

Abstract
We have reported that the major endogenous estrogen, 17β-estradiol (E2), protects against oxidative injury during ethanol withdrawal (EW) in a cultured hippocampal cell line (HT22). Here, we investigated whether the pro-oxidant nature of EW mediates opening of the mitochondrial membrane permeability transition pore (PTP) in a manner protected by E2. Excess PTP opening provokes mitochondrial membrane swelling (MMS) and the collapse of membrane potential (ΔΨm). HT22 cells were collected at the end of ethanol exposure (100 mM) for 24 h or at 4 h of EW to assess MMS by monitoring absorbance decline at 540 nm and to assess ΔΨm using flow cytometry. Protective effects of E2 on PTP were compared with an antioxidant butylated hydroxytoluene (BHT) and an E2 analog, ZYC26 [(3-hydroxy-2-adamantyl(1)-4-methyl-estra-1,3,5(10)-17-one], with higher antioxidant potency than E2. To assess cellular consequences of PTP opening, effects of a PTP inhibitor (cyclosporin A) on EW-induced cell death were assessed using the calcein assay. Major findings were that: 1) EW resulted in rapid MMS and ΔΨm collapse; 2) cyclosporin A attenuated EW-induced cell death; and 3) E2 treatment restricted to the EW phase protected against the PTP opening more prominently than BHT and to a similar degree to ZYC26. These findings suggest that EW provokes PTP opening partly but not entirely through the pro-oxidant nature and that E2 counteracts EW-associated factors to protect against the PTP opening.
Ethanol withdrawal (EW) refers to abrupt termination of long-term ethanol abuse. EW motivates alcoholics to relapse into ethanol abuse because of its discomfort and disorders, but there is little mechanistic insight into this important clinical problem. This study was undertaken to gain a better understanding of the mechanisms by which EW damages mitochondrial membranes in a cellular model of ethanol/EW. We used the immortalized hippocampal cell line (HT22 cells) because this cell line is advantageous to access an oxidative mechanism. HT22 cells lack ionotropic glutamate receptors; thus, the cellular injury is unlikely through excitotoxicity involving glutamate receptors (Zaulyanov et al., 1999). Instead, HT22 cells contain the glutamate/cystine antiporter, which is required for the delivery of cystine into neuronal cells for the synthesis of an endogenous antioxidant glutathione. Therefore, HT22 cellular injury is often associated with a reduction in endogenous antioxidant capacity (Tan et al., 1998). Using this cell line, we have demonstrated that EW is pro-oxidant by provoking protein oxidation and lipid peroxidation to a greater degree than ethanol per se and that 17β-estradiol (E2) treatment protects against the oxidative stress (Jung et al., 2006). In the current study, we tested whether EW damages mitochondrial membranes through its pro-oxidant nature in an E2-preventable manner.
We focused on mitochondrion because this organelle is one of the major subcellular targets of ethanol intoxication and withdrawal (Mansouri et al., 2001; Miñana et al., 2002). Reactive oxygen species (ROS) produced during ethanol metabolism altered mitochondrial function (Mansouri et al., 2001; Miñana et al., 2002), and EW increased mitochondrial permeability in response to a superoxide generator (phenazine methosulfate) and oxidative stress (French and Todoroff, 1971; Hosein et al., 1980). In addition, acute administration of ethanol (5 g/kg i.p.) to mice depleted mitochondrial DNA in heart, liver, and brain (Mansouri et al., 2001). The mitochondrial susceptibility seems to be in part because of compromised endogenous antioxidant capacity based on a study in which ethanol-treated hepatocytes showed depletion of glutathione (Fernández-Checa et al., 1998).
Mitochondrial membranes contain a group of proteins that forms mitochondrial permeability transition pores (PTPs). PTPs regulate permeability to electrolytes, nucleotides, and metabolic substrates, all of which are essential for ATP production. Prolonged opening of PTP perturbs the electrochemical gradient for H+ (ΔΨm), driving force for oxidative phosphorylation, resulting in disrupted ATP production. Oxidative or apoptotic stress causes excess opening of PTP, which in turn permits diffusion of water and electrolytes across mitochondrial membranes. As a consequence, ΔΨm collapses, and oxidative phosphorylation of ATP fails (Stuart, 2002). Because of such an intimate relationship between PTP and ΔΨm, ΔΨm is often measured as a typical marker of PTP. Mitotoxic effects of ethanol have been reported in a study where the inhibition of PTP opening by cyclosporin A prevented ethanol-induced cell death (Miñana et al., 2002). However, most studies have not systematically differentiated effects of ethanol exposure and EW on mitochondria; it is not clear whether mitochondrial damages were because of ethanol per se, or EW, or both. The differentiation is important because toxic effects of EW are not necessarily identical to those of ethanol and can cause more brain damage than ethanol per se (Phillips and Cragg, 1983; Jung et al., 2004).
We recently observed that EW provoked oxidation of mitochondrial proteins in ovariectomized rats and E2 implantation markedly prevented the protein oxidation (Jung et al., 2008). Others also reported that ovariectomy caused an increase in peroxide production by mitochondria in a manner prevented by E2 (Borrás et al., 2003), suggesting that estrogen plays a role in reducing oxidative burden in mitochondria (Simpkins et al., 2005). Despite abundant evidence of mitoprotection by estrogen, a mechanism by which E2 protects against mitotoxic EW remains uncertain. Therefore, in part I of this study, we assessed whether the pro-oxidant nature of EW mediates mitotoxicity. In part II of this study, we examined mitoprotective effects of E2 and its potential antioxidant mechanisms by comparing with an antioxidant butylated hydroxytoluene (BHT) and an E2 analog, ZYC26, which previously showed higher antioxidant potency than E2 during EW (Jung et al., 2006).
