in oncology drug development. KIT is a receptor tyrosine-kinase that is activated by its ligand, SCF (1–3). Gain-of-function mutations in c-kit have been observed in MCL, systemic mastocytosis (9) and gastrointestinal stromal tumors (11), and KIT mutation is considered to be a major mechanism underlying oncogenesis in these diseases. The KIT inhibitor imatinib is widely used in treatment of these diseases (27, 35–37). However, imatinib fails to inhibit cells that exhibit the D816V mutation, the most common gain-of-function mutation in systemic mastocytosis (27, 35, 36). The tyrosine kinase inhibitor PKC 412 has been reported to inhibit D816V KIT activation in vivo and in vitro (29, 30). In a patient with MCL who had associated myelodysplastic syndrome/myeloproliferative disorder and a D816V KIT mutation, PKC412 resulted in a significant reduction in the peripheral blood mast cell count. Interestingly, although this effect was transient, KIT phosphorylation was suppressed at the time of relapse (29), suggesting that other mechanisms for driving cell proliferation might exist in relapsed MCL.
Wnt signaling is required for normal hematopoiesis, and deregulated Wnt signaling has been implicated in the etiology and progression of various malignancies (38–43). In colorectal cancer, truncation or loss of the APC protein or mutation of the GSK-3 β phosphorylation sites in β-catenin are thought to be critical mechanisms underlying β-catenin cytoplasmic and nuclear accumulation, promoting the expression of β-catenin-regulated pro-proliferative and survival genes (16, 43). However, β-catenin signaling was reported to be increased in acute myeloid leukemia and multiple myeloma without mutation of APC or β-catenin (39, 41), suggesting that alternative mechanisms might contribute to β-catenin upregulation. Previous studies have suggested that aberrant tyrosine-phosphorylation of β-catenin in tumor cells characterized by abnormal expression of the tyrosine-kinases ErbB2 (33) or MET/RON (22, 25) might be related to tumorigenesis. Recently, we found that activated FMS like tyrosine-kinase 3 (FLT3) directly phosphorylates tyrosine-residues of β-catenin in acute myeloid leukemia cells, resulting in nuclear localization of β-catenin and upregulation of β-catenin target genes (T.K. et al., manuscript submitted). To date, no study has investigated the relationship between KIT activation and β-catenin. Moreover, tyrosine-phosphorylation of β-catenin in mast cell diseases has not been examined. Our results show that activated KIT promotes tyrosine-phosphorylation of β-catenin, while KIT inhibition reverses this phenomenon. Tyrosine-phosphorylation of β-catenin is strongly associated with β-catenin’s nuclear localization and the expression of its target genes. Furthermore, co-immunoprecipitation assay revealed that activated KIT binds to β-catenin in MCL, and in vitro kinase assay demonstrated that active KIT can phosphorylate tyrosine-residues of β-catenin directly. Although KIT activates PI3K (6), and signaling via PI3K/AKT stabilizes β-catenin protein level through inhibition of GSK-3 β (18, 19), our data show that KIT-dependent regulation of both MCL cell growth and tyrosine-phosphorylation of β-catenin is not mediated by KIT activation of the PI3K/AKT axis. Indeed, our findings suggest that loss of nuclear β-catenin accurately predicts cell growth inhibition in MCL.
The data presented here suggest that enhanced β-catenin tyrosine-phosphorylation, nuclear retention, and transcriptional activity may be a shared downstream event among various hematologic malignancies with deregulated protein tyrosine kinase activity, including MCL expressing de-regulated KIT. Nuclear β-catenin retention and signaling may thus represent a significant therapeutic target in these neoplasms.
Acknowledgments
Grant Support note: This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Support was also provided to TK by the Sumitomo Life Social Welfare Sciences Foundation.
The authors thank Kristin Beebe, for her excellent technical assistance.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Support was also provided to TK by the Sumitomo Life Social Welfare Sciences Foundation. There are no conflicts of interest.
Footnotes
Statement of authorship: Designed research (TK, JBT, LN), performed research (TK, SL, MJ), wrote the paper (TK).
