

Abstract
Structures have been obtained for the complexes tiamulin, homoharringtonine and bruceatin form with the large ribosomal subunit of Haloarcula marismortui at resolutions ranging from 2.8 to 3.2 Å. They show that these inhibitors all block protein synthesis by competing with the amino acid side chains of incoming aminoacyl-tRNAs for binding in the A-site cleft in the peptidyl transferase center, which is universally conserved. In addition these structures support the hypothesis that the species-specificity exhibited by the A-site cleft inhibitors is determined by the interactions they make, or fail to make, with a single nucleotide, U2504 (E. coli). In the ribosome, the position of U2504 is controlled by its interactions with neighboring nucleotides, whose identities vary among kingdoms.
Keywords: antibiotic specificity, large ribosomal subunit, tiamulin, homoharringtonine, bruceantin
Introduction
Since the systematic search for antibiotics began, thousands of natural products have been discovered that inhibit protein synthesis by binding to the ribosome. Additional ribosomal inhibitors have emerged from cancer chemotherapy screens. Many of them block the activity of the large ribosomal subunit by interfering with the binding of either aminoacyl-tRNA or peptidyl-tRNA to the peptidyl transferase center (PTC), where peptide bonds form (Figure 1). 1–3
Figure 1.

Cross-sectional view of the large ribosomal subunit. The subunit has been cut open to expose the peptidyl transferase center and the polypeptide exit tunnel. Ribosomal RNA is grey and proteins are blue. A P-site-bound tRNA (brown) is shown with an A-site-bound tRNA (purple) just visible behind it, and a nascent peptide chain (red) shown schematically extending down the exit tunnel. The site where the antibiotics of interest bind to the ribosome is also shown. The inset provides an enlarged view of the drug-binding site. tRNAs are shown as surfaces, color-coded as described above. The two nucleotides that define the A-site cleft are shown in gray, and the three drugs discussed here are shown superimposed on each other (tiamulin(yellow), homoharringtonine(green) and bruceantin(orange)).
Even though the structure of the ribosome is highly conserved, some ribosome inhibitors are orders of magnitude more active against eubacterial ribosomes than eukaryotic ribosomes, while others either have the opposite specificity or no specificity at all. Thus the obvious questions to ask about all of them are: (1) why do they inhibit protein synthesis, and (2) why do they have the specificities they do. Since 2000, our understanding of both issues has been revolutionized by the availability of atomic resolution crystal structures of the ribosome with various inhibitors.
Here we present crystal structures of the large ribosomal subunit from Haloarcula marismortui (H. ma) with three such inhibitors bound: tiamulin, bruceantin, and homoharringtonine (Figure 2a, b, c). Tiamulin (Figure 2a) is a derivative of the naturally-occurring antibiotic pleuromutilin. 4,5 It specifically inhibits eubacterial protein synthesis, and is used in veterinary medicine to treat gram-positive infections. 4,6 In vitro, pleuromutilins inhibit both poly(U)-directed polyphenylalanine synthesis and the puromycin reaction, and compete with chloramphenicol for binding to the large ribosomal subunit. 6,7 Thus, the biochemical data suggest that pleuromutilins bind to the A-site of the large ribosomal subunit consistent with the structure that has been obtained of that drug bound to the large ribosomal subunit from Deinococcus radiodurans8,9.
Figure 2.

The chemical structures of tiamulin, homoharringtonine and bruceantin, and the corresponding difference electron density. Panels (a), (b), and (c) show the chemical structures of these inhibitors. Panels (d), (e), and (f) show the feature in the appropriate (Fo-Fo) difference electron density map that were assigned to the three drugs with the structures of the drugs superimposed on them. Difference electron density maps were contoured at 3σ. Residues forming the A-site cleft are shown in cyan, and 23S rRNA bases are numbered to correspond with the 23S rRNA of E. coli.
The alkaloid homoharringtonine (Figure 2b) was first isolated by Paudler from Cephalotaxus in 1963, and gained attention a decade later because of its antitumor activity. 10–12 Biochemical data suggest that homoharringtonine also binds to the A-site of the large ribosomal subunit and, like other A-site specific compounds, e.g. anisomycin, inhibits the binding of aminoacyl-tRNA to the ribosome. 3,13 However, unlike tiamulin, homoharringtonine preferentially inhibits eukaryotic protein synthesis14,15.
The third antibiotic discussed here, bruceantin (Figure 2c) is also an anti-tumor agent that specifically inhibits eukaryotic protein synthesis14,16. It is obtained from Brucea antidysenterica, and biochemical experiments have shown that it inhibits the binding of other A-site drugs such as trichodermin and anisomycin to yeast and rabbit reticulocyte ribosomes. 13 Like homoharringtonine, bruceantin has a low affinity for polysomes, and interferes with the elongation step of translation. 3,13
Tiamulin, bruceantin and homoharringtonine were soaked into the large ribosomal subunit crystals of H. marismortui at high drug concentrations, and consistent with the biochemical data, they all bind in the A-site cleft, which is the target of many, better known antibiotics, e.g. anisomycin, chloramphenicol and linezolid 1,3,6–9,10,13,16–23. The A-site cleft is the wedge-shaped gap in the large ribosomal subunit formed by the bases of A2451(2486) and C2452(2487) of 23S rRNA24. (The convention used here for numbering nucleotides is Escherichia coli number first, followed by the corresponding H. ma number in parentheses). A-site cleft antibiotics appear to inhibit protein synthesis by competing with the amino-acid side chains of A-site bound aminoacyl-tRNAs for binding to that same site.