Materials and Methods
Reagents
E2 was purchased from Steraloids (Newport, RI). ZYC26 was made in our laboratories using methods described previously (Perez et al., 2006). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless indicated otherwise.
Cell Culture
HT22 cells were obtained from David Schubert (Salk Institute, San Diego, CA). The HT22 cell line was originally selected from HT4 cells based on glutamate sensitivity. HT4 cells were immortalized from primary hippocampal neurons using a temperature-sensitive simian virus-40 T antigen (Morimoto and Koshland, 1990). HT22 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% charcoal-stripped fetal bovine serum (HyClone Laboratories, Logan, UT) and gentamicin (50 μg/ml), at 37°C in an atmosphere containing 5% CO2 and 95% air. Protein concentrations were determined by the Bradford method (Bradford, 1976) using bovine serum albumin at concentrations ranging from 0.063 to 1 mg/ml as a standard curve.
Ethanol Treatment
HT22 cells (4000 cells/well) in 100 μl of cell culture medium were plated into 96-well culture plates or Petri dishes. On the following day, the cells were exposed to control media dimethyl sulfoxide (DMSO) or 100 mM ethanol for 24 h. The culture plates or Petri dishes were tightly sealed with parafilm immediately after ethanol treatment to prevent ethanol evaporation. We have maintained an intended ethanol concentration for 24 h using this method (Jung et al., 2006) (see Results). For the continuous ethanol exposure condition, cells were collected at the end of the 24-h ethanol exposure. In withdrawal experiments, the ethanol-containing medium was replaced with control media for 4 h after the 24-h ethanol exposure (Mostallino et al., 2004). At the end of the 4 h of EW, cells were collected to assess PTP opening and cell viability. An E2, BHT, or ZYC26 stock solution was prepared at a concentration of 0.1 or 1 μM in DMSO, and the compounds were administered to cells either during the entire period of ethanol exposure/withdrawal or during the EW phase. There were no measurable effects of DMSO on PTP or cell viability under these conditions.
Assessment of PTP
Flow Cytometric Analysis of ΔΨm. HT22 cells (2 × 104 cells/ml) were cultured in 12-well culture plates. Cells were divided into four groups: control media, ethanol EW, and EW + E2 groups. On the following day, cells were treated with ethanol (100 mM) as mentioned above, and E2 (1 μM) was given when ethanol-containing media were replaced with control media to restrict E2 treatment to the EW phase. At the end of 24-h ethanol exposure or at 4 h of EW, the cells were washed with phosphate-buffered saline (PBS) twice. JC-1 solution (0.5 ml; 2.5 μg/ml) was then transferred into each well. The cells were incubated at 37°C in a 5% CO2 incubator for 20 min. Subsequently, the cells were digested with trypsin to lyse cells. Precipitates obtained from the suspension after centrifugation (3 min, room temperature, 800g) were resuspend in 2 ml of PBS followed by centrifugation. The cell pellets were suspended in 0.5 ml of PBS, which was ready for flow cytometric analysis. JC-1 aggregates in healthy mitochondria emit red fluorescence at 590 nm. JC-1 monomers that were leaked from stressed mitochondria emit green fluorescence at 530 nm. The red and green fluorescence were measured in the green (FL-1) and red (FL-2) channels of the flow cytometer, respectively. The cells were then immediately observed with a fluorescence microscope using a “dual-band pass” filter designed to simultaneously detect fluorescein and rhodamine or fluorescein and Texas Red.
Spectrometric Analysis of Mitochondrial Membrane Swelling. Mitochondrial membrane swelling (MMS) is often measured as an indication of PTP opening by monitoring absorbance decline at 540 nm (Ruiz-Meana et al., 2006). Intact mitochondria scatter light at 540-nm wavelength; mitochondrial swelling and rupture because of prolonged or excessive PTP opening reduce mitochondrial light scattering and absorbance. Using a differential centrifugation method, mitochondria were isolated from HT22 cells that were collected at the end of 24-h ethanol exposure or at 4 h of EW. The cells were homogenized in ice-cold isolation buffer (320 mM sucrose, 1 mM K2EDTA, 10 mM Tris-HCl) and centrifuged at 1330g for 5 min at 4°C. The pellets were resuspended in 0.5 volumes of isolation buffer and recentrifuged. The two supernatants were combined and centrifuged at 21,200g for 5 min. The resulting pellets were resuspended in 12% Percoll solution and centrifuged at 6900g for 10 min. The resulting soft pellets were then washed once with mitochondrial isolation buffer and centrifuged again at 6900g for 10 min. The precipitant mitochondrial pellets were suspended in medium containing phosphate, which induces MMS, and they ruptured more rapidly in vulnerable mitochondrial membranes than healthy mitochondrial membranes (Menze et al., 2005). The medium contained 250 mM sucrose, 10 mM Tris-MOPS, 0.05 mM EGTA, 5 mM pyruvate, 5 mM malate, and 1 mM phosphate, pH 7.4. Absorbance by this suspension was measured at 540 nm in a Beckman DU 640 spectrophotometer (Beckman Coulter, Fullerton, CA).
