

Abstract
New developments regarding the structure and in vivo dynamics of protein kinase B (PKB/Akt) have been recently exposed. Here, we specifically review how the use of multi-disciplinary approaches has resulted in reaching the recent progress made to relate the quaternary structure of PKB to its in vivo function. Using X-ray crystallography, the structure of PKB pleckstrin homology (PH) and kinase domains was determined separately. The molecular mechanisms involved in (a) the binding of the phosphoinositides to the PH domain and (b) the activation of the kinase with the rearrangement of the catalytic site and substrate binding were determined. In vitro, nuclear magnetic resonance and circular dychroism studies gave complementary information on the interaction of the PH domain with the phosphoinositides. However, the molecular nature and the function of the interactions between the PKB domains could not be deduced from the X-ray data since the full-length PKB has not been crystallised. In vitro, dynamic information on the inter-domain conformational changes related to PKB activation states emerged with the use of tandem mass spectrometry. Cell imaging and Förster resonance energy transfer provided in vivo dynamics. Molecular modelling and dynamic simulations in conjunction with mutagenesis and biochemical analysis were used to investigate the complex interactions between the PKB domains in vivo and understand at the molecular level how it linked to its activity. The compilation of the information obtained on the 3-D structure and the spatiotemporal dynamics of this widely studied oncogene could be applied to the study of other proteins. This inter-disciplinary approach led to a more profound understanding of PKB complex activation mechanism in vivo that will shed light onto new ideas and possibilities for modulating its activity.
Keywords: Protein kinase B, Akt, 3-D structure, Molecular modelling, Dynamics, Multi-disciplinary approach, FRET/FLIM
Introduction
Protein kinase B (PKB/Akt) belongs to the AGC super family of related serine/threonine protein kinases including protein kinase A (PKA), p90 ribosomal S6 kinase, p70 ribosomal S6 kinase and serum and glucocorticoid-induced protein kinase (for review, see [35, 45]). Three isoforms of PKB exist in mammals (PKBα/Akt1, PKBβ/Akt2 and PKBγ/Akt3) that contain an N-terminal pleckstrin homology (PH) domain, a catalytic (kinase) domain and a C-terminal regulatory part containing a hydrophobic motif (HM) [12, 54]. PKB is a key regulator downstream of various growth factors and hormones [18]. It activates numerous proteins involved in metabolism, proliferation, growth and survival ([48] and for review, see [42]). Several lines of evidence indicate that PKB pathway is involved in human cancer [13, 49, 57] and in particular, its over expression induces malignant transformation and chemoresistance (for review, see [20, 60]). A critical step in PKB activation is its translocation to the plasma membrane due to the interaction of its PH domain with the lipid products of the PI3-kinase (PtdIns (3,4,5) P3 and PtdIns (3,4) P2) [6]. Two regulatory sites essential for PKB activation have been identified; Thr 308 in the activation loop of PKBα /Akt 1 kinase domain and Ser 473 in the hydrophobic motif of the C-terminal part (Thr 309 and Ser 474 in PKBβ/Akt 2). The phosphorylation of both regulatory sites leads to the full activation of PKB [2] and whether Thr 308 needs to be phosphorylated prior to Ser 473, or vice versa, remains to be elucidated. Independently of these critical residues, the phosphorylations of Thr 450 of the turn motif [11, 30] as well as phosphorylations on tyrosines have also been reported to play a role in PKB activation [19, 23].
Phosphoinositide-dependent protein kinase-1 (PDK1) is another PH domain containing kinase that belongs to the AGC protein kinases family [45]. It is activated by PtdIns (3,4,5) P3 and has been shown to be responsible for the phosphorylation of PKB on Thr 308 [3]. The identity of the Ser 473 kinase has been highly debated and it is possible that more than one protein might be responsible for Ser 473 phosphorylation depending on the context. Both DNA-dependent protein kinase [24] and the target of Rapamycin complex 2 (TORC2) [52] were recently described as Ser 473 kinases. The dephosphorylation of PKB on the two regulatory residues Thr 308 and Ser 473 has been shown to lead to its deactivation [43]. The PP2A family of okadaic-sensitive phosphatases were proposed to be responsible for the dephosphorylation of Thr 308 and Ser 473 [14, 7, 43]. More recently, a family of okadaic acid-insensitive phosphatases, PHLLP, was also shown to promote the dephosphorylation of Ser 473 [27].
