

Abstract
Doxorubicin (Dox) is known to cause cardiomyopathy and congestive heart failure upon chronic administration. The mechanisms underlying these toxicities remain uncertain but have been attributed, at least in part, by induction of cardiac cell apoptosis. Fas ligation with its cognate ligand (FasL) induces apoptosis and activates cellular inflammatory responses associated with tissue injury. We determined whether interruption of Fas/FasL interaction by cardiac-targeted expression of soluble Fas (sFas), a competitive inhibitor of FasL, would protect against Dox chronic cardiotoxicity in mice. Wild-type (WT) and sFas transgenic mice were administrated intravenously with 4 mg/kg Dox or with an equivalent volume of saline twice a week for a total of 10 injections. There were 25% mortality in WT mice, but no death was observed in sFas mice during the period of Dox treatment. Echocardiographic evaluation revealed a significant decrease in left ventricle fractional shortening after Dox treatment in WT mice but not in sFas mice. WT mice treated with Dox developed extensive myocardial cytoplasmic vacuolization, apoptosis, and interstitial fibrosis, which were much less or absent in sFas mice. The increased inducible nitric oxide synthase expression, nitric oxide production, superoxide generation, and peroxynitrite formation after Dox treatment in WT mice were attenuated by sFas expression. sFas expression also attenuated Dox-mediated induction of proinflammatory cytokines, tumor necrosis factor-α, interleukin (IL)-1β, and IL-6 in the myocardium. These observations indicate that FasL is an important mediator in Dox-associated cardiotoxicity by generating reactive oxygen and nitrogen species.
Doxorubicin (Dox) is a widely used chemotherapeutic agent in the treatment of a variety of cancers. However, the major limitation of Dox in the clinical application is its dose-related cardiotoxicity that may cause irreversible myocardial damage, leading to dilated cardiomyopathy with congestive heart failure (Singal and Iliskovic, 1998). Although the precise mechanisms of Dox cardiotoxicity remain elusive, there is increasing evidence that Dox exposure can trigger myocyte apoptosis and that this type of cell death represents the predominant form of myocyte damage seen in this setting (Kalyanaraman et al., 2002; Takemura and Fujiwara, 2007).
Fas ligation with its cognate ligand (FasL) is one of the key regulators of the apoptotic pathway and has been shown to exist in the heart (Setsuta et al., 2004). Fas consists of two isoforms, membrane-anchored Fas and soluble (sFas). The membrane isoform (membrane-anchored Fas) is a 45-kDa cell surface protein containing a single transmembrane region and induces apoptosis upon FasL binding, whereas the soluble isoform (sFas) lacks the transmembrane domain because of alternative splicing of the transcript and is thought to block Fas-mediated apoptosis by sequestering FasL (Suda et al., 1993; Cheng et al., 1994). In experimental models, Fas is overexpressed by cardiac myocytes in response to Dox administration (Nakamura et al., 2000; Lien et al., 2006). Both in vitro and in vivo studies demonstrated that blocking of the Fas/FasL interaction with an FasL-neutralizing antibody inhibited Dox-induced toxicity in cardiomyocytes (Nakamura et al., 2000; Wu et al., 2002). In a recent study, an elevated level of sFas has been found in patients with several different cancers (Tamakoshi et al., 2008), and studies have shown that patients with increased levels of sFas during chemotherapy had a better overall survival (Perik et al., 2006; Pichon et al., 2006). The recent reports from others and our laboratory demonstrate that expression of sFas leads to improvement in cardiac function and overall survival in mice with ischemic myocardial injury (Li et al., 2004; Niu et al., 2006). To investigate whether sFas has a beneficial effect on chronic Dox cardiotoxicity and elucidate possible underlying mechanisms, transgenic mice with cardiac-targeted expression of sFas were administrated repeatedly with a low dose of Dox over a period of 7 weeks. Data presented here demonstrate that cardiac-targeted expression of sFas attenuates Dox-induced generation of reactive oxygen and nitrogen species, formation of peroxynitrite, apoptotic cell death, and production of proinflammatory cytokines in the heart, leading to the inhibition of chronic Dox cardiotoxicity.
Materials and Methods
Animals. Transgenic mice with cardiac-targeted expression of sFas were generated from the FVB/N strain under the control of α-myosin heavy-chain promoter, and the homozygous sFas transgenic mice were produced by interbreeding and maintained in our animal facility. Detailed description for the development and characterization of sFas mice were reported previously (Niu et al., 2006). Wild-type (WT) mice from the same background (FVB/N strain) were purchased from Harlan (Indianapolis, IN) and served as controls. Male mice, 12 weeks old, were used for experiments. The experimental procedures in mice and protocol used in this study were approved by Animal Care and Use Committee of the University of Central Florida, in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996).
Experimental Protocol. Both WT and sFas mice were randomly assigned to two groups (saline and Dox; n = 10-12 for each group) and injected intravenously with 4 mg/kg Dox (Sigma-Aldrich, St. Louis, MO) dissolved in sterile saline (Dox-treated groups) or with an equivalent volume of pathogen-free saline only (saline-treated groups) via tail vein twice a week for a total of 10 injections. After the first four injections, the animals were not treated for 2 weeks to allow the recovery of bone marrow depression. The dosage of Dox and treatment protocol were based on the previous report that showed the clinically relevant cardiomyopathy in the mouse (Sun et al., 2001). The animals were kept in individual cages in temperature-controlled rooms. They had free access to tap water and food during the experimental period. The animals were observed daily and weighed weekly throughout the duration of the study and were euthanized under anesthesia after echocardiographic examination in the 2 weeks after the last Dox injection.