Calcein-Acetoxymethyl Ester Viability Assay
Cell viability was quantitated using the membrane-permeant calcein-acetoxymethyl ester (AM) dye (Invitrogen, Carlsbad, CA). Calcein-AM is a fluorogenic esterase substrate that easily permeates live cells that have esterase activity and membranes. Once hydrolysis of calcein-AM by intracellular esterases begins, it produces calcein, a strongly fluorescent compound that is well retained in the cell cytoplasm, which enables us to measure relative fluorescent units. HT22 cells were treated with ethanol and withdrawn as described above. Cyclosporin A (0.2 μM) treatment was restricted to the EW phase such that cyclosporin A was treated when ethanol-containing media were replaced with control media to test whether EW-induced PTP opening mediates cell death. Calcein-AM was treated 30 min before the end of EW. After the removal of the medium from the 96-well plates, the cells were rinsed once with PBS, pH 7.4, and incubated in a solution of 2.5 μM calcein-AM in PBS. Twenty minutes later, fluorescence was determined using a BioTek FL600 microplate reader (BioTek Instruments, Winooski, VT) with an excitation/emission filter set at 485/530 nm. Cell culture wells treated with methanol served as blanks. The results, obtained in relative fluorescent units, were expressed as the percentage of control media values. In HT22 cells, we have observed a linear relationship between the number of viable cells per well and measured calcein-AM fluorescence when viable cell numbers were between 300 and 5000 cells/well (r2 = 0.9991). HT22 cell death induced by excitotoxin in the calcein assay is consistent with previous reports using alternative viability methods, such as the colorimetric methyl-thiazole-tetrazolium test (Tan et al., 1998).
Ethanol Concentrations in the HT22 Cell Culture
Ethanol concentrations were measured to test whether E2, BHT, or ZYC26 per se alters ethanol kinetics. For ethanol exposure conditions, the cells were treated with control medium (DMSO) or 100 mM ethanol for 24 h. E2 (1 μM), BHT (1 μM), or ZYC26 (1 μM) was simultaneously treated with ethanol for 24 h. Cells were then collected at the end of the ethanol exposure. For EW conditions, the compounds were readministered when ethanol-containing media were replaced with control media, and the cells were collected at 4 h of EW. The 10 μl of sample solution was added to 200 μl of ice-cold 0.55 M perchloric acid. The media were neutralized with 200 μl of 0.6 M KOH containing 50 mM acetic acid. This solution precipitates the perchlorate ion and buffers the solution to approximately pH 5. After the samples were centrifuged, the resulting supernatant was used to measure ethanol concentrations using an enzymatic assay (Smolen et al., 1986). In brief, a 20-μl aliquot of the supernatant solution was pipetted into duplicate assay tubes and one blank tube. The assay solution consisted of 2.29 mM nicotinamide adenine, 30 units of yeast alcohol dehydrogenase, and 500 mM Tris-HCl, pH 8.8, in a total volume of 140 μl. The blank tube contained no alcohol dehydrogenase. The reaction mixture was incubated at room temperature for 30 min before measuring the absorbance of nicotinamide adenine dinucleotide formed at 340 nm in a Gilford 240 spectrophotometer. Ethanol concentrations were calculated from linear regression analysis of a standard curve of known ethanol concentrations.
Sample Size
Three sample replicates (n = 3) were used for the mitochondrial swelling assay in each group for each test. Five sample replicates (n = 5) were used for calcein-AM viability assay and ethanol concentrations. These experiments were repeated two to four times. Data are presented as average ± S.E.M. obtained from the sample replicates of the most recent test. For the flow cytometric analysis of ΔΨm and the fluorescence microscopic imaging, two sample replicates (n = 2) were used in each group. The experiments were repeated twice. Data from the most recent experiments are presented.
Statistical Analysis
For data illustrating absorbance decline (mitochondrial swelling), average absorbance values from three sample replicates were used for repeated (by minutes) measures one-way ANOVA. For calcein-AM cell viability assay, one-way ANOVA (by treatment) was conducted using five sample replicates in each group. When significance was found (P < 0.05), a post hoc Tukey's multiple comparison was conducted to determine difference between treatment conditions. The significance level for all data analysis was set at P < 0.05.
Results
Part I: Effects of Ethanol/EW on PTP
Effects of Ethanol/EW on MMS. Unhealthy mitochondrial membranes that have prolonged opening of PTP lose ability to scatter lights, resulting in absorbance decline at 540 nm. Repeated (by minutes) measures ANOVA indicated that MMS significantly differs between treatment conditions [F(2,38) = 19, P < 0.001] (Fig. 1). MMS occurred more rapidly in ethanol-withdrawn mitochondria than mitochondria under ethanol exposure (P < 0.01) or control media (P < 0.001). No significant difference was observed between control media and ethanol exposure. These data suggest that EW renders mitochondrial membranes vulnerable to a greater degree than ethanol per se.
Fig. 1.

Effects of ethanol/EW on MMS. HT22 cells were exposed to control media (DMSO) or ethanol (100 mM) for 24 h. At the end of the 24 h, ethanol-containing media were replaced with control media for 4 h. Cells were collected at the end of the 24-h ethanol exposure (ethanol condition) or at 4 h of EW (EW condition). Absorbance at 540 nm was recorded for 12 min. More rapid absorbance decline indicates more severe MMS. EW provoked MMS more rapidly than control media (P < 0.001) or ethanol exposure cells (P < 0.01). Depicted are mean ± S.E.M. for n = 3/group. Some S.E.M. values were too small to be depicted.
Fluorescence Microscopic Observation of ΔΨm during Ethanol/EW. To examine whether EW provokes PTP opening, we measured ΔΨm using JC-1 dye fluorescence. Healthy mitochondria that have high ΔΨm readily uptake JC-1, which results in the formation of JC-1 aggregates inside the mitochondria, and the JC-1 aggregates emit red fluorescence at 590 nm. Damaged mitochondrial membranes that have low ΔΨm cause JC-1 to leak to the cytosol, which results in a monomeric form of JC-1, emitting green fluorescence at 530 nm. When cells were observed with a fluorescence microscope (Fig. 2), EW cells exhibited stronger green fluorescence and less red fluorescence than control cells or ethanol exposure cells, indicating that EW results in more depolarized mitochondria.