Comparison of the crystal structures described here with other ribosome crystal structures suggests that the species specificities of tiamulin, homoharringtonine and bruceantin are determined primarily by the propensity of a single base, U2504, to adopt a conformation compatible with their binding to the ribosome. The conformational preferences of U2504 are strongly influenced by its interactions with two bases, A2096(C2055) and U2607 (A2572), which differ between eubacteria and eukaryotes. This hypothesis, which has been articulated before8,25, is supported by observations that have been made with other A-site cleft inhibitors.
Results
Crystal structures
The structures of the complexes that tiamulin, homoharringtonine, and bruceantin form with the large ribosomal subunit from H. ma were determined by X-ray crystallography. The complexes studied were prepared by soaking preformed crystals of the large ribosomal subunit into solutions containing each compound. Table 1 provides statistics characterizing the quality of the data obtained.
Table 1.
Crystallographic statistics of data collection
All data were collected at wavelengths ranging from 1.07Å to 1.1Å.
| Crystal soak | Antibiotic concentration | Resolution (Å) | I/σ | Rmerge (%) | Completeness (%) | Redundancy |
|---|---|---|---|---|---|---|
| Tiamulin | 1 mM | 50.0–3.20 (3.31–3.20) | 9.09 (0.62) | 10.3 (85) | 93.3 (57.4) | 7.1 (2.3) |
| Homoharringtonine | 1 mM | 50.0–2.65 (2.74–2.65) | 17.8 (1.17) | 5.6 (82.4) | 99.5 (97.5) | 5.2 (3.3) |
| Bruceantine | 1 mM | 50.0–2.85(2.95–2.85) | 13.2 (1.12) | 7.5 (>100) | 98.6 (98.6) | 5.9 (5.6) |
In each case, structural analysis began with the computation of difference electron density maps. The difference amplitudes used were obtained by subtracting the amplitudes measured from a crystal lacking the drug from the amplitudes measured from a crystal containing the drug, i.e. ΔF(h,k,l)dif = (F(h,k,l)+drug − F(h,k,l)−drug). The phases used to compute difference maps were obtained by rigid-body refinement of the apo-structure of the large ribosomal subunit into the data obtained from the crystals containing the drug. In all three maps there is only one positive electron density feature large enough to accommodate the relevant drug, and in every case that feature is of appropriate size and shape (Figure 2d, e, f). Once the location and orientation of each inhibitor had been determined, models were constructed and refined as described in Materials and Methods (Table 2).
Table 2.
Statistics of model refinement
| Crystal soak | Antibiotic concentration | Resolution (Å ) | Rcryst (%) | Rfree (%) | Bond (Å) | Angle (°) | PDB ID |
|---|---|---|---|---|---|---|---|
| Tiamulin | 1 mM | 50.0–3.20 | 21.3 | 28.9 | 0.008 | 1.2 | 3G4S |
| Homoharringtonine | 1 mM | 50.0–2.65 | 19.4 | 22.7 | 0.007 | 1.1 | 3G6E |
| Bruceantine | 1 mM | 50.0–2.85 | 19.1 | 22.9 | 0.007 | 1.1 | 3G71 |
Tiamulin
Tiamulin binds to the large ribosomal subunit with the three rings that form its core stacking on the base of G2061(2102), the base of C2452(2487) and the ribose of G2505(2540) (Figure 3a). These three moieties delimit a largely hydrophobic cavity in the ribosome that the drug almost completely fills. Tiamulin has a surface area of 878.8 Å2 and 70% of it is solvent-inaccessible when it is bound to the ribosome. In addition, the “tail” of tiamulin extends far enough into the P-site to interfere with the binding of substrates to that site also.
Figure 3.

Drug interactions with the ribosome, and the conformational changes associated with drug binding. Panels (a), (c), and (e) show how tiamulin, homoharringtonine, and bruceantin interact with the ribosome. Drugs are shown with C gold, N blue, O red, and S green. Water molecules are shown as red balls and hydrogen bonds are shown as red dashes. Panels (b), (d), and (f) show the conformational changes associated with drug binding. The drugs are color coded as just described. The drug-bound conformation of surrounding bases is shown in cyan, superimposed on the apo- conformation (gray).
Small shifts in the positions of nearby nucleotides are associated with tiamulin binding (Figure 3b). A2451(2486) moves away from the drug by a modest amount, and the base of C2452(2487) moves towards A2453(2488), a change in location that improves its stacking on both the 6-membered ring of tiamulin and A2453(2488). Similar shifts are seen when anisomycin binds to the ribosome. A2062 also moves away to avoid a steric clash with the bound tiamulin. It is interesting to note that the K+ ion that is found coordinated by the base triple (A2451(2486), G2061(2102), G2447(2482)) in the native structure appears to be lost upon tiamulin binding.