Structural studies have been critical to establish a deeper understanding of PKB activation mechanism. The isolated PKB PH and kinase domains structures were resolved by X-ray crystallography and gave key insights into the understanding of the domains function at a molecular level. However, full-length PKB protein failed to crystallise. The use of alternative methods to crystallography such as protein mass spectrometry, Förster resonance energy transfer (FRET) by fluorescence lifetime imaging microscopy, molecular dynamics together with classical biochemical approaches was essential to understand the relationship between the domains and to determine the overall topology of the protein. The in vivo conformational change of PKB at different stages of its activation was described and its in situ molecular behaviour relative to the activation by PDK1 was exposed.
Structure of isolated PKB domains by X-ray crystallography and NMR—insights into their function
Domain prediction software has allowed the identification of three domains in PKB. The N-terminal PH domain comprises the residues 1–133; the kinase domain, the residues 150–408 and the C-terminal regulatory part that contains a HM, the residues 449–480 (numbering for PKBα). Flexible linkers connect the domains. Their structures have, however, not been crystallised and could not be predicted by modelling (Fig. 1). The resolution of crystal structures of isolated kinase and PH domains of PKB as well as the use of circular dichroism and nuclear magnetic resonance (NMR) spectroscopy have given insights into the mechanism of PKB allosteric activation by phosphorylations and HM interaction and into the role for the plasma membrane interaction with PtdIns (3,4,5) P3.
Fig. 1.

PKB/Akt primary structure. a Structural alignment of selected members of the AGC superfamily of protein kinases: the three protein kinase B (PKB/Akt) isoforms, phosphoinositides-dependent protein kinase 1 (PDK1) and protein kinase A (PKA). A high homology exists at the level of the kinase domains (grey). The residues highlighted in pink represent the phosphorylation sites important for the activity of the kinases. PH domains are represented in blue. b Amino acid sequence of PKBα/Akt1. The isolated sections of the protein that have been crystallised are highlighted in blue (PH domain), orange (kinase domain) and green (hydrophobic motif). The red stars indicate the residues Thr 308 and Ser 473. The domains are separated by linker regions that have not been crystallised. The linker 1 is between the PH domain and the kinase domain and the linker 2 is between the kinase domain and the C terminus
Mechanism of PKB kinase domain activation
Structure of the PKB kinase domain
The structure of inactive unphosphorylated PKBβ kinase domain was described independently by Yang et al. [62] and Huang et al. [32]. Yang et al. presented the high resolution crystal structures of the recombinant kinase domain of PKBβ (residues 146–460) deleted of the PH domain and of the last 21 residues of the C-terminal (ΔPH-PKBβ-ΔCt) and of ΔPH-PKBβ (residues 146–481). In the study of Huang et al., the kinase domain of PKBβ (residues 143–481) was analysed.
PKB quaternary structure resembled the first crystallised AGC kinase, PKA kinase domain, in its closed (active) conformation. It comprised an N-terminal lobe formed of five-stranded β sheets with two α helices (αB and αC helices) and a larger C-terminal lobe containing mainly α helices and the activation loop. The ATP-binding site was situated in the cleft at the interface of the two lobes and the peptide-binding site in the C-terminal lobe. However, important differences between the two kinases were observable. A rotation of about 20° of the relative orientation of the N- and C-lobes in PKBβ kinase domain induced a misalignment of the active site residues. Both studies also found that the αB and αC helices of the N-lobe of PKBβ kinase domain were disordered. In addition, most of the activation loop was also disordered and the remaining ordered segment adopted a conformation that sterically blocked the ATP and peptide-binding sites. Furthermore, an intra-molecular disulphide bond in the active site as well as residues in a region between the N- and C-terminal lobes contributed to the inactive conformation [32]. These findings indicated a complex mechanism of auto-inhibition of protein kinases not described before that could possibly be generalised to other members of the AGC kinase family. It is interesting to note that in the crystal structures of PKBβ containing the C-terminal, the last 40 residues of the protein (comprising the HM) were disordered. These data suggested that the HM motif did not bind to the kinase. This is contrary to what happens in PKA, where the HM motif was buried inside of a hydrophobic groove in the kinase domain and interacted strongly with the αB and αC helices [37]. Therefore, the lack of binding of the PKB HM could explain the disorder of the helices αB and αC detected in the PKB structure, and hence, the role of the C-terminal HM was investigated.