Assessment of Cardiac Function. At the beginning of Dox treatment and on the day that animals were euthanized, cardiac function was assessed by echocardiography. In brief, mice were lightly anesthetized via a nose cone and maintained with 0.5 to ∼2.0% isoflurane (AErrane; Baxter, McGaw Park, IL) mixed with oxygen. The chest was shaved, and animals were placed in a supine position with a slight tilt to the left decubitus position. Thermoregulation was achieved by using an autoregulated heating pad. A 15-MHz high-frequency transducer connected to an Agilent Technologies SONOS 4500 ultrasound machine (Philips Medical System; Agilent Technologies, Santa Clara, CA) was used. A two-dimensional short-axis view of the left ventricle was obtained at the level of the papillary muscles, and two-dimensionally targeted M-mode tracings were recorded at a sweep speed of 100 mm/s. The left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension were measured using online analyzing system to calculate the left ventricular fractional shortening, an index of cardiac function, by the equation: fractional shortening = [(LVEDD - left ventricular end-systolic dimension)/LVEDD] × 100%. All measurements were made according to the guidance of the American Society of Echocardiography leading edge-to-leading edge technique. Data from three to five consecutive selected cardiac cycles were analyzed and averaged.
Cardiac Histology and Histomorphometric Analysis. After echocardiographic measurements and collection of blood samples, the hearts were removed, weighed, and fixed by immersion in 10% phosphate-buffered formaldehyde or snap-frozen in liquid nitrogen for further examination as described below. A few photographs of whole hearts were taken with a Spot Insight digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI) with a computerized Nikon SMZ1000 dissecting microscope (Nikon, Tokyo, Japan). Equatorial regions of the heart were routinely processed and paraffin embedded. Sections were stained with hematoxylin and eosin and Masson's trichrome using standard protocols for histomorphometric analysis. Quantitative assessments for myocardial fibrotic area were performed on five sections in five randomly selected fields per section and expressed as interstitial collagen volume fraction. The collagen volume fraction was calculated as a percentage of the sum of all blue-stained areas to the total ventricular areas by using the Metamorph Series 6.2 image analysis program (Molecular Devices, Sunnyvale, CA) as described previously (Niu et al., 2006).
Immunohistochemical Detection of Peroxynitrite Formation and Poly(ADP-Ribose) Polymerase Activation. Immunohistochemistry was performed in sections to determine expression levels of 3-nitrotyrosine (3-NT; a biomarker of peroxynitrite formation and oxidative stress) and cleaved poly(ADP-ribose) polymerase (PARP p85 fragment, a marker for detection of apoptosis) in the myocardium. In brief, after deparaffinization and rehydration, sections (5 μm) were treated with 3% H2O2/methanol solution to quench endogenous peroxidase activity and incubated with blocking buffer (PerkinElmer Life and Analytical Sciences, Waltham, MA) to block nonspecific binding. Sections were then incubated with polyclonal rabbit anti-nitrotyrosine (1:100; Millipore, Billerica, MA) and polyclonal rabbit anti-PARP p85 fragment (1:00; Promega, Madison, WI) antibodies overnight at 4°C, respectively, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:200; Santa Cruz Biochemicals, Santa Cruz, CA). Peroxidase activity was visualized with diaminobenzidine. The counterstain was developed with hematoxylin. Incubation without primary antibody was performed as controls. On control sections, no specific immunoreactivity was detected. Photomicrographs were obtained at 400× magnification. The positive areas of staining were measured and expressed as the percentage of total LV area by using the Metamorph Series 6.2 image analysis program. The immunoreactivity for 3-NT and PARP p85 fragment were quantified from five randomly selected sections for each animal, and five animals were studied per group.
Measurement of Superoxide Generation in Hearts. Superoxide anion generation in response to phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) in freshly prepared ventricular homogenates was assessed spectrophotometrically by luminol-mediated chemiluminescence, as described previously (Iwata et al., 1995), with the Superoxide Anion Detection Kit (Calbiochem, San Diego, CA) according to the manufacturer's instructions. PMA is an activator of the intracellular enzyme protein C and is a potent stimulator of NADPH oxidase. Luminol is sensitive to the presence of superoxide anion and was used to monitor NADPH-stimulated superoxide generation. In brief, cardiac tissues were homogenized and centrifuged at 1000g for 10 min. The supernatant (100 μl) was added to 100 μl of the SuperoxideAnion assay medium reagent mixture containing 200 μM luminol, 250 μM enhancer, and 200 ng/ml PMA, then incubated for 30 min at room temperature. Chemiluminescence was measured with a spectrophotometer, and the background luminescence was subtracted from the readings with PMA. Values were standardized to the amount of protein present and expressed as relative light intensity per milligram of protein.
Measurement of the ROS Generation in the Dox-Treated H9c2 Cells. The redox-sensitive fluorophore hydroethidium has been used to monitor the intracellular oxidative stress (Benov et al., 1998). In brief, the H9c2 cardiomyocyte cell line (CRL-1446; American Type Culture Collection, Manassas, VA) was maintained in Dulbecco's modified Eagle's medium containing 10% (v/v) heat-inactivated fetal bovine serum, 100 U/ml penicillin G, 100 mg/ml streptomycin, and 2 mM l-glutamine. After pretreatment of H9c2 with sFas (hBA157, 10 μg/ml; Santa Cruz Biochemicals) and then 2 μM Dox for 6 h, culture medium was aspirated, and cells were washed with Dulbecco's phosphate-buffered saline and incubated in fresh culture medium without fetal bovine serum. Hydroethidine (10 μM) was added to the cells and incubated for 30 min, during which hydroethidine was oxidized to the fluorophore dihydro-β-erythroidine (DHE). Fluorescence imagines were obtained using a Nikon fluorescence microscope equipped with a rhodamine filter. The fluorescence intensity values were calculated from three different wells using a fluorescence spectrophotometer, and the average values were represented.
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling. Cell death in the myocardium was detected with the use of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) technique by using the Cardio TACS in situ apoptosis detection kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instruction. In brief, sections were incubated with proteinase K for 20 min at room temperature and then washed with phosphate-buffered saline. Endogenous peroxidase was inactivated by 3% H2O2 for 5 min at room temperature, and sections were incubated with labeling buffer containing terminal deoxynucleotidyl transferase (TdT), Mn2+, and biotinylated-deoxyuridine 5-triphosphate at 37°C for 60 min. Sections were then incubated with streptavidin-horseradish peroxidase for 10 min, and the signals were visualized with TACS blue. Quantitative analysis was performed by using the Metamorph Series 6.2 image analysis program. The percentage of TUNEL-positive cells was calculated as a percentage of total cells viewed in five randomly selected fields for each animal, and three animals were studied per group.