The complex of tiamulin with the ribosome is stabilized by a large number of hydrogen bonds, as well as by hydrophobic interactions (Figure 3a). The keto-group of the cyclo-pentanone ring of tiamulin is part of a hydrogen-bond network that involves the O2 of C2452(2487), the phosphate oxygens of U2506(2541), and a water molecule (O197). The keto group in the tail of tiamulin is within hydrogen bonding distance of both the N1 and the N2 of G2061(2102). The OH group of the 8-membered ring of tiamulin forms a hydrogen bond with a water molecule (O277), which is hydrogen-bonded to the O2′ of A2503(2538) and the O2P of G2505(2540). The same tiamulin hydroxyl group makes a hydrogen bond with a second water (O6452) that in turn forms a hydrogen bond to the N6 of A2059(2100). Finally, the sulfur of the thioether in the tail of tiamulin is within hydrogen bonding distance of a water molecule (O4261), which is held in place by a hydrogen bond to the O2′ of C2063(2104).
Homoharringtonine
The interaction of homoharringtonine with the ribosome is stabilized by both hydrophobic interactions and hydrogen bonding (Figure 3c). Its 6-membered aromatic ring stacks on the base of C2452(2487), and the A-site cleft is completely filled by it and the 5-membered dioxylane ring attached to it. Homoharringtonine has a surface area of 998.9 Å2 and when bound to the ribosome, 73% of its surface becomes solvent inaccessible. The tertiary amine of homoharringtonine, which is protonated at neutral pH, forms a hydrogen bond with the O2 of C2452(2487). This bond is 2.66Å long and positive electron density connecting the O2 of C2487 and the tertiary amine of homoharringtonine can be seen in difference electron density maps calculated using observed amplitudes, (Fo-Fo), contoured at 2.5 σ. The hydroxyl group at the tip of the “tail” of the drug is hydrogen bonded to the O6 of G2058(2099) through an intervening water molecule, and the carbonyl group at the other end of the “tail”, forms a hydrogen bond with the N2 of G2061(2102).
The binding of homoharringtonine to the large ribosomal subunit is accompanied by several conformational changes (Figure 3d). The most dramatic of them is a roughly 50° reorientation of the base of U2506(2541), which enables it to stack on top of the drug. In the drug complex U2506(2541) forms a symmetric, O4-N3/N3-O4 base pair with U2585(2620). In all other crystal structures of the large ribosomal subunit from H. ma U2506(2541) forms a GU base pair with G2583(2618). A number of more subtle conformational changes are seen in the A-site cleft region. C2452(2487) undergoes a slight rotation away from A2451(2486), and the base of A2453(2488) rotates about 40° about its glycosidic bond. This rotation allows A2453(2488) to form a base pair with U2500(2535) and improves its stacking on C2452(2487).
Bruceantin
Like homoharringtonine, the core of bruceantin is a fused, polycyclic system, and when bound to the ribosome, the ring on its left end in Figure 2c is inserted into the A-site cleft (Figure 3e). The cleft opens to accommodate bruceantin. A2451(2486) moves away from C2452(2487) by a small amount while C2452(2487) moves away from A2451(2486) by 1.8 Å, as measured by the change in the position of its C2 (Figure 3f). The ring of bruceantin that fills the cleft is oriented more or less parallel to the base of A2451(2486), and the fit is so tight that despite our best efforts at refinement, there are many short (< 3 Å) non-bonded contacts between the drug and the ribosome in the cleft region. The motion of C2452(2487) propagates through the backbone for several nucleotides, having its largest effect on the placement of the base of A2453(2488), on which the base of C2452(2487) stacks. Drug binding is also associated with a movement of A2062(2103) away from the drug, which alleviates what would otherwise be a steric clash.
Like tiamulin and homoharringtonine, bruceantin interacts with the ribosome extensively and 66% of its surface area of 870.9 Å2 is buried when it is bound to the ribosome. Several hydrogen bonds also stabilize the bruceantin-ribosome complex (Figure 3e). The N6 of A2062(2103) forms a hydrogen bond with the carbonyl oxygen of the ester group in the “tail” of the drug. The carbonyl oxygen of the carboxymethyl side chain of the molecule hydrogen bonds with the O2′ of A2503(2538). Finally, the ribose of G2505(2540) interacts extensively with the 5-membered oxycylcopentane ring and the adjacent 6-membered lactone ring of the drug further stabilizing the complex.
Discussion
Since halophilic archaea are much less sensitive to many inhibitors of protein synthesis than eubacteria and eukaryotes, 14,26 high inhibitor concentrations (1 mM) were used in this study to ensure the formation of drug-ribosome complexes. Spurious binding is always a concern when macromolecules are exposed to high concentrations of small molecules. However, dozens of structures have been obtained of inhibitors bound to the large ribosomal subunit from H. ma using inhibitor concentrations in the mM range, 1,25,27–30 and in that entire set of structures there is only one example where the inhibitor may have bound to a spurious site. 28 With that one possible exception, these structures explain why the inhibitors in question block protein synthesis, and rationalize the genetic and biochemical data obtained using more sensitive organisms. Finally, structures have been obtained for several of these inhibitors bound to the ribosomes from other species. In almost all cases, these structures agree well with the corresponding H. ma structures and the few exceptions have interesting biochemical implications. 1,31
When tiamulin, homoharringtonine and bruceantin bind to the large ribosomal subunit from H. marismortui, they completely fill its A-site cleft, precluding the occupation of that same space by the amino acid side chains of A-site-bound aminoacyl-tRNAs (Figure 2d, e, f). Thus even if an aminoacyl-tRNA were to bind to the A-site under these conditions, its amino acid portion would be sufficiently displaced from its proper position to prevent the participation of its α-amino group in peptide bond formation. These observations indicate that at the (high) concentrations used in these experiments, all three compounds should inhibit protein synthesis in H. ma.