Role of the activation loop and hydrophobic motif phosphorylations
Similar to PKA, the interaction of the C-terminal HM with the hydrophobic groove of PKB in addition to the active site phosphorylation was essential for its re-organisation. Using a series of peptides derived from the sequence of the PKBβ HM or of another AGC kinase PKC-related kinase 2 (PDK1-interacting fragment (PIF)tide), Yang et al. demonstrated that the HM phosphorylated on Ser 474 (or containing the phosphorylation mimicking amino acid Asp instead) induced the allosteric activation of the recombinant ΔPH-PKBβ-ΔCt mono phosphorylated on Thr 309 [62]. In fact, the sole phosphorylation of PKB Thr 309 of the activation loop was shown to be insufficient to allow the re-organisation of the catalytic site. In addition, phenylalanine residues from the PKBβ HM sequence Phe470-X-X-Phe473-Ser/Thr-Tyr (sequence present in many AGC kinases [35, 45]) were also essential for the binding of the HM. Furthermore, the loss of HM binding upon mutation of hydrophobic groove residues of PKBβ (in the αC helix or β5-strand) led to the loss of re-organisation of the active site. This result confirmed the role of the HM binding in the stabilisation of the αC helix and β5-strand and the rearrangement of PKBβ active site.
In conclusion, these studies proved the essential role of the binding of the phosphorylated C-terminal HM in the allosteric control of PKBβ kinase activity. However, the molecular mechanism involved in the activation of PKBβ by Thr 309 activation loop phosphorylation could not be investigated. Therefore, the comparison of the inactive and active crystal structures of PKB shed light onto this mechanism.
The fundamental role of the activation loop phosphorylations in the activation of protein kinases has been well documented (for reviews, see [47, 55]). By comparing the crystal structure of inactive [62] and active PKBβ [61], the role of Thr 309 phosphorylation in addition to the role of the HM in PKBβ activation was demonstrated. The structures of PKBβ residues 146 to 467 fused with the PIF fragment (PKB-PIF) and PKBβ residues 146 to 481 mutated on Ser 474 to Asp (PKB-S474D) were determined to a resolution of 1.6 and 1.7 Å [61]. The recombinant proteins were phosphorylated on Thr 309 and crystallised in complex with AMP-PNP/Mn2+ and 12 residues of GSK3β corresponding to the phosphorylation site of PKB.
Unlike previously described in inactive PKBβ kinase domain crystal structure [32, 62], the αB and αC helices of the N-lobe as well as the activation segment and the HM were ordered in both activated PKB ternary complexes. The active site was properly re-arranged with specifically Glu 200 and His 196 of the αC helix interacting with the Lys 181 and the phosphate of Thr 309 respectively allowing catalytic activity. In addition, the re-ordering of the αC helix induced a change in conformation together with the phosphorylation of Thr 309 that alleviated the steric blockage on the ATP-binding site.
As expected by comparing with PKA, an intra-molecular interaction of the phosphorylated HM with a hydrophobic groove on the N-lobe of PKBβ kinase domain was detected. The groove was formed at the interface of the ordered αB and αC helices and the β4 and β5 strands of the central β sheets. Extensive hydrophobic interactions between two phenylalanines of the HM (Phe470-X-X-Phe473-Ser-Tyr) and hydrophobic residues induced the stabilisation of the αB and αC helices. These results explained at a molecular level the importance of the HM conserved phenyalanines in PKBβ activation. The role of the Ser 474 phosphorylation in PKBβ or of a negatively charged amino acid at the equivalent position in other AGC kinases was shown to increase the binding affinity of the HM for the kinase domain hydrophobic groove.