Reverse Transcriptase-Polymerase Chain Reaction. Expression of FasL and several proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and iNOS in the myocardium, were examined using reverse transcriptase (RT)-polymerase chain reaction (PCR). Total RNA was extracted from frozen cardiac tissues obtained from WT and sFas mice using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA quantity was monitored at 260 nm. Total RNA (2 μg) was reverse transcribed to cDNA using the SuperScript First-Strand Synthesis System (Bio-Rad-IScript; Bio-Rad, Hercules, CA). Subsequently, the resulting cDNA was amplified with the following primers for detecting FasL, TNF-α, IL-1β, IL-6, and iNOS genes expression. Primer pairs used were as follows: FasL: forward, 5′-GCCCATGAATTACCCATGTG-3′ and reverse, 5′-GCCAGAGATTTGTGTTGTGG-3′; TNF-α: forward 5′-ACTCAACAAACTGCCCTTCTGAG-3′ and reverse, 5′-TTACAGCTGGTTTCGATCCATTT-3′; IL-1β: forward, 5′-TGTGGCTGTGGAGAAGCTGT-3′ and reverse, 5′-CAGCTCATATGGGTCCGAGA-3′; IL-6: forward, 5′-CACGGCCTTCCCTACTTCAC-3′ and reverse, 5′-TGCAAGTGCATCATCGTTGT-3′; and iNOS: forward, 5′-ACATCGACCCGTCCACAGTAT-3′ and reverse, 5′-CAGAGGGGTAGGCTTGTCTC-3′. Likewise, β-actin primers (forward, 5′-AAATCGTGCGTGACATCAAAG-3′ and reverse, 5′-TGTAGTTTCATGGATGCCACAG-3′) were used in PCR reactions under the same conditions, which included an initial denaturation (94°C/5 min), followed by a cycle of denaturation (94°C/30 s), annealing (60°C/30 s), and extension (72°C/30 s). Each sample was subjected to 35 cycles followed by a final extension (72°C/10 min). PCR products were separated and visualized on 1.5% agarose ethidium bromide-stained gel. Band intensity was assessed using imaging software (Alphaimager 2200). The transcript levels of FasL, TNF-α, IL-1β, IL-6, and iNOS were normalized to β-actin in each sample and expressed as the ratio of expression of β-actin.
Measurement of Serum Nitrite and Nitrate. Whole blood was collected from Dox- and saline-treated WT and sFas mice on the day that animals were euthanized. Circulating levels of nitrite and nitrate, the major metabolites of nitric oxide (NO), were measured by Griess reagent kit (Invitrogen) according to the manufacturer's instructions. The absorbance at 550 nm was then measured using microplate reader, and the levels of nitrite and nitrate were determined from standard curves.
Data and Statistical Analysis. All values are presented as mean ± S.E.M. of n observations; n represents the number of animals studied. The results were analyzed by one-way analysis of variance (ANOVA) followed by a post-test for multiple comparisons. A p value of less than 0.05 was considered significant.
Results
Clinical Manifestations in Mice after Dox Treatment. As reported by others (Sun et al., 2001), during the course of Dox treatment, a significant inhibition of body weight gain in WT mice has been noted after 6 weeks of injections compared with the saline-treated controls, whereas this Dox-induced inhibition of body weight gain was suppressed in sFas mice (data not shown). WT animals also showed signs of heart failure-like syndrome including lethargy, ruffled hair, and respiratory stress at week 9, and pleural effusions began to appear in five of 12 Dox-treated WT animals at week 10, and three of 12 Dox-treated WT animals died spontaneously at week 11 after first Dox administration. In contrast, sFas animals remained healthy at this time point. Gross anatomical examinations at the end of study period showed that the hearts were dilated in both the atrium and the ventricle, especially in the left atrium, in the Dox-treated WT mice (Fig. 1A). The heart/body weight ratio was markedly increased in the Dox-treated WT mice but not in the sFas transgenic mice (Fig. 1B).
Fig. 1.

Targeted cardiac expression of sFas protects against Dox-induced cardiomyopathy. A, gross anatomical inspection reveals marked cardiomegaly in Dox-treated WT heart (left) compared with Dox-treated sFas heart (right). B, quantitative data for heart weight index of the saline- and Dox-treated WT and sFas mice. White bars, saline injection; shaded bars, Dox injection. *, p < 0.05 versus saline-injected WT mice and Dox-treated sFas mice; n = 10 to 12 per group.
Histological Changes. Histological examination showed that, in comparison with the saline-treated controls (Fig. 2, A and B), the myocardium from Dox-treated WT mice showed myocardial fiber swelling and diffuse myocyte vascularization (Fig. 2C), whereas these histological degenerative changes were barely found in Dox-treated sFas mice (Fig. 2D). Cardiac sections were stained with Masson's trichrome for detection of collagen deposition (Fig. 2, E and F), and myocardial interstitial fibrosis was determined by measurement of collagen volume fraction. WT animals treated with Dox demonstrated increased collagen volume fraction (2.4 ± 0.16%), which was much less in Dox-treated sFas animals (0.52 ± 0.09%, p < 0.05). There was no difference in the collagen volume fraction between saline-treated WT and sFas mice (0.48 ± 0.07 versus 0.46 ± 0.09%, p > 0.05).
Fig. 2.

Representative photomicrographs demonstrating the effect of cardiac-specific expression of sFas on Dox-induced cardiac histopathology. A to D, hematoxylin and eosin-stained sections of paraffin-embedded hearts reveals prominent and diffuse vascularization in the Dox-treated WT mice (C) but not saline control (A and B) and Dox-treated (D) sFas mice. E and F, Masson's trichrome-stained sections showing interstitial fibrosis (blue) in Dox-treated WT (E) but not Dox-treated sFas mice (F).
sFas Transgenic Mice Are Resistant to Dox-Induced Cardiac Dysfunction. At the starting of the Dox administration, all the experimental animals showed the same of left ventricular dimension and the percentage of fraction shortening, and there were no differences in these parameters between saline-treated WT and sFas mice (data not shown). Shown in Fig. 3A are representative two-dimensional M-mode tracings of LV wall motion from a WT and a sFas animal at baseline and after Dox treatment, respectively. In contrast to the LV wall motion at baseline, the waveforms from the Dox-treated animal clearly demonstrated blunted anterior and posterior wall motion, consistent with decreased fractional shortening (p < 0.05; Fig. 3B), indicating a decrease in myocardial contractility in WT mice after Dox treatment. In contrast, fractional shortening from the Dox-treated sFas mice showed a slight decrease but was comparable with that of baseline (Fig. 3B). In contrast to the LV end-diastolic dimension at baseline, Dox treatment induced a slight increase but did not result in significant change in LV end-diastolic dimension in both WT and sFas mice (Fig. 3C). These results indicate that mice with cardiac-specific expression of sFas seem to be resistant to Dox-induced cardiac dysfunction.