It is much more challenging to explain the species specificities of these inhibitors. However, if it is assumed that the structure of the A-site cleft region of the H. ma ribosome is eukaryotic in character, analysis of the structural data available leads to a hypothesis that explains the species specificities of all A-site cleft protein synthesis inhibitors. The rationale for assuming that archaeal ribosomes have eukaryotic character is clear at the level of ribosomal proteins. 32–34 Comparison of ribosomal RNA sequences also indicate that the archaea, as a group, are more closely related to eukaryotes than to eubacteria. 35 In addition, and particularly pertinent here, archaea, like eukaryotes, are sensitive to the A-site cleft inhibitor anisomyin, but insensitive to another A-site cleft inhibitor chloramphenicol, to which eubacteria are sensitive. 3
Tiamulin
Comparison of the crystal structures oftiamulin bound to the large ribosomal subunit of the eubacterium Deinococcus radiodurans 9 and the archaebacterium H. ma, suggests an explanation for why drugs of this class are specific for eubacteria. Within experimental error, the fused ring system of the drug binds in the same location and with the same orientation to both species of ribosomes.
The most striking difference between the binding sites for tiamulin in the two species is the placement of the base of U2504(2539) in the unliganded structure, and we suspect that this difference accounts for the species specificity of this drug as well as other A-site cleft drugs (Figure 4a). In the D. radiodurans subunit, as well as in other eubacteria, in the absence of the drug, the base of U2504 (2539) is approximately positioned to stack on the ring system of tiamulin when that drug binds8,9, while in the large ribosomal subunit of H. ma, it is not (Figure 4d). That difference in placement appears to be a consequence of the interaction the base of U2504(2539) with the nucleotide at position 2055(2096), which is a C in eubacteria and an A in archaea and eukaryotes (Figure 5). In H. ma, the base of U2504(2539) stacks on the base of A2055(2096), which is in the syn conformation, and that helps to stabilize the noninteractive positioning of U2504. In D. radiodurans and other eubacteria, the base at 2055(2096) is a C, and there is no stacking. Instead, the U2504 adopts a conformation that favors tiamulin binding, which is stabilized by base pairing with C2452(2487).
Figure 4.

Comparison of the interactions of tiamulin and homoharringtonine with archaeal and eubacterial ribosomes. (a) The structure of the complex tiamulin (gold) forms with the large ribosomal subunit from H. ma (gold) is shown superimposed on the structure of tiamulin (beige) bound to the large ribosomal subunit from D. radiodurans (beige) (PDB# 1XBP). 9(b) The structure of homoharringtonine (gold) bound to the H. ma large ribosomal subunit (gold) is shown superimposed on the structure of the E. coli large ribosomal subunit (PDB# 2AW4, magenta,). 39(c) The structure of bruceantine (gold) bound to H. ma large ribosomal subunit (gold) is shown superimposed on the structure of the E. coli large ribosomal subunit (PDB# 2AW4, magenta,). 39(d) The apo structures of H. ma (PDB# 1S72, gray,), 50T. thermophilus (PDB# 2J03, orange,), 51E. coli (PDB# 2AW4, magenta) and D. radiodurans (PDB# 1NKW, beige,). 41(e) The apo (PDB# 1S72, gray), anisomycin- bound (PDB# 1K73, green,) 1 and linezolid-bound (PDB# 3CPW, gold,) 17 structures of the H. marismortui large ribosomal subunit are superimposed. Anisomycin is green, and linezolid is gold. (f) A superposition of the apo structure of H. ma (PDB# 1S72, gray), the structure of the complex the H. ma ribosome forms with linezolid (PDB# 3CPW, gold), and the structure of linezolid bound to the D. radiodurans large ribosomal subunit (PDB# 3DLL, beige,). 52 All superpositions in (a), (b), (c),(d), (e) and (f) were done using PTC phosphorus atom positions.
Figure 5.
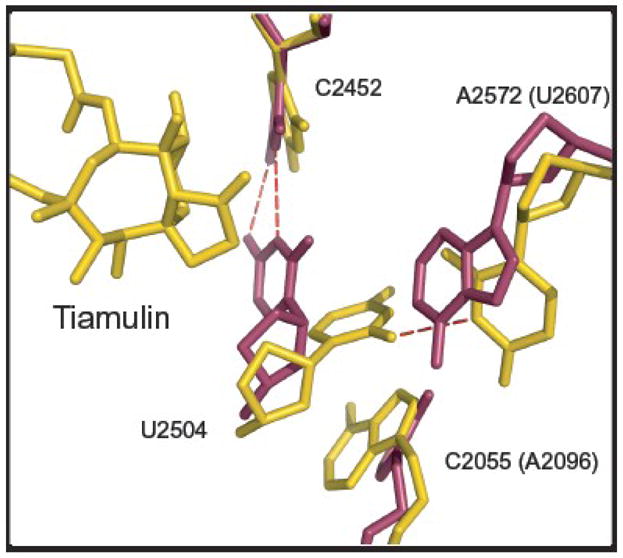
Interactions that control the conformation of U2504(2539) in H. ma and E. coli. The H. ma structure shown is the tiamulin-ribosome complex structure described here, and the E. coli structure is PDB# 2AW439. The two have been superimposed using phosphorus atom positions in the PTC. Tiamulin and the H. ma ribosome are shown in gold and the E. coli ribosome is in magenta.