Fig. 2.

Fluorescence microscopic observation of ΔΨm during ethanol/EW. HT22 cells were exposed to control media or 100 mM ethanol for 24 h and were withdrawn for 4 h. JC-1 dye (2.5 μg/ml) was treated at 24 h of ethanol exposure (ethanol exposure condition) or 4 h of EW. Cells were then observed with a fluorescence microscope. EW resulted in more intense green fluorescence emitted from JC-1 monomers and less red fluorescence emitted from JC-1 aggregates because of low ΔΨm than control or ethanol exposure cells. Magnification, 100 ×.
Cellular Consequences of EW-Induced PTP Opening. Whether or not EW-induced excess PTP opening mediates cell death was tested (Fig. 3). HT22 cells were treated with control media or 100 mM ethanol for 24 h and withdrawn for 4 h. Cyclosporin A (0.2 μM), an inhibitor of PTP opening (Halestrap et al., 1997), was treated when ethanol-containing media were replaced with control media to determine whether EW-induced PTP, not ethanol-induced PTP, is responsible for cell death during EW. Cell viability was assessed at the end of the ethanol exposure or at 4 h of EW and reported as relative values to ethanol-free cells. As expected, cell viability significantly differed between treatments [F(3, 20) = 206, P < 0.001]. Ethanol-withdrawn cells with control media treatment had lower cell survival than ethanol-free cells (100% dash line), ethanol exposure cells (75 ± 2%; †, P < 0.001) or EW cells + cyclosporin A (68 ± 1.4%; ‡, P = 0.014). These data indicate that EW-induced PTP opening accounts, at least in part, for cell death.
Fig. 3.

Cellular consequences of EW-induced PTP opening. HT22 cells were treated with control media or 100 mM ethanol for 24 h and withdrawn for 4 h. Cyclosporin A (0.2 μM), an inhibitor of PTP opening was treated when ethanol-containing media were replaced with control media. Cell viability was assessed at the end of the ethanol exposure or at 4 h of EW and reported as relative values to ethanol-free cells (100% dash line). EW cells treated with control media had lower cell survival than ethanol exposure cells (†, P < 0.001) or EW cells treated with cyclosporin A (‡, P = 0.014). *, P < 0.001 versus ethanol-free cells. Depicted are mean ± S.E.M. for n = 5/group.
Part II: Estrogen Protection against PTP Opening
E2 Protection against EW-Induced MMS. Whether or not E2 protects against EW-induced MMS was assessed (Fig. 4). E2 (0.1 or 1 μM) was treated during the entire period of ethanol exposure and EW. Repeated (by minutes) measures one-way ANOVA indicated that the rate of absorbance decline differs between treatment groups (rapid decline indicates more severe MMS) [F(3,51) = 17, P < 0.001]. At 4 h of EW,EW cells without E2 treatment showed the most rapid MMS (P < 0.01) among the treatment conditions. In comparison, E2 treatment at 1 (P < 0.001) and 0.1 (P < 0.05) μM delayed the MMS toward a control condition in a dose-dependent manner (P < 0.05), indicating protective effects of E2 on MMS.
Fig. 4.

E2 protection against EW-induced MMS. HT22 cells were exposed to control media or ethanol (100 mM) for 24 h and withdrawn for 4 h. E2 (0.1 or 1 μM) was simultaneously treated with ethanol and readministered when ethanol-containing media were replaced with control media at the end of the ethanol exposure. Cells were collected at 4 h of EW (EW cells) for assessment of MMS. EW cells showed more rapid absorbance decline (more MMS) than cells treated with control (P < 0.001), 1 μME2 (P < 0.001), or 0.1 μME2 (P < 0.05). Depicted are mean ± S.E.M. for n = 3/group. Some S.E.M. values were too small to be depicted.
E2 Treatment Window for E2 Protection. To determine whether E2 protection against EW results from E2 effects on the ethanol exposure phase or the EW phase, E2 (1 μM) treatment was restricted to the EW phase and compared with E2 treatment during the entire period of ethanol exposure (100 mM) and EW (Fig. 5). When measured at 4 h of EW, repeated (by minutes) measures one-way ANOVA indicated that MMS significantly differs between treatment groups [F(3,51) = 29, P < 0.001]. At 4 h of EW, EW cells without E2 treatment had a more rapid MMS than cells treated with control media (P < 0.001), E2 treatment during the ethanol + EW phase (P < 0.001) or E2 treatment during the EW phase (P < 0.01). There was no significant difference between the two treatment conditions of E2: E2 treatment during the combined period of ethanol exposure + EW or E2 treatment that was restricted to the EW phase. These data indicate that E2 counteracts EW-associated factors and that E2 treatment during the EW phase per se is sufficient to protect against EW.
Fig. 5.

E2 treatment window for E2 protection. HT22 cells were exposed to control media or ethanol (100 mM)-containing media. Ethanol-treated cells were further divided into three groups: 1) ethanol + control media, 2) E2 treatment (1 μM) during the entire period of ethanol exposure and EW, and 3) E2 treatment that was restricted to the EW phase. Cells were collected at 4 h of EW to assess MMS. EW cells had the most rapid MMS (absorbance decline) among treatments (P < 0.001). E2 treatment that was restricted to the EW phase significantly delayed (P < 0.05) MMS toward control cells or toward EW cells that were treated with E2 during ethanol exposure and EW. No difference was observed between E2 treatments during the two different time windows. Depicted are mean ± S.E.M. for n = 3/group. Some S.E.M. values were too small to be depicted.