Fig. 3.

Echocardiographic analysis of cardiac function of mice treated with Dox administration. A, representative two-dimensional M-mode tracings of LV wall motion in WT and sFas mice given Dox. B, fractional shortening normalized to the baseline measurements in WT mice showing progressively declined after Dox injection, whereas no significant effects were observed in Dox-treated sFas mice. C, LVEDDs did not significantly change in the two groups after Dox treatment. *, p < 0.05 versus baseline; #, p < 0.05 versus Dox-treated sFas mice; n = 5 per group.
Dox-Mediated Induction of FasL Is Reduced in sFas Mice. Dox has been shown recently to activate FasL transcription via the nuclear factor of activated T-lymphocytes (NFAT) signaling mechanism in vitro (Kalivendi et al., 2005). We examined whether FasL was induced after Dox administration in vivo. RT-PCR analysis was performed to assess FasL mRNA levels in cardiac tissue of WT and sFas mice treated with saline or Dox. As demonstrated in Fig. 4, FasL mRNA levels are significantly induced in Dox-treated WT animals in comparison with saline-treated controls, whereas sFas mice showed less levels of FasL mRNA transcript in comparison with those in WT mice after Dox administration.
Fig. 4.

Dox-mediated induction of FasL is attenuated in sFas transgenic mice. A, FasL mRNA in the myocardium of saline- and Dox-treated WT and sFas mice were assayed by RT-PCR. B, RT-PCR data were quantitated by densitometric analysis and normalized against β-actin. n = 5 per group; *, p < 0.01 versus saline-treated WT and sFas mice; #, p < 0.05 versus Dox-treated sFas mice.
Dox-Induced Cell Death in the Myocardium Is Attenuated by Expression of sFas. Administration of Dox significantly increased the number of TUNEL-positive cells in the myocardium of WT mice (Fig, 5A), whereas only a few of the apoptotic cells were present in the myocardium of Doxtreated sFas mice (Fig. 5B). Quantification of TUNEL-positive cells in the myocardium demonstrated that the number of apoptotic cells was significantly reduced in the myocardium of Dox-treated sFas mice compared with Dox-treated WT animals (Fig. 5E). In hearts from saline-treated mice, there were only a few apoptotic cells observed in the myocardium of both WT and sFas mice (data not shown).
Fig. 5.

Dox-induced cell death in the myocardium is attenuated by expression of sFas. A and B, representative photomicrographs of TUNEL-stained sections obtained from Dox-treated WT mice and Dox-treated sFas mice. Blue staining, TUNEL-positive cells. C and D, representative photomicrographs of immunohistochemical staining demonstrated a strong immunoreactivity for PARP p85 fragment in the myocardial sections from Dox-treated WT mice compared with very little immunoreactivity in the myocardium of Dox-treated sFas mice. Immunoreactivity was visualized with diaminobenzidine (brown). E and F, histograms showing a marked decrease in TUNEL-positive cells and PARP p85 fragment immunoreactivity, respectively, in the myocardium of Dox-treated sFas mice compared with Dox-treated WT mice. n = 5 per group; *, p < 0.01 versus saline-treated WT and sFas mice; #, p < 0.05 versus Dox-treated sFas mice.
The proapoptotic signal triggered by FasL is very rapidly propagated through sequential activation of caspases that result in cleavage of PARP into two fragments, i.e., p85 and p25. Detection of caspase-3 cleavage fragments of PARP has been established as a hallmark of apoptosis (Duriez and Shah, 1997). We observed a significant increase in PARP p85 fragment-positive cells after Dox administration in the myocardium of WT mice (Fig. 5, C and F). This increase was significantly reduced in the myocardium of sFas mice (Fig. 5, D and F). Therefore, these data indicate that myocardial histological damage induced by Dox is significantly attenuated by targeted cardiac-specific expression of sFas.
Expression of sFas Ameliorated Dox-Induced Cardiac Oxidative-Nitrosative Stress. Previous work suggested that Dox cardiotoxicity involved the increased formation of NO, superoxide, and, therefore, peroxynitrite, enhancing oxidative-nitrosative stress to the heart (Weinstein et al., 2000; Chaiswing et al., 2004; Andreadou et al., 2007). Therefore, superoxide generation in the hearts of each group was estimated as an indicator of tissue oxidative stress. As shown in Fig. 6A, the myocardial superoxide contents were similar in saline-treated WT and sFas mice. In contrast, there was a marked elevation of superoxide generation in WT hearts treated with Dox, and this elevation was significantly inhibited by cardiac-targeted expression of sFas. To determine the effect of sFas on the generation of intracellular reactive oxygen species, the H9c2 cells were pretreated with 10 μg of sFas and incubated with 2 μM Dox for 6 h. We used the fluorescent probe, dihydroethidium, which has been used to detect intracellular superoxide formation (Benov et al., 1998). Figure 6B shows the intracellular red fluorescence because of the intercalation of ethidium into DNA. Dox-induced enhancement in ethidium fluorescence was inhibited by the presence of sFas (Fig. 6C), indicating an overall reduced oxidative stress.
Fig. 6.