Contributors
Designed research (TK, JBT, LN), performed research (TK, SL, MJ), wrote the paper (TK).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. Embo J. 1987;6:3341–51. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DM, Lyman SD, Baird A, Wignall JM, Eisenman J, Rauch C, et al. Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms. Cell. 1990;63:235–43. doi: 10.1016/0092-8674(90)90304-w. [DOI] [PubMed] [Google Scholar]
- 3.Martin FH, Suggs SV, Langley KE, Lu HS, Ting J, Okino KH, et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203–11. doi: 10.1016/0092-8674(90)90301-t. [DOI] [PubMed] [Google Scholar]
- 4.Miettinen M, Lasota J. KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol. 2005;13:205–20. doi: 10.1097/01.pai.0000173054.83414.22. [DOI] [PubMed] [Google Scholar]
- 5.Blechman JM, Lev S, Givol D, Yarden Y. Structure-function analyses of the kit receptor for the steel factor. Stem Cells. 1993;11 (Suppl 2):12–21. doi: 10.1002/stem.5530110804. [DOI] [PubMed] [Google Scholar]
- 6.Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, et al. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461–4. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- 7.Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- 8.Akin C, Metcalfe DD. Systemic mastocytosis. Annu Rev Med. 2004;55:419–32. doi: 10.1146/annurev.med.55.091902.103822. [DOI] [PubMed] [Google Scholar]
- 9.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995;92:10560–4. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20:1692–703. doi: 10.1200/JCO.2002.20.6.1692. [DOI] [PubMed] [Google Scholar]
- 11.Moriyama Y, Tsujimura T, Hashimoto K, Morimoto M, Kitayama H, Matsuzawa, et al. Role of aspartic acid 814 in the function and expression of c-kit receptor tyrosine kinase. J Biol Chem. 1996;271:3347–50. doi: 10.1074/jbc.271.7.3347. [DOI] [PubMed] [Google Scholar]
- 12.Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. Embo J. 1989;8:1711–7. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peifer M, McCrea PD, Green KJ, Wieschaus E, Gumbiner BM. The vertebrate adhesive junction proteins beta-catenin and plakoglobin and the Drosophila segment polarity gene armadillo form a multigene family with similar properties. J Cell Biol. 1992;118:681–91. doi: 10.1083/jcb.118.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–42. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 16.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–20. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 17.Barker N, Clevers H. Catenins, Wnt signaling and cancer. Bioessays. 2000;22:961–5. doi: 10.1002/1521-1878(200011)22:11<961::AID-BIES1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 18.Easwaran V, Song V, Polakis P, Byers S. The ubiquitin-proteasome pathway and serine kinase activity modulate adenomatous polyposis coli protein-mediated regulation of beta-catenin-lymphocyte enhancer-binding factor signaling. J Biol Chem. 1999;274:16641–5. doi: 10.1074/jbc.274.23.16641. [DOI] [PubMed] [Google Scholar]
- 19.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. Embo J. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 21.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–43. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 22.Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol. 2001;21:5857–68. doi: 10.1128/MCB.21.17.5857-5868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris TJ, Peifer M. Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol. 2005;15:234–7. doi: 10.1016/j.tcb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Camp ER, Yang A, Gray MJ, Fan F, Hamilton SR, Evans DB, et al. Tyrosine kinase receptor RON in human pancreatic cancer: expression, function, and validation as a target. Cancer. 2007;109:1030–1039. doi: 10.1002/cncr.22490. [DOI] [PubMed] [Google Scholar]
- 25.Zinser GM, Leonis MA, Toney K, Pathrose P, Thobe M, Kader SA, et al. Mammary-specific Ron receptor overexpression induces highly metastatic mammary tumors associated with beta-catenin activation. Cancer Res. 2006;66:11967–74. doi: 10.1158/0008-5472.CAN-06-2473. [DOI] [PubMed] [Google Scholar]
- 26.Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. Embo J. 2007;26:1456–66. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99:1741–4. doi: 10.1182/blood.v99.5.1741. [DOI] [PubMed] [Google Scholar]
- 28.Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, et al. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res. 2003;27:677–82. doi: 10.1016/s0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 29.Gotlib J, Berube C, Growney JD, Chen CC, George TI, Williams C, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106:2865–70. doi: 10.1182/blood-2005-04-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Bohm A, et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–9. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- 31.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 32.Piedra J, Miravet S, Castano J, Palmer HG, Heisterkamp N, Garcia de Herreros A, et al. p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol Cell Biol. 2003;23:2287–97. doi: 10.1128/MCB.23.7.2287-2297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonvini P, An WG, Rosolen A, Nguyen P, Trepel J, Garcia de Herreros A, et al. Geldanamycin abrogates ErbB2 association with proteasome-resistant beta-catenin in melanoma cells, increases beta-catenin-E-cadherin association, and decreases beta-catenin-sensitive transcription. Cancer Res. 2001;61:1671–7. [PubMed] [Google Scholar]
- 34.Lengyel E, Sawada K, Salgia R. Tyrosine kinase mutations in human cancer. Curr Mol Med. 2007;7:77–84. doi: 10.2174/156652407779940486. [DOI] [PubMed] [Google Scholar]
- 35.Akin C, Brockow K, D’Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, et al. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol. 2003;31:686–92. doi: 10.1016/s0301-472x(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 36.Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002;1:1115–24. [PubMed] [Google Scholar]
- 37.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 38.Chung EJ, Hwang SG, Nguyen P, Lee S, Kim JS, Kim JW, et al. Regulation of leukemic cell adhesion, proliferation, and survival by beta-catenin. Blood. 2002;100:982–90. doi: 10.1182/blood.v100.3.982. [DOI] [PubMed] [Google Scholar]
- 39.Derksen PW, Tjin E, Meijer HP, Klok MD, MacGillavry HD, van Oers MH, et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc Natl Acad Sci U S A. 2004;101:6122–7. doi: 10.1073/pnas.0305855101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2004;101:3118–23. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene. 2005;24:2410–20. doi: 10.1038/sj.onc.1208431. [DOI] [PubMed] [Google Scholar]
- 42.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 43.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]