Other interactions also contribute to the difference in the placement of U2504(2539) between archaeal ribosomes and eubacterial ribosomes. In eubacteria, the base at 2572(2607) is an A, and unless it moves, it will sterically prevent U2504(2539) from adopting the archaeal position. In archaea as well as in eucaryotes the base at that position is a U, which is smaller, and it forms a H-bond with U2504(2539) securing it in its place (Figure 5).
The recently published structure of linezolid, another drug that binds in the A-site cleft, complexed with the H. ma ribosome shows that it is possible for U2504(2539) to adopt the eubacterial conformation. 17(Figure 4e and f). However, the fact that this placement of U2504 (2539) is not normally seen in H. ma ribosomes indicates that the conformational rearrangement required to put it there has an appreciable free energy cost. Either the system “pays” that cost and then benefits from stacking interactions with U2504 (2539), which is what happens when linezolid binds (Figure 4e), or it does not, which is the outcome when tiamulin binds (Figure 4a). In either case, both linezolid and tiamulin should bind more strongly to eubacterial ribosomes than they do to eukaryotic/archaeal ribosomes, 36,37 and hence preferentially inhibit the former. [Preliminary data obtained in the course of this work indicate that the MIC for H. ma for tiamulin is ~ 400 μg/ml (~0.66 mM), which is about 8 times its MIC for E. coli and 8000 times its MIC for S. aureus 7(Gurel, unpublished observations). Thus H. ma is highly resistant to tiamulin, consistent with the structural findings discussed here.]
The two structures available for tiamulin/ribosome complexes reveal a number of less important differences in the way the drug interacts with archaeal and eubacterial ribosomes. For example, the conformation of the tail of the drug is not the same (Figure 4a), and in the D. radiodurans complex the hydroxyl group on the 8-membered ring of tiamulin appears to hydrogen-bond directly to the O1P of G2505(2540), instead of interacting with that same O1P through an intervening water molecule. Furthermore, in D. radiodurans, the binding of pleuromutilin derivatives induces a rotation of the U2506(2541) base towards the tricyclic core of these drugs 8 similar to that seen when homoharringtonine binds to the H. ma.
Homoharringtonine
The refinement of the homoharringtonine structure bound to the large ribosomal subunit was not straightforward because the structure of the drug deposited in the Cambridge Structural Database (CSD) (http://www.ccdc.cam.ac.uk/products/csd/) appears to have the wrong hand. 38 The structure from the CSD could not be fit into the corresponding difference electron density in any orientation, but its mirror image could. Interestingly, the stereochemistry of the chiral centers of homoharringtonine implied by its chemical name 10 indicates that the structure deposited in the CSD is inverted, which suggests that use of its mirror image here is justified (Figure 6).
Figure 6.

Choosing the correct homoharringtonine enantiomer. A stereo pair is provided that compares the fit of the refined homoharringtonine structure (gold), structure to the electron density for that drug with the fit obtained for the homoharringtonine structure obtained from Cambridge Structural Database (green), which is its enantiomer. The electron density shown is Fo-Fo difference electron density contoured at 3σ.
The structure of the ribosome complex with homoharringtonine presented above rationalizes the structure-activity relationship data that have been obtained for the drug. Activity assays of several derivatives of homoharringtonine done using HeLa cells, reticulocytes and reticulocyte lysates indicate that neither the ring system nor the tail moiety of homoharringtonine are effective inhibitors by themselves15. Consistent with these observations, both the ring system and the tail of homoharringtonine participate in interactions with the ribosome that contribute to drug binding. The most striking interaction made by the tail is its hydrophobic interaction with the base of U2506(2541), which locks the drug in its binding site. (As noted earlier, the movement of that base observed when homoharringtonine binds is similar to the one that occurs when pleuromutilins bind to ribosomes for D. radiodurans (Figure 3d))
Insight into the structural differences that determine the species specificity of homoharringtonine were obtained by superimposing the structure of the E. coli large ribosomal sububunit 39 on the structure of the ribosome-drug complex reported here (Figure 4b). E. coli is resistant to the drug, 40 and H. ma, whose ribosomes resemble eukaryotic ribosomes, is likely to be more sensitive. The E. coli ribosome differs from the H. ma ribosome in two ways that could explain its reduced affinity for homoharringtonine. First, in the E. coli structure the orientation of the base of U2506(2541) is different from what it is in H. ma, but we do not think this observation is significant (Figure 4b), because the placement of U2506(2541) appears to be highly variable. Five different orientations of that base can be observed in reported crystal structures (Figure 4). Figure 4d shows three of them, namely the one observed in the apo- structures of E. coli and D. radiodurans ribosomes, 41 the positioning reported for that base in the apo- structure of the T. thermophilus ribosome, and finally the placement normally observed in H. ma. The fourth orientation is seen in the complex that homoharringtonine forms with H. ma large ribosomal subunit (Figure 4b), and the fifth is the rotated orientation found in complexes of pleuromutilin derivatives with eubacterial ribosomes 8(not shown). The second difference between the structures of E. coli and H. ma, and far more significant in our view, is once again a difference in the placement of U2504(2539). If homoharringtonine were to bind to a eubacterial ribosome the way it binds to the ribosomes of H. ma, it would clash with U2504 (2539) (Figure 4b).