Dox-induced generation of ROS is inhibited by cardiac-specific expression of sFas. A, Dox-induced superoxide generation in myocardial homogenates was determined by luminol-mediated chemiluminescence assay after addition of 200 ng/ml PMA. Superoxide production was expressed as the light intensity per milligram of protein. *, p < 0.01 versus saline-treated WT and sFas controls; #, p < 0.05 versus Doxtreated WT mice; n = 3 per group. B and C, sFas inhibited Dox-induced oxidative stress as measured by DHE fluorescence. H9c2 cells treated with Dox (2 μM) with or without sFas (10 μg/ml) as described under Materials and Methods. After 6 h of incubation, the medium was aspirated, and cells were washed with Dulbecco's phosphate-buffered saline and incubated with 10 μM DHE for 30 min. Representative microphotographs of differential interference contrast (DIC) images and DHE-stained images are shown in B. The red fluorescence indicates positive staining. C, intensity of fluorescence was measured by a fluorescence spectrophotometer and averaged from three samples for each group. *, p < 0.01 versus untreated control; #, p < 0.05 versus Dox-treated cells.
Peroxynitrite, a potent oxidant and nitrating intermediate, is a product of the reaction of superoxide and NO derived from iNOS. To evaluate the possible involvement of nitrosative damage in the pathogenesis of the cardiomyopathy after Dox treatment and the effect of cardiac-targeted expression of sFas on this process, we performed immunohistochemistry to examine the production of peroxynitrite by detecting nitrotyrosine residues in proteins with the 3-NT antibody. As shown in Fig. 7A, 3-NT levels were highly elevated in the myocardium of Dox-treated WT mice compared with the myocardium of the Dox-treated sFas mice, where only a very weak signal of 3-NT could be detected. There was no positive staining of 3-NT in the myocardium of both saline-treated WT and sFas mice. Quantification of the 3-NT staining revealed a drastic reduction in nitrotyrosine formation in the myocardium of the Dox-treated sFas mice compared with Dox-treated WT mice (Fig. 7B).
Fig. 7.

DOX-induced nitrotyrosine formation and iNOS expression in heart are attenuated by cardiac-specific expression of sFas. A, representative photomicrographs of immunohistochemical staining revealed a strong immunoreactivity for 3-NT in the myocardial section obtained from Dox-treated WT mice compared with less immunoreactivity in the myocardial section from Dox-treated sFas mice. Immunoreactivity was visualized with diaminobenzidine (brown). There was no positive staining for 3-NT in myocardial sections from saline-treated controls. B, histogram showing a significant decrease of the immunoreactivity for nitrotyrosine in the myocardium of Dox-treated sFas mice compared with Dox-treated WT mice. #, p < 0.01, n = 5 per group. C, iNOS mRNA expression in the myocardium of saline-, Dox-treated WT and sFas mice were assayed by RT-PCR, and the bands of iNOS expression were quantitated by densitometric analysis and normalized by β-actin. n = 5 per group; *, p < 0.01 versus saline-treated WT and sFas mice; #, p < 0.05 versus Dox-treated WT mice. D, circulating levels of nitrite and nitrate, in saline-, Dox-treated WT and sFas mice. Total nitrated proteins significantly increased in the serum of Dox-treated WT mice compared with the saline-treated controls and Dox-treated sFas mice. *, p < 0.01 versus saline-treated controls; #, p < 0.05 versus Dox-treated WT mice; n = 5 per group.
To elucidate the changes of iNOS expression in the hearts of each group, total cardiac RNA was subjected to RT-PCR for mouse-specific iNOS and β-actin as a control. Cardiac iNOS mRNA was significantly increased in the myocardium of WT mice after Dox treatment in comparison with saline-treated controls, whereas this increase in iNOS expression was significantly suppressed in the myocardium of Dox-treated sFas mice (Fig. 7C). Nitrate and nitrite analysis of the plasma showed that the levels of nitrate and nitrite after Dox treatment were significantly less in sFas mice than the levels found in WT mice, although the magnitude of the reduction in sera levels of nitrate and nitrite did not reach exactly the levels observed in saline-treated controls (Fig. 7D). Together, these results indicate that the neutralization of FasL by cardiac-specific expression of sFas attenuated Dox-induced cardiac oxidative-nitrosative stress.
Dox-Mediated Induction of Cytokines is Suppressed by Expression of sFas. Cytokines, such as TNF-α, IL-1β, and Il-6, have been shown to be involved in different cardiac diseases and heart failure (Mann, 2002). To assess whether these cytokines were induced after Dox administration, we performed RT-PCR analysis to evaluate TNF-α, IL-1β, and IL-6 mRNA levels in cardiac tissue of Dox-treated WT and sFas mice and saline-treated controls. As shown in Fig. 8, TNF-α, IL-1β, and IL-6 mRNA levels are significantly elevated in the myocardium of WT mice after Dox administration in comparison with those of the saline-treated controls. The mRNA levels of TNF-α, IL-1β, and IL-6 in the Dox-treated sFas mice were significantly lower in comparison with those of Dox-treated WT mice, suggesting that the induction of these cytokines caused by Dox treatment involved Fas/FasL signaling.
Fig. 8.

Cardiac-specific expression of sFas suppresses myocardial proinflammatory cytokine production after Dox treatment. A, expression of TNF-α, IL-1β, and IL-6 mRNA in the myocardium of saline-, Dox-treated WT and sFas mice were assayed by RT-PCR. B to D, RT-PCR Bands were quantitated by densitometric analysis and normalized by β-actin. n = 5 per group; *, p < 0.05 versus saline-treated controls; #, p < 0.05 versus Dox-treated sFas mice.
Discussion
Cardiac toxicity is a major dose-limiting factor for application of doxorubicin as a cancer chemotherapeutic agent. Although the precise biochemical mechanisms of Dox cardiotoxicity remain uncertain, it has been demonstrated that the cardiac toxicity associated with Dox administration is mediated, at least in part, by induction of oxidative stress (Weinstein et al., 2000; Chaiswing et al., 2004; Andreadou et al., 2007) and cardiac cell apoptosis (Kalyanaraman et al., 2002; Takemura and Fujiwara, 2007). In a recent study, in vitro experiments demonstrated that ROS generated from Dox metabolism in mitochondrial activates NFAT signaling, which leads to the initiation of the apoptotic cascade in cardiac cells with FasL transcription (Kalivendi et al., 2005). In the present study, we demonstrated that expression and secretion of sFas by the myocardium to decoy of FasL results in lower production of proinflammatory cytokines, fewer TUNEL-positive cells, and less formation of superoxide and peroxynitrite in the myocardium, leading to preserved cardiac function in the experimental mouse model of Dox cardiotoxicity. This study provides direct in vivo evidence for the involvement of Fas/FasL signaling in Dox cardiotoxicity.