Flow Cytometric Analysis of E2 Protection against ΔΨm Collapse during EW. We quantitated ΔΨm using flow cytometry that measures intensity of green and red fluorescence. The green and red fluorescences are emitted from a monomeric form of JC-1 in damaged mitochondria and emitted from JC-1 aggregates in healthy mitochondria, respectively. We computed a ratio of green/red (a higher ratio indicates greater ΔΨm collapse) at the end of ethanol exposure or at 4 h of EW with or without E2 treatment that was restricted to the EW phase. EW resulted in a higher ratio of green/red (68/32% = 2.1) than control (51/44% = 1.1), ethanol exposure (53/46% = 1.2), or EW + E2 (44/56% = 0.8) conditions, indicating that EW depolarizes mitochondria in a manner that is protected by E2 treatment (Fig. 6).
Fig. 6.

Flow cytometric analysis of E2 protection against ΔΨm collapse during EW. HT22 cells were treated with control media or ethanol (100 mM) for 24 h and were withdrawn for 4 h with or without E2 treatment that was restricted to the EW phase. JC-1 was treated at 24 h of ethanol exposure or 4 h of EW. Cells were then immediately analyzed with flow cytometry in which a higher ratio of green (bottom right quadrant) to red (top right quadrant) indicates more severe ΔΨm collapse. EW resulted in a higher ratio of green/red fluorescence (68/32% = 2.1) than control (51/44% = 1.1), ethanol exposure (53/46% = 1.2), or EW + E2 (44/56% = 0.8) conditions. The number in each quadrant indicates cell population (%) in the quadrant out of total cells (100%).
Effects of BHT or ZYC26 on MMS. To determine whether the pro-oxidant stimuli of EW provoke MMS, we used an antioxidant BHT (1 μM) with which an inhibited PTP opening had already been shown (Colell et al., 2004). When measured at 4 h of EW, there was a significant difference in MMS between treatment groups [F(3,51) = 27, P < 0.0001] (Fig. 7). As was the case for E2 treatment, BHT treatment that was restricted to the EW phase delayed MMS (P < 0.05) of EW cells. Because ZYC26 previously exerted protection against EW-induced cell death and oxidation of HT22 cells, we tested effects of ZYC26 (1 μM) on MMS. ZYC26 that was treated during the EW phase per se also delayed the MMS of EW cells (P < 0.001).
Fig. 7.

Effects of BHT or ZYC26 on MMS. HT22 cells were exposed to control media or ethanol (100 mM) for 24 h and withdrawn for 4 h. BHT (1 μM) or ZYC26 (1 μM) treatment was restricted to the EW phase. Cells were collected at 4 h of EW to assess MMS. BHT (P < 0.05) or ZYC26 (P < 0.001) treatment delayed EW-induced MMS. Depicted are mean ± S.E.M. for n = 3/group. Some S.E.M. values were too small to be depicted.
We compared the degree of protection afforded by the three compounds (E2, BHT, and ZYC26) by computing how much these compounds delayed the absorbance decline (an indicator of MMS) of EW cells (data not shown). E2 protection was greater than BHT (P < 0.01) and similar to ZYC26 [F(2,36) = 4.1, P = 0.031]. Based on this and our previous observation that ZYC26 showed higher antioxidant potency than E2 during EW (Jung et al., 2006), these data indicate that pro-/antioxidant effects of EW/E2 are part of, but not a sole mechanism of, EW/E2-induced mitochondrial injury/protection.
Ethanol Concentrations. Finally, we measured ethanol concentrations to test whether the observed protection by E2, BHT, or ZYC26 results from altered ethanol kinetics. HT22 cells were exposed to ethanol (100 mM) for 24 h and withdrawn for 4 h. E2 (1 μM), BHT (1 μM), or ZYC26 (1 μM) was treated simultaneously with ethanol. Cells were collected at the end of the 24-h ethanol exposure or at 4 h of EW. Cells treated with 100 mM ethanol per se contained 4.5 ± 0.01 mg/ml ethanol at 24-h ethanol exposure. Cells cotreated with E2, BHT, or ZYC26 had 4.48 to 4.54 mg/ml ethanol when administered ethanol concentration was 100 mM. No measurable ethanol was detected at 4 h of EW. Collectively, none of E2, BHT, or ZYC26 altered ethanol concentrations, and cells maintained intended ethanol concentrations for 24 h that were eliminated at 4 h of EW, resembling in vivo situations. These results indicate that the kinetics of ethanol does not account for protection by E2, BHT, or ZYC26 against the EW-induced PTP opening.
Effects of E2, BHT, or ZYC26 Per Se on PTP Opening. If treatment with E2, BHT, or ZYC26 per se alters mitochondrial membrane integrity, the observed effects of E2, BHT, or ZYC26 on ethanol withdrawn cells do not necessarily indicate protection against EW. This possibility was examined by treating ethanol-free cells with E2, BHT, or ZYC26 (0.1 and 1 μM) for 24 h and another 4 h when media were replaced with fresh media. When measured at 4 h of withdrawal from control media exposure, none of the three compounds significantly altered MMS. These data indicate that E2, BHT, or ZYC26 counteracts factors associated with EW, exerting protection against EW-induced PTP opening (data not shown).
Discussion
We demonstrated for the first time that EW provokes PTP opening in HT22 cells partly through, but not entirely through, its pro-oxidant nature. E2 treatment restricted to the EW phase ameliorated the mitochondrial membrane injury, suggesting that E2 counteracts factors associated with EW, exerting the mitoprotection.