Since the conformation of the ribosome in the A-site cleft region exhibits some plasticity, a sufficiently high concentration of the drug might enable homoharringtonine to bind to eubacterial ribosomes, just as tiamulin, a drug specific for eubacteria binds to archaeal ribosomes. However, the conformational change required to relieve the clash between U2504(2539) and homoharringtonine in eubacterial ribosomes would have a free energy cost, which eukaryotic/archaeal ribosomes do not have to “pay”, and which should make its affinity for eubacterial ribosomes lower than its affinity for eukaryotic/archaeal ribosomes. The specificity of anisomycin for eukaryotic/archaeal ribosomes, which is another A-site cleft inhibitor, can be explained the same way (Figure 4e). 25
Bruceantin
Poly(U) directed polyphenylalanine synthesis systems derived from halophilic archaea are less sensitive to bruceantine than similar systems derived from yeast and more sensitive than the in vitro systems derived from E. coli. 14 The structure of the large ribosomal subunit with bruceantin bound can explain the structure-activity data available. For example, the reason a double bond is required in the 6-membered ring at the left end of the bruceantin ring system (Figure 2c) to obtain inhibition of protein synthesis is the insertion of the ring system into the A-site cleft (Figure 3f). 16 If that double bond were replaced by a single bond, the ring would be non-planar, and the drug would no longer fit in the cleft due to steric clashes between the oxygen atoms in that ring and the ribosomal bases that form the cleft.
Once again the species specificity of bruceantin can be explained by species-dependent differences in the position of U2504(2539). If bruceantin were to bind to the ribosome of E. coli there would be steric clashes between the drug and the bases of U2504 (2539) and U2506 (2541) as well as with the phosphate oxygens of G2505 (2540) unless a significant conformational change occurred in that region (Figure 4c). Thus the species specificity of bruceantin can be explained exactly the same way as the species specificity of homoharringtonine.
The here presented structural studies provide an explanation for the mechanism of action and for the species-specificities exhibited by three inhibitors -tiamulin, homoharringtonine and tiamulin- that bind to the binding site of the aminoacid side chain of the A-site tRNA. The affinity of these inhibitors for the A-site of ribosomes from different kingdoms arises from the interactions they can or cannot make with the base of a single nucleotide, U2504(2539), whose variable position is controlled by its interactions with neighboring nucleotides. Since the identities of these controlling neighboring nucleotides are the same in the ribosomes of H. ma and in eukaryotes but different in eubacteria, we conclude that these studies of the H. ma ribosome complexes with these inhibitors provide the structural bases for explaining their different specificities for eukaryotes and eubacteria.
Materials and Methods
Inhibitors
BM Cyclin 1 (Tiamulin) was purchased from Roche. Homoharringtonine (NSC: 141633) and bruceantin (NSC: 165563) were obtained from the Open Chemical Repository of the Developmental Therapeutics Program sponsored by the National Cancer Institute (http://dtp.nci.nih.gov).
Preparation of Crystals
50S ribosomal subunits were crystallized and stabilized as described previously34,42,43. Crystals were soaked in solutions containing the inhibitors in buffer B [12% polyethylene glycol 6000, 20% ethylene glycol, 1.7M NaCl, 0.5 M NH4Cl, 1 mM CdCl2, 100 mM KCH3COO, 6.5 mM CH3COOH (pH 6.0), 5mM β-mercaptoethanol, and with 30 mM MgCl2, or 21 mM MgCl2 and 30 mM SrCl2] for several hours, with one exchange of soaking solution at 4 °C. All three inhibitors were used at a concentration of 1mM.
Crystallography
Data were collected at the National Synchrotron Light Source (Brookhaven National Laboratory) on beamline X29 and at the Advanced Photon Source (Argonne National Laboratory) on beamline 24-ID-C, as previously described (Table 1). 34 After data processing with HKL200044, difference electron density maps were calculated for each complex using amplitudes (|Fo(hkl)| drug bound ribosome-|Fo (hkl) | best apo) and phases obtained from an apo- large ribosomal subunit structure (PDB 3CC2,). 25 The starting models for homoharringtonine 38 and tiamulin45 were obtained from Cambridge Structure Database (http://www.ccdc.cam.ac.uk/products/csd/). The starting model for bruceantin was created using the Dundee PRODRG2 server (http://davapc1.bioch.dun-dee.ac.uk/prodrg/). The refinement in CNS included rigid-body refinement, minimization, and B-factor refinement 46(Table 2). Structures were visualized in the program O and figures were generated using the program PyMol47,48. Buried surface areas were calculated in CNS 46 using the Lee and Richards algorithm49.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with the following accession numbers: 3G4S (tiamulin), 3G6E (homoherringtonine), and 3G71 (bruceantin).