Dox-induced cardiotoxicity has been attributed, at least in part, to cardiac cell apoptosis (Kalyanaraman et al., 2002; Takemura and Fujiwara, 2007). FasL is a well characterized apoptosis inducer that binds to its cell surface receptor Fas, resulting in sequential activation of caspases, forming the death-inducing signaling complex that triggers apoptosis (Suda et al., 1993). Previous studies have demonstrated a dose-related increase in the expression of Fas antigen and cardiac myocyte and endothelial cell apoptosis with Dox treatment (Nakamura et al., 2000; Lien et al., 2006). For our investigation, a cumulative Dox dose of 40 mg/kg was given to the mice over 7 weeks. The animals showed heart failure-like syndrome, including lethargy, ruffled hair, respiratory stress, and pleural effusions. To confirm the presence of apoptosis, we measured cell death using the TUNEL assay and immunohistochemistry. Large numbers of TUNEL-positive cells were found in the myocardium of Dox-treated WT mice, whereas fewer TUNEL-positive cells were observed in the myocardium of Dox-treated sFas mice. The TUNEL-positive cells were cardiomyocytes, endothelial cells, and infiltrating inflammatory cells that were consistent with the findings of previous in vivo studies (Nakamura et al., 2000; Wu et al., 2002). To further assess a link between the induction of apoptosis and caspase-3-activated PARP cleavage, we performed immunostaining of myocardial sections using antibody against the p85 fragment of PARP. The appearance of the 85-kDa fragment of PARP in the cardiomyocyte nuclei coincided with a significant increase of TUNEL-positive cells in the myocardium of Dox-treated WT mice. In contrast, fewer positive cells were observed in the myocardial sections obtained from Dox-treated sFas mice. This observation is in concordance with our recent data showing sFas inhibition of caspase-3/7 activities and activation of PARP in the myocardium (Niu et al., 2008). These data together indicate that sFas expression protects against Dox-induced cardiac dysfunction, at least in part by preventing apoptotic cell death of cardiac cells, further supporting the important role of FasL expression in Dox cardiotoxicity.
Dox-induced cardiac cell apoptosis has been attributed to the production of ROS (Chaiswing et al., 2004), which has been shown to activate FasL transcription (Kalivendi et al., 2005). Activation of the Fas receptor has been shown to be associated with rapid generation of ROS (Gulbins et al., 1996; Sato et al., 2004; Medan et al., 2005). The generation of ROS such as peroxynitrite, the reaction product of NO and superoxide, has been implicated in the pathogenesis of Dox-induced myocardial dysfunction (Weinstein et al., 2000; Chaiswing et al., 2004; Andreadou et al., 2007). Although studies suggest that NO by itself is an antioxidant molecule (Wink et al., 1993), increased NO production via iNOS has been implicated in cardiomyocyte oxidative damage, apoptosis, and/or necrosis through peroxynitrite formation (Ferdinandy et al., 2000; Zhang et al., 2007; Niu et al., 2008). Peroxynitrite-related cardiac protein nitration, myofibrillar thinning, and irregular striations patterns have been documented to be responsible for cardiac dysfunction in doxorubicin-treated mice (Mihm et al., 2002). Our results showed that superoxide generation in the heart was greatly enhanced after Dox treatment. This was confirmed in cultured H9c2 cells in vitro by measurement of intracellular reactive species using fluorescence probe dihydroethidium. We also found increased iNOS expression, elevation of nitrite and nitrate, and formation of peroxynitrite in WT mice after Dox treatment. These findings are in accordance with the previous reports that NO-derived oxidative species and particularly peroxynitrite are formed during Dox induced cardiotoxicity (Weinstein et al., 2000; Chaiswing et al., 2004; Andreadou et al., 2007). In contrast, these changes were inhibited by neutralization of FasL through cardiac-targeted expression of sFas, resulting in protection of the heart against the toxic effect of Dox. This suggests that neutralization of FasL by cardiac-targeted expression of sFas decreased the levels of Dox-induced cardiac oxidative stress. Our findings are supported by recent studies showing that FasL induces ROS generation by activation of NADPH oxidase (Wang et al., 2008). Thus, we present the first in vivo evidence that FasL is an important mediator in Dox-associated cardiotoxicity by generating reactive oxygen and nitrogen species.
Increased iNOS expression and nitrotyrosine formation have been observed in mice cardiomyocytes after Dox treatment (Mihm et al., 2002). Higher NO production via iNOS has been shown to be associated with the pathogenesis of cardiomyopathy and heart failure. However, the concept that deficiency of iNOS may enhance Dox-induced acute cardiotoxicity in mice was also documented recently (Cole et al., 2006). We did detect an increase in iNOS mRNA and nitrotyrosine formation in the hearts of WT mice treated with Dox, and these increases in iNOS expression, NO production, superoxide generation, and nitrotyrosine formation were accompanied by a marked loss of cardiac mechanical function, whereas these changes were attenuated in sFas mice with preservation of cardiac function. In the presence of Dox, not only NO synthesis is elevated, but also the generation of myocardial superoxide. Superoxide can react with NO to reduce its bioavailability, leading to formation of peroxynitrite, a highly toxic reactive species. Based on our data, it seems likely that elevated NO and reactive oxygen species production would contribute to chronic Dox cardiotoxicity, suggesting that inhibition of production of either of the peroxynitrite precursors (NO or superoxide) would have a beneficial effect on myocardial oxidative stress and contractile function.