We observed previously that EW inactivated a key mitochondrial enzyme, cytochrome c oxidase, and provoked oxidation of mitochondrial proteins in rats (Jung et al., 2007, 2008). In the current study, we used an in vitro HT22 cell model because this cell line has the advantage of assessing oxidative mechanisms. As mentioned earlier, HT22 cells lack ionotropic glutamate receptors (Zaulyanov et al., 1999) but contain the glutamate/cystine antiporter, necessary for the synthesis of an endogenous antioxidant glutathione. Therefore, HT22 cellular injury reflects compromised endogenous antioxidant capacity (Tan et al., 1998). In accordance, EW-induced PTP opening is unlikely because of excitotoxicity associated with glutamate receptors but may result from oxidative insults. This hypothesis is supported by the finding that an antioxidant BHT treatment restricted to the EW phase attenuated the EW-induced PTP opening (Fig. 7) and cell death (unpublished data). Vulnerability of mitochondrial membranes to ethanol has been reported in a previous study where ethanol treatment resulted in excess PTP opening in mice lacking superoxide dismutase (endogenous antioxidant enzyme) (Kessova and Cederbaum, 2007). The study suggests that loss of endogenous antioxidant capacity mediates PTP opening. In agreement, an early study (French and Todoroff, 1971) noted that EW increased mitochondrial permeability to a superoxide generator (phenazine methosulfate) in male rats. Ethanol-dependent rats showed swollen mitochondria in liver, demonstrating the pathological consequence of PTP opening (Yan et al., 2007). Taken together, our findings agree with these studies and provide empirical evidence that pro-oxidant EW injures mitochondrial membranes, perturbs the regulatory function of PTP, and ultimately results in detrimental cellular consequences (Fig. 3).
Our model of EW resembles alcoholism, in which withdrawal plays a key role
in neuronal and cellular damage. Using this model, we previously demonstrated
that EW evoked greater lipid peroxidation and protein oxidation than ethanol
per se in rats and cells (Jung et al.,
2004,
2006). The pro-oxidant nature
of EW also has been demonstrated in a clinical situation in which EW-induced
hyperexcitability was associated with oxidative damages; cerebrospinal fluid
of withdrawn alcoholics contained higher concentrations of
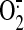 and excitatory amino
acids than control subjects (Tsai et al.,
1998). These studies suggest that deleterious interaction between
pro-oxidants and excitatory neurotransmission accounts for EW-distress. As
such, if pro-oxidants contribute to EW injury and to PTP opening
(Kessova and Cederbaum, 2007),
one can speculate that EW renders more severe PTP opening than ethanol per se
in part because of its greater pro-oxidant activity than ethanol exposure
(Fig. 1).
and excitatory amino
acids than control subjects (Tsai et al.,
1998). These studies suggest that deleterious interaction between
pro-oxidants and excitatory neurotransmission accounts for EW-distress. As
such, if pro-oxidants contribute to EW injury and to PTP opening
(Kessova and Cederbaum, 2007),
one can speculate that EW renders more severe PTP opening than ethanol per se
in part because of its greater pro-oxidant activity than ethanol exposure
(Fig. 1).
We next assessed whether PTP opening contributes to cell death during EW. PTP intimately affects cell survival/death because if PTP remains open, cells cannot maintain optimal ATP levels for respiration (Halestrap, 2006). If excess opening of PTP mediates cell death, preventing the pore opening should attenuate the cell death. This hypothesis was tested using a PTP inhibitor cyclosporin A. Cyclosporin A is a prototype inhibitor of PTP opening (Norenberg and Rao, 2007) by interfering with protein (cyclophilin D)-protein (adenine nucleotide translocator) interaction that is essential for PTP opening (Sullivan et al., 2005). Cyclosporin A protected against apoptotic and necrotic cell death associated with PTP (Ishida, 2004). In our results, although only 49% of cells survived EW toxicity, 68% of cells survived EW after treatment with cyclosporin A. Therefore, our data extend cytotoxic PTP opening to a cellular model of EW.
We and others (Viña et al., 2007) have shown that estrogens have substantial effects on mitochondrial function, particularly in the face of stress. For instance, estrogen exerted protection against β-amyloid- or ROS-induced mitotoxicity (Viña et al., 2007). E2 protected against H2O2-induced decline in ATP synthesis in human neuroblastoma cells (Wang et al., 2006). Estrogens have been shown to exert their anti-apoptotic effects through maintenance of ΔΨm in the face of stresses (Nilsen and Brinton, 2003). Furthermore, nonfeminizing estrogens shared this ability to protect ATP production (Wang et al., 2006). These data indicate collectively that estrogens potently stabilize bioenergetics functions of mitochondria during stress, such as oxidative stress.