Acknowledgments
We thank Larysa Vasylenko for technical assistance, Jimin Wang for useful discussions during structure refinement, the staff at the Center for Structural Biology Core Laboratory for computational help, and the staffs of beamline X29 (National Synchrotron Light Source, Brookhaven National Laboratory) and beamline 24-ID-C (Advanced Light Source, Argonne National Laboratory) for their support during data collection. This work was supported by a grant from the National Institutes of Health to PBM and TAS (GM022778).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J Mol Biol. 2003;330:1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- 2.Wilson DN, Harms JM, Nierhaus KH, Schlunzen F, Fucini P. Species-specific antibiotic-ribosome interactions: implications for drug development. Biol Chem. 2005;386:1239–1252. doi: 10.1515/BC.2005.141. [DOI] [PubMed] [Google Scholar]
- 3.Vazquez D. Molecular Biology Biochemistry and Biopyhsics. Springer Verlag; New York: 1979. Inhibitors of Protein Biosynthesis. [DOI] [PubMed] [Google Scholar]
- 4.Drews J, Georgopoulos A, Laber G, Schutze E, Unger J. Antimicrobial Activities of 81.723 Hfu, a New Pleuromutilin Derivative. Antimicrob Agents Chemother. 1975;7:507–516. doi: 10.1128/aac.7.5.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kavanagh F, Hervey A, Robbins JW. Antibiotic Substances from Basidiomycetes. VIII Pleurotus Multilus (Fr.) Sacc and Pleurotus Passeckerianus Pilat. Proc Natl Acad Sci USA. 1951;37:570–574. doi: 10.1073/pnas.37.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodgin LA, Hogenaue G. Mode of Action of Pleuromutilin Derivatives - Effect on Cell-Free Polypeptide-Synthesis. Eur J Biochem. 1974;47:527–533. doi: 10.1111/j.1432-1033.1974.tb03721.x. [DOI] [PubMed] [Google Scholar]
- 7.Hogenauer G. Mode of Action of Pleuromutilin Derivatives - Location and Properties of Pleuromutilin Binding-Site on Escherichia-Coli Ribosomes. Eur J Biochem. 1975;52:93–98. doi: 10.1111/j.1432-1033.1975.tb03976.x. [DOI] [PubMed] [Google Scholar]
- 8.Davidovich C, Bashan A, Auerbach-Nevo T, Yaggie RD, Gontarek RR, Yonath A. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc Natl Acad Sci USA. 2007;104:4291–4296. doi: 10.1073/pnas.0700041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlunzen F, Pyetan E, Fucini P, Yonath A, Harms JM. Inhibition of peptide bond formation by pleuromutilins: the structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol Microbiol. 2004;54:1287–1294. doi: 10.1111/j.1365-2958.2004.04346.x. [DOI] [PubMed] [Google Scholar]
- 10.Kantarjian HM, Talpaz M, Santini V, Murgo A, Cheson B, O’Brien SM. Homoharringtonine - History, current research, and future directions. Cancer. 2001;92:1591–1605. doi: 10.1002/1097-0142(20010915)92:6<1591::aid-cncr1485>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 11.Powell RG, Weislede D, Smith CR. Antitumor Alkaloids from Cephalotaxus-Harringtonia - Structure and Activity. J Pharm Sci. 1972;61:1227. doi: 10.1002/jps.2600610812. [DOI] [PubMed] [Google Scholar]
- 12.Paudler WW, Kerley GI, McKay JB. Alkoloids of Cephalotaxus Drupacea and Cephalotaxus Fortunei. J Org Chem. 1963;28:2194–2197. [Google Scholar]
- 13.Fresno M, Gonzales A, Vazquez D, Jimenez A. Bruceantin, a Novel Inhibitor of Peptide-Bond Formation. Biochim Biophys Acta. 1978;518:104–112. doi: 10.1016/0005-2787(78)90120-x. [DOI] [PubMed] [Google Scholar]
- 14.Sanz JL, Marin I, Urena D, Amils R. Functional-Analysis of 7 Ribosomal Systems from Extremely Halophilic Archaea. Can J Microbiol. 1993;39:311–317. [Google Scholar]
- 15.Huang MT. Harringtonine, an Inhibitor of Initiation of Protein-Biosynthesis. Mol Pharmacol. 1975;11:511–519. [PubMed] [Google Scholar]
- 16.Liao LL, Kupchan SM, Horwitz SB. Mode of Action of Antitumor Compound Bruceantin, an Inhibitor of Protein-Synthesis. Mol Pharmacol. 1976;12:167–176. [PubMed] [Google Scholar]
- 17.Ippolito JA, Kanyo ZF, Wang DP, Franceschi FJ, Moore PB, Steitz TA, et al. Crystal structure of the oxazolidinone antibiotic linezolid bound to the 50S ribosomal subunit. J Med Chem. 2008;51:3353–3356. doi: 10.1021/jm800379d. [DOI] [PubMed] [Google Scholar]
- 18.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 19.Liao LL, Grollman AP, Horwitz SB. Bruceantin - Inhibitor of Protein-Biosynthesis. Fed Proc. 1974;33:581–581. [Google Scholar]
- 20.Coutsoge C. On Mechanism of Action of Chloramphenicol in Protein Synthesis. Biochim Biophys Acta. 1966;129:214. doi: 10.1016/0005-2787(66)90027-x. [DOI] [PubMed] [Google Scholar]
- 21.Coutsoge C. Amino Acylaminonucleoside Inhibitors of Protein Synthesis. Effect of Amino Acyl Ribonucleic Acid on Inhibition. Biochemistry. 1967;6:1704. doi: 10.1021/bi00858a019. [DOI] [PubMed] [Google Scholar]
- 22.Jardetzky O. Studies on Mechanism of Action of Chloramphenicol. 1 Conformation of Chloramphenicol in Solution. J Biol Chem. 1963;238:2498. [PubMed] [Google Scholar]