Although Fas is best known for its involvement in the regulation of apoptosis, Fas ligation also contributes to activation of NF-κB (Ponton et al., 1996), and it has been shown that interruption of Fas/FasL signaling suppressed NF-κB activation and cytokine expression (Shiraishi et al., 2002). We recently showed that NF-κB activation and production of proinflammatory cytokines were suppressed in sFas transgenic mice (Niu et al., 2008). A recent study has reported that NF-κB activation is necessary for cardiomyocyte apoptosis evoked by Dox (Wang et al., 2002). In the present study, we found that Dox administration leads to a substantial increase in the levels of TNF-a, IL-1β, and IL-6 in the myocardial tissues of WT animals after Dox administration. In contrast, the levels of these proinflammatory cytokines were significantly lower in the myocardial tissues of Dox-treated sFas animals. Although there is increasing evidence that short-term expression of proinflammatory cytokines, such as TNF-α, can be beneficial to the heart (Wilson et al., 2004), our data demonstrated that increases in proinflammatory cytokine production are accompanied by a marked loss of cardiac mechanical function in chronic Dox cardiotoxicity. Taken together, these results suggest that not only the direct effect of oxidative stress but also the inflammatory response induced by FasL plays a significant role in Dox-associated cardiotoxicity. Because iNOS expression is induced upon cytokine stimulation (Zeng et al., 2005), the inhibition of NF-κB activation and proinflammatory cytokine production by neutralization of FasL may explain the decreased levels of NO in sFas mice. In agreement with this finding, it has been reported that impairment of the Fas-FasL interaction resulted in decreased NO production (Martins et al., 2001). In this study, we also examined FasL gene expression, and there was a substantial decrease of FasL transcript in the hearts of sFas mice compared with WT mice after Dox treatment. The mechanism(s) responsible for suppression of FasL expression in sFas mice is not clear. It is likely that reduced ROS generation and inactivation of NF-κB may attenuate Dox-induced activation of NFAT signaling mechanism, resulting in decreased FasL expression in the hearts of sFas animals.
In conclusion, the present study clearly demonstrates that sFas-induced protective effect against Dox-induced cardiac toxicity in mice is attributable to attenuation of ROS generation, peroxynitrite formation, inhibition of myocyte apoptosis, and reduction of proinflammatory cytokine production. These findings demonstrate that FasL is a key mediator of Dox-induced cardiac toxicity and, therefore, may provide a novel therapeutic strategy in clinic to reduce Dox-induced cardiotoxicity in patients.
This work was supported by the National Institutes of Health [Grant HL69458].
doi:10.1124/jpet.108.146423.
ABBREVIATIONS: Dox, doxorubicin; FasL, Fas ligand; sFas, soluble Fas; WT, wild-type; LVEDD, left ventricular end-diastolic dimension; 3-NT, 3-nitrotyrosine; PARP, poly(ADP-ribose) polymerase; PMA, phorbol 12-myristate 13-acetate; ROS, reactive oxygen species; DHE, dihydro-β-erythroidine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; TNF, tumor necrosis factor; IL, interleukin; iNOS, inducible NO synthase; RT, reverse transcriptase; PCR, polymerase chain reaction; NO, nitric oxide; NFAT, nuclear factor of activated T-lymphocytes; NF, nuclear factor; LV, left ventricle.
References
- Andreadou I, Sigala F, Iliodromitis EK, Papaefthimiou M, Sigalas C, Aligiannis N, Savvari P, Gorgoulis V, Papalabros E, and Kremastinos DT (2007) Acute doxorubicin cardiotoxicity is successfully treated with the phytochemical oleuropein through suppression of oxidative and nitrosative stress. J Mol Cell Cardiol 42 549-558. [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, and Fridovich I (1998) Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25 826-831. [DOI] [PubMed] [Google Scholar]
- Chaiswing L, Cole MP, St Clair DK, Ittarat W, Szweda LI, and Oberley TD (2004) Oxidative damage precedes nitrative damage in Adriamycin-induced cardiac mitochondrial injury. Toxicol Pathol 32 536-547. [DOI] [PubMed] [Google Scholar]
- Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, and Mountz JD (1994) Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 263 1759-1762. [DOI] [PubMed] [Google Scholar]
- Cole MP, Chaiswing L, Oberley TD, Edelmann SE, Piascik MT, Lin SM, Kiningham KK, and St Clair DK (2006) The protective roles of nitric oxide and superoxide dismutase in Adriamycin-induced cardiotoxicity. Cardiovasc Res 69 186-197. [DOI] [PubMed] [Google Scholar]
- Duriez PJ and Shah GM (1997) Cleavage of poly (ADP-ribose) polymerase: a sensitive parameter to study cell death. Biochem Cell Biol 75 339-349. [PubMed] [Google Scholar]
-
Ferdinandy P, Danial H, Ambrus I, Rothery RA, and Schulz R
(2000) Increased production of NO and superoxide
(
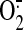 ) that interact to
produce peroxynitrite, a powerful oxidizing and nitrating agent that is a
major contributor to cytokine-induced myocardial contractile failure.
Circ Res 87
241-247. [DOI] [PubMed] [Google Scholar]
) that interact to
produce peroxynitrite, a powerful oxidizing and nitrating agent that is a
major contributor to cytokine-induced myocardial contractile failure.
Circ Res 87
241-247. [DOI] [PubMed] [Google Scholar] -
Gulbins E, Brenner B, Schlottmann K, Welsch J, Heinle H,
Koppenhoefer U, Linderkamp O, Coggeshall KM, and Lang F (1996)
Fas-induced programmed cell death is mediated by a Ras-regulated
 synthesis.
Immunology 89
205-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
synthesis.
Immunology 89
205-212. [DOI] [PMC free article] [PubMed] [Google Scholar] - Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals, 7th ed, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC.