Despite a plethora of evidence of mitoprotective estrogen, the mechanisms by which E2 protects mitochondria from EW insults remain uncertain. We first tested whether E2 protection against EW is because of its residual effects on ethanol per se. However, this possibility is unlikely because E2 exerted protection even when E2 treatment was restricted to the EW phase, suggesting that E2 directly or indirectly regulates factors associated with EW, thereby exerting mitoprotection. Because BHT protected against EW-induced PTP opening, it is reasonable to speculate that antioxidant activity of E2 counteracts pro-oxidants EW, thereby inhibiting PTP opening. In support of this view, PTP opening was inhibited by vitamin E but was induced by H2O2 (Lee et al., 2005; Sokol et al., 2005). It is unfortunate that it is challenging to differentiate an antioxidant mechanism from other mechanisms of E2 that protect PTP. In addition, we are not aware of an estrogen analog that has physiologically and pharmacologically identical properties to E2 but lacking only antioxidant property. As an indirect strategy to test whether E2 protects PTP through an antioxidant mechanism, we compared the degree of protection against PTP opening among E2, an antioxidant BHT, and an E2 analog ZYC26. ZYC26 previously showed a 10-fold higher antioxidant potency than E2 in the HT22 cell model of EW (Jung et al., 2006). ZYC26 contains an adamantyl group at the C2 position of estrone. This structural configuration allows ZYC26 to scavenge ROS more effectively than E2. The adamantyl group enhances the stability of the adjacent phenolic radical, which is an essential element of scavenging ROS (Dhandapani and Brann, 2002). We computed the magnitude by which these compounds prevent MMS of ethanol withdrawn cells at a given time point. Had antioxidant action, especially ROS-scavenging activity, been a major mechanism of E2 protection against PTP opening, ZYC26 that has higher ROS-scavenging potency than E2 should have been more protective than E2. However, the magnitude of E2 protection against MMS was greater than BHT (P < 0.05) and did not differ from ZYC26 (data not shown). These observations indicate that other factors in addition to ROS-scavenging activity of E2 also mediate protection against PTP opening. For instance, E2 might have activated glutathione peroxidase that directly reduces membrane-bound lipid hydroperoxides (Ha and Smith, 2003; Lapointe et al., 2005). In addition, because HT22 cell injury is associated with compromised redox status, it is also possible that E2 might have maintained glutathione homeostasis in the face of EW insults, thereby reducing the oxidative burden of mitochondrial membranes and subsequently attenuating PTP opening. Alternatively, because high levels of mitochondrial Ca2+ impede the regulatory function of the PTP proteins (Halestrap and Brennerb, 2003) and because E2 induces mitochondrial tolerance to Ca2+ load (Nilsen and Diaz Brinton, 2003), E2 might have increased a threshold of mitochondrial Ca2+ that activates PTP. At the very least, it seems that E2 orchestrates multiple mechanisms in addition to antioxidant action, effectively protecting PTP from EW.
Taken together, our findings permit a conclusion that EW perturbs mitochondrial membrane integrity partly but not entirely through its pro-oxidant nature and that E2 exerts mitoprotection through multiple factors perhaps, involving an antioxidant mechanism, independent of ethanol metabolism. We extended EW injury to mitochondrial membranes, attributing to PTP opening and cell death in the HT22 cell model of EW. Our findings may provide new insights into mitochondrial mechanistic pathways underlying counteraction between EW and E2.
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grants AA013864, AA015982].
doi:10.1124/jpet.108.146829.
ABBREVIATIONS: EW, ethanol withdrawal; E2, 17β-estradiol; ROS, reactive oxygen species; PTP, mitochondrial membrane permeability transition pore; BHT, butylated hydroxytoluene; ZYC26, (3-hydroxy-2-adamantyl(1)-4-methyl-estra-1,3,5(10)-17-one; DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; MMS, mitochondrial membrane swelling; AM, acetoxymethyl ester; ANOVA, analysis of variance.
References
- Borrás C, Sastre J, García-Sala D, Lloret A, Pallardó FV, and Viña J (2003) Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med 34 546-552. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72 248-254. [DOI] [PubMed] [Google Scholar]
- Colell A, García-Ruiz C, Mari M, and Fernández-Checa JC (2004) Mitochondrial permeability transition induced by reactive oxygen species is independent of cholesterol-regulated membrane fluidity. FEBS Lett 560 63-68. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM and Brann DW (2002) Protective effects of estrogen and selective estrogen receptor modulators in the brain. Biol Reprod 67 1379-1385. [DOI] [PubMed] [Google Scholar]
- Fernández-Checa JC, García-Ruiz C, Colell A, Morales A, Marí M, Miranda M, and Ardite E (1998) Oxidative stress: role of mitochondria and protection by glutathione. Biofactors 8 7-11. [DOI] [PubMed] [Google Scholar]
- French SW and Todoroff T (1971) Effect of chronic ethanol ingestion and withdrawal on brain mitochondria. Res Commun Chem Pathol Pharmacol 2 206-215. [PubMed] [Google Scholar]
- Ha EJ and Smith AM (2003) Plasma selenium and plasma and erythrocyte glutathione peroxidase activity increase with estrogen during the menstrual cycle. JAm Coll Nutr 22 43-51. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2006) Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34 232-237. [DOI] [PubMed] [Google Scholar]
- Halestrap AP and Brennerb C) (2003) The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 10 1507-1525. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Connern CP, Griffiths EJ, and Kerr PM (1997) Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem 174 167-172. [PubMed] [Google Scholar]
- Hosein EA, Lee H, and Hofmann I (1980) The influence of chronic ethanol feeding to rats on liver mitochondrial membrane structure and function. Can J Biochem 58 1147-1155. [DOI] [PubMed] [Google Scholar]