- 23.Fresno M, Jimenez A, Vazquez D. Inhibition of Translation in Eukaryotic Systems by Harringtonine. Eur J Biochem. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- 24.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 25.Blaha G, Gurel G, Schroeder SJ, Moore PB, Steitz TA. Mutations outside the anisomycin-binding site can make ribosomes drug-resistant. J Mol Biol. 2008;379:505–519. doi: 10.1016/j.jmb.2008.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cammarano P, Teichner A, Londei P, Acca M, Nicolaus B, Sanz JL, et al. Insensitivity of Archaebacterial Ribosomes to Protein-Synthesis Inhibitors - Evolutionary Implications. Embo J. 1985;4:811–816. doi: 10.1002/j.1460-2075.1985.tb03702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol Cell. 2002;10:117–128. doi: 10.1016/s1097-2765(02)00570-1. [DOI] [PubMed] [Google Scholar]
- 28.Schroeder SJ, Blaha G, Moore PB. Negamycin binds to the wall of the nascent chain exit tunnel of the 50S ribosomal subunit. Antimicrob Agents Ch. 2007;51:4462–4465. doi: 10.1128/AAC.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeder SJ, Blaha G, Tirado-Rives J, Steitz TA, Moore PB. The structures of antibiotics bound to the E site region of the 50 S ribosomal subunit of Haloarcula marismortui: 13-deoxytedanolide and girodazole. J Mol Biol. 2007;367:1471–1479. doi: 10.1016/j.jmb.2007.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tu D, Blaha G, Moore PB, Steitz TA. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005;121:257–270. doi: 10.1016/j.cell.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Tenson T, Mankin A. Antibiotics and the ribosome. Mol Microbiol. 2006;59:1664–1677. doi: 10.1111/j.1365-2958.2006.05063.x. [DOI] [PubMed] [Google Scholar]
- 32.Hill WE, Dahlberg A, Garrett RA, MoorePBSchlessinger D, Warner JR. American Society for Microbiology Washington DC. 1990. The Ribosome:Structure, Function and Evolution; pp. 617–633. [Google Scholar]
- 33.Danson MJ, Hough DW, Lunt GG. The Archaebacteria: Biochemistry and Biotechnology. Portland Press; 1992. pp. 89–98. [Google Scholar]
- 34.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 35.Dahlberg AE, Zimmermann RA. Ribosomal RNA: Structure, Evolution, Processing, and Function in Protein Synthesis. C.R.C. Press; Boca Raton: 1995. [Google Scholar]
- 36.Hogenauer G, Ruf C. Ribosomal-Binding Region for the Antibiotic Tiamulin - Stoichiometry, Subunit Location, and Affinity for Various Analogs. Antimicrob Agents Chemother. 1981;19:260–265. doi: 10.1128/aac.19.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandi L, Fabbretti A, La Teana A, Abbondi M, Losi D, Donadio S, et al. Specific, efficient, and selective inhibition of prokaryotic translation initiation by a novel peptide antibiotic. Proc Natl Acad Sci USA. 2006;103:39–44. doi: 10.1073/pnas.0507740102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao G, Xu C, He C, Yao J. The Crystal Structures of Harringtonine and Homoharringtonine. Acta Acad Med Sin. 1988;19:283–287. [PubMed] [Google Scholar]
- 39.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, et al. Structures of the bacterial ribosome at 3.5 angstrom resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 40.Oliver JL, Sanz JL, Amils R, Marin A. Inferring the Phylogeny of Archaebacteria - the Use of Ribosomal Sensitivity to Protein-Synthesis Inhibitors. J Mol Evol. 1987;24:281–288. [Google Scholar]
- 41.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, et al. High resolution structure of the large ribosomal subunit from a mesophilic Eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 42.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Ban N, Freeborn B, Nissen P, Penczek P, Grassucci RA, Sweet R, et al. A 9 angstrom resolution x-ray crystallographic map of the large ribosomal subunit. Cell. 1998;93:1105–1115. doi: 10.1016/s0092-8674(00)81455-5. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Cao XJ, Sun CR, Pan YJ. Tiamulin-hydrogen fumarate-methanol (1/1/1) Acta Crystallogr E. 2004;60:o1546–o1548. [Google Scholar]
- 46.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.DeLano WL. The Pymol Molecular Graphics System. De Lano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 48.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved Methods for Building Protein Models in Electron-Density Maps and the Location of Errors in These Models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 49.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 50.Klein DJ, Moore PB, Steitz TA. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J Mol Biol. 2004;340:141–177. doi: 10.1016/j.jmb.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 51.Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Weir JR, Kelley AC, Ramakrishnan V. Structure of the Thermus Thermophilus 70S Ribosomecomplexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 52.Wilson DN, Schluenzen F, Harms JM, Starosta AL, Connell SR, Fucini P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc Natl Acad Sci USA. 2008;105:13339–13344. doi: 10.1073/pnas.0804276105. [DOI] [PMC free article] [PubMed] [Google Scholar]