- Iwata R, Ito H, Hayashi T, Sekine Y, Koyama N, and Yamaki M (1995) Stable and general-purpose chemiluminescent detection system for horseradish peroxidase employing a thiazole compound enhancer and some additives. Anal Biochem 231 170-174. [DOI] [PubMed] [Google Scholar]
- Kalivendi SV, Konorev EA, Cunningham S, Vanamala SK, Kaji EH, Joseph J, and Kalyanaraman B (2005) Doxorubin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. Biochem J 389 527-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, and Kotamraju S (2002) Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem 234-235 119-124. [PubMed] [Google Scholar]
- Li Y, Takemura G, Kosai K, Takahashi T, Okada H, Miyata S, Yuge K, Nagano S, Esaki M, Khai NC, et al. (2004) Critical roles for the Fas/Fas ligand system in postinfarction ventricular remodeling and heart failure. Circ Res 95 627-636. [DOI] [PubMed] [Google Scholar]
- Lien YC, Daosukho C, and St Clair DK (2006) TNF receptor deficiency reveals a translational control mechanism for adriamycin-induced Fas expression in cardiac tissues. Cytokine 33 226-230. [DOI] [PubMed] [Google Scholar]
- Mann DL (2002) Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res 91 988-998. [DOI] [PubMed] [Google Scholar]
- Martins GA, Petkova SB, MacHado FS, Kitsis RN, Weiss LM, Wittner M, Tanowitz HB, and Silva JS (2001) Fas-FasL interaction modulates nitric oxide production in Trypanosoma cruzi-infected mice. Immunology 103 122-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medan D, Wang L, Toledo D, Lu B, Stehlik C, Jiang BH, Shi X, and Rojanasakul Y (2005) Regulation of Fas (CD95)-induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J Cell Physiol 203 78-84. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Yu F, Weinstein DM, Reiser PJ, and Bauer JA (2002) Intracellular distribution of peroxynitrite during doxorubicin cardiomyopathy: evidence for selective impairment of myofibrillar creatine kinase. Br J Pharmacol 135 581-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H, and Koh E (2000) Fasmediated apoptosis in adriamycin-induced cardiomyopathy in rats: in vivo study. Circulation 102 572-578. [DOI] [PubMed] [Google Scholar]
- Niu J, Azfer A, Deucher MF, Goldschmidt-Clermont PJ, and Kolattukudy PE (2006) Targeted cardiac expression of soluble Fas prevents the development of heart failure in mice with cardiac-specific expression of MCP-1. J Mol Cell Cardiol 40 810-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Azfer A, and Kolattukudy PE (2008) Protection against lipopolysaccharide-induced myocardial dysfunction in mice by cardiac-specific expression of soluble Fas. J Mol Cell Cardiol 44 160-169. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, and Yorek MA (2005) Oxidative-nitrosative stress and poly (ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes 54 3435-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perik PJ, Van der Graaf WT, De Vries EG, Boomsma F, Messerschmidt J, Van Veldhuisen DJ, Sleijfer DT, and Gietema JA (2006) Circulating apoptotic proteins are increased in long-term disease-free breast cancer survivors. Acta Oncol 45 175-183. [DOI] [PubMed] [Google Scholar]
- Pichon MF, Labroquère M, Rezaï K, and Lokiec F (2006) Variations of soluble Fas and cytokeratin 18-Asp 396 neo-epitope in different cancers during chemotherapy. Anticancer Res 26 2387-2392. [PubMed] [Google Scholar]
- Ponton A, Clément MV, and Stamenkovic I (1996) The CD95 (APO-1/Fas) receptor activates NF-κB independently of its cytotoxic function. J Biol Chem 271 8991-8995. [DOI] [PubMed] [Google Scholar]
- Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K, Takada K, Oku T, Kawano Y, Okamoto T, et al. (2004) Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol 173 285-296. [DOI] [PubMed] [Google Scholar]
- Setsuta K, Seino Y, Ogawa T, Ohtsuka T, Seimiya K, and Takano T (2004) Ongoing myocardial damage in chronic heart failure is related to activated tumor necrosis factor and Fas/Fas ligand system. Circ J 68 747-750. [DOI] [PubMed] [Google Scholar]
- Shiraishi H, Toyozaki T, Tsukamoto Y, Saito T, Masuda Y, Hiroshima K, Ohwada H, Kobayashi N, and Hiroe M (2002) Antibody binding to Fas ligand attenuates inflammation cell infiltration and cytokine secretion, leading to reduction of myocardial infarct area and reperfusion injury. Lab Invest 82 1121-1129. [DOI] [PubMed] [Google Scholar]
- Singal PK and Iliskovic N (1998) Doxorubicin-induced cardiomyopathy. N Engl J Med 339 900-905. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Golstein P, and Nagata S (1993) Molecular cloning and expression of the Fas ligand: a novel member of the tumor necrosis factor family. Cell 75 1169-1178. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhou Z, and Kang YJ (2001) Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res 61 3382-3387. [PubMed] [Google Scholar]
- Takemura G and Fujiwara H (2007) Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis 49 330-352. [DOI] [PubMed] [Google Scholar]
- Tamakoshi A, Nakachi K, Ito Y, Lin Y, Yagyu K, Kikuchi S, Watanabe Y, Inaba Y, and Tajima K (2008) Soluble Fas level and cancer mortality: findings from a nested case-control study within a large-scale prospective study. Int J Cancer 123 1913-1916. [DOI] [PubMed] [Google Scholar]
- Wang L, Azad N, Kongkaneramit L, Chen F, Lu Y, Jiang BH, and Rojanasakul Y (2008) The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J Immunol 180 3072-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, and Kalyanaraman B (2002) Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem J 367 729-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein DM, Mihm MJ, and Bauer JA (2000) Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther 294 396-401. [PubMed] [Google Scholar]
- Wilson EM, Diwan A, Spinale FG, and Mann DL (2004) Duality of innate stress responses in cardiac injury, repair, and remodeling. J Mol Cell Cardiol 37 801-811. [DOI] [PubMed] [Google Scholar]
- Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, and Mitchell JB (1993) Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc Natl Acad Sci U S A 90 9813-9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Ko YS, Teng MS, Ko YL, Hsu LA, Hsueh C, Chou YY, Liew CC, and Lee YS (2002) Adriamycin-induced cardiomyocyte and endothelial cell apoptosis: in vitro and in vivo studies. J Mol Cell Cardiol 34 1595-1607. [DOI] [PubMed] [Google Scholar]
- Zeng C, Lee JT, Chen H, Chen S, Hsu CY, and Xu J (2005) Amyloid-beta peptide enhances tumor necrosis factor-alpha-induced iNOS through neutral sphingomyelinase/ceramide pathway in oligodendrocytes. J Neurochem 94 703-712. [DOI] [PubMed] [Google Scholar]
- Zhang P, Xu X, Hu X, van Deel ED, Zhu G, and Chen Y (2007) Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res 100 1089-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]