- Ishida H (2004) [The research method for investigating the role of the mitochondrial permeability transition pore in cell death]. Nippon Yakurigaku Zasshi 123 329-334. [DOI] [PubMed] [Google Scholar]
- Jung ME, Agarwal R, and Simpkins JW (2007) Ethanol withdrawal posttranslationally decreases the activity of cytochrome c oxidase in an estrogen reversible manner. Neurosci Lett 416 160-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung ME, Rewal M, Perez E, Wen Y, and Simpkins JW (2004) Estrogen protects against brain lipid peroxidation in ethanol-withdrawn rats. Pharmacol Biochem Behav 79 573-586. [DOI] [PubMed] [Google Scholar]
- Jung ME, Wilson AM, and Simpkins JW (2006) A nonfeminizing estrogen analog protects against ethanol withdrawal toxicity in immortalized hippocampal cells. J Pharmacol Exp Ther 319 543-550. [DOI] [PubMed] [Google Scholar]
- Jung ME, Yan LJ, Forster MJ, and Simpkins JW (2008) Ethanol withdrawal provokes mitochondrial injury in an estrogen preventable manner. J Bioenerg Biomembr 40 35-44. [DOI] [PubMed] [Google Scholar]
- Kessova IG and Cederbaum AI (2007) Mitochondrial alterations in livers of Sod1-/-mice fed alcohol. Free Radic Biol Med 42 1470-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Kimmins S, Maclaren LA, and Bilodeau JF (2005) Estrogen selectively up-regulates the phospholipid hydroperoxide glutathione peroxidase in the oviducts. Endocrinology 146 2583-2592. [DOI] [PubMed] [Google Scholar]
- Lee CS, Kim YJ, Ko HH, and Han ES (2005) Synergistic effects of hydrogen peroxide and ethanol on cell viability loss in PC12 cells by increase in mitochondrial permeability transition. Biochem Pharmacol 70 317-325. [DOI] [PubMed] [Google Scholar]
- Mansouri A, Demeilliers C, Amsellem S, Pessayre D, and Fromenty B (2001) Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmacol Exp Ther 298 737-743. [PubMed] [Google Scholar]
- Menze MA, Hutchinson K, Laborde SM, and Hand SC (2005) Mitochondrial permeability transition in the crustacean Artemia franciscana: absence of a calcium-regulated pore in the face of profound calcium storage. Am J Physiol Regul Integr Comp Physiol 289 R68-R76. [DOI] [PubMed] [Google Scholar]
- Miñana JB, Gómez-Cambronero L, Lloret A, Pallardó FV, Del Olmo J, Escudero A, Rodrigo JM, Pellíin A, Viña JR, Viña J, et al. (2002) Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology 35 1205-1214. [DOI] [PubMed] [Google Scholar]
- Morimoto BH and Koshland DE Jr (1990) Excitatory amino acid uptake and N-methyl-d-aspartate-mediated secretion in a neural cell line. Proc Natl Acad Sci U S A 87 3518-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostallino MC, Mascia MP, Pisu MG, Busonero F, Talani G, and Biggio G (2004) Inhibition by miltirone of up-regulation of GABAA receptor alpha4 subunit mRNA by ethanol withdrawal in hippocampal neurons. Eur J Pharmacol 494 83-90. [DOI] [PubMed] [Google Scholar]
- Nilsen J and Diaz Brinton R (2003) Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc Natl Acad Sci U S A 100 2842-2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg MD and Rao KV (2007) The mitochondrial permeability transition in neurologic disease. Neurochem Int 50 983-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EJ, Cai ZY, Covey DF, and Simpkins JW (2006) Neuroprotective effects of estratriene analogs: structure-activity relationships and molecular optimization. Drug Dev Res 66 78-92. [Google Scholar]
- Phillips SC and Cragg BG (1983) Chronic consumption of alcohol by adult mice: effect on hippocampal cells and synapses. Exp Neurol 80 218-226. [DOI] [PubMed] [Google Scholar]
- Ruiz-Meana M, Garcia-Dorado D, Miró-Casas E, Abellán A, and Soler-Soler J (2006) Mitochondrial Ca2+ uptake during simulated ischemia does not affect permeability transition pore opening upon simulated reperfusion. Cardiovasc Res 71 715-724. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Wang J, Wang X, Perez E, Prokai L, and Dykens JA (2005) Mitochondria play a central role in estrogen-induced neuroprotection. Curr Drug Targets CNS Neurol Disord 4 69-83. [DOI] [PubMed] [Google Scholar]
- Smolen A, Marks MJ, Smolen TN, and Collins AC (1986) Dose and route of administration alter the relative elimination of ethanol by long-sleep and short-sleep mice. Alcohol Clin Exp Res 10 198-204. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Dahl R, Devereaux MW, Yerushalmi B, Kobak GE, and Gumpricht E (2005) Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr 41 235-243. [DOI] [PubMed] [Google Scholar]
- Stuart R (2002) Insertion of proteins into the inner membrane of mitochondria: the role of the Oxa1 complex. Biochim Biophys Acta 1592 79-87. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, and Springer JE (2005) Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 79 231-239. [DOI] [PubMed] [Google Scholar]
- Tan S, Wood M, and Maher P (1998) Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem 71 95-105. [DOI] [PubMed] [Google Scholar]
- Tsai GE, Ragan P, Chang R, Chen S, Linnoila VM, and Coyle JT (1998) Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am J Psychiatry 155 726-732. [DOI] [PubMed] [Google Scholar]
- Viña J, Lloret A, Vallés SL, Borrás C, Badía MC, Pallardó FV, Sastre J, and Alonso MD (2007) Effect of gender on mitochondrial toxicity of Alzheimer's Abeta peptide. Antioxid Redox Signal 9 1677-1690. [DOI] [PubMed] [Google Scholar]
- Wang X, Dykens JA, Perez E, Liu R, Yang S, Covey DF, and Simpkins JW (2006) Neuroprotective effects of 17beta-estradiol and nonfeminizing estrogens against H2O2 toxicity in human neuroblastoma SK-N-SH cells. Mol Pharmacol 70 395-404. [DOI] [PubMed] [Google Scholar]
- Yan M, Zhu P, Liu HM, Zhang HT, and Liu L (2007) Ethanol induced mitochondria injury and permeability transition pore opening: role of mitochondria in alcoholic liver disease. World J Gastroenterol 13 2352-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaulyanov LL, Green PS, and Simpkins JW (1999) Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in vitro model for assessment of the neuroprotective effects of estrogens. Cell Mol Neurobiol 19 705-718. [DOI] [PMC free article] [PubMed] [Google Scholar]