Substrate binding
The ternary complex structures of PKB with GSK3 peptide and an ATP analogue allowed also the determination of the molecular mechanism for substrate recognition and binding. The optimal substrate sequence for PKB—Arg-X-Arg-X-X-Ser/Thr-ϕ (ϕ being a large hydrophobic residue)—had already been identified from the peptide screen and from the physiological site phosphorylation [48]. Important residues for the recognition were two arginines at positions P(−3) and P(−5); numbering starting from the Ser/Thr phosphorylation site. Glu 236 and Asp 440 were shown to interact with substrate Arg positioned at P(−3) and Glu 279 and Glu 342 with the Arg at position P(−5). From these data, the differences in substrate specificity between PKA and PKB could be explained.
In conclusion, prior to activation and phosphorylation, the PKB catalytic pocket is disordered. It is maintained in an Apo form that does not allow the binding of ATP and substrate due to steric hindrance by the activation loop. Therefore, the phosphorylation of the Thr 309 is needed to induce a change in conformation allowing the opening of the ATP-binding site. The re-ordering of the active site is also dependent on the interaction of the phosphorylated HM motif with the hydrophobic groove in the N-lobe of the kinase domain. This interaction induces a disorder to order transition allowing the realignment of the catalytic residues of the kinase active site. It is quite difficult to assess in which sequence the phosphorylations may occur from these data but it explained and also emphasised the role of the dual phosphorylations in the allosteric activation of PKBβ. The allosteric activation of AGC and non-AGC kinases by domain interaction and phosphorylation was recently compared [28]. Since PKB activation had been shown to be strictly dependent on its translocation to the plasma membrane and the interaction of its PH domain with the phosphoinositides products of PI3-kinase, it was important to understand the underling mechanism of the PH domain binding to its substrate.
Function of the PKB PH domain
Mechanism of PKB PH domain interaction with PtdIns(3,4,5)P3
Two crystal structures of PKBα PH domain interacting with Ins (1,3,4,5) P4 were resolved at 1.4 [58] (residues 1–113) and 0.98 Å [44] (residues 1–123). They showed that the PH domain interacted specifically with the phosphates in position D3 and D4 (but not D5) of the inositol ring, explaining why PKB bound similarly to PtdIns (3,4,5) P3 and PtdIns (3,4) P2. This interaction involved a cluster of basic residues on the PH domain known as the phosphoinositide-binding pocket. The D3 phosphate was shown to interact with Lys 14, Arg 23, Arg 25 and Asn 53 and the D4 phosphate with Lys 14, Asn 53 and Arg 86. The D5 phosphate did not interact with any residues in the binding pocket and was solvent exposed.
A second cluster of basic residues was identified—Arg 15, Lys 20, Arg 67 and Arg 69—that was not involved in the binding with Ins (1,3,4,5) P4. These basic residues maybe involved in stabilising the PH domain at the plasma membrane by interacting with negatively charged lipids [26]. In agreement with a role of these residues in the activation of PKB, a mutation of Arg 15 Ala impaired the platelet-derived growth factor (PDGF)-stimulated PKB activation [11].
The relative positioning of the PtdIns (3,4,5) P3 head group in PKBα PH domain pocket appeared to be very different from its positioning in other PH domain containing proteins. Overall, PKBα PH domain formed fewer interactions with Ins (1,3,4,5) P4 than the phosphoinositide-binding PH domains of dual adaptor of phosphotyrosine and 3-phosphoinositides (DAPP1), general receptor for phosphoinositides isoform 1 (GRP1) or Bruton tyrosine kinase (BTK). This may explain why the affinity of PKBα PH for the phosphoinositides had previously been shown to be tenfold lower than the affinity of other PH domains [3, 25, 56]. It is possible that the maintenance of a tight balance between activated and non-activated forms of PKB would explain the necessity for a weaker affinity for PtdIns (3,4,5) P3 compared to other PtdIns (3,4,5) P3 PH domains.
As expected, the mutagenesis of Lys 14, Arg 25 and Arg 86 totally abolished the interaction of the PH domains with PtdIns (3,4) P2 and PtdIns (3,4,5) P3 in an overlay assay and also prevented the activation of the equivalent full-length mutant proteins in vitro. It is interesting to note that although the mutation of Asn 53 reduced significantly the interaction of the PH domain with the phosphoinositides in the overlay assay, it did not preclude the full activation of PKB in an in vitro kinase assay. A hypothesis would be that this residue might be destabilising the interface between PH and kinase domain allowing an easier access to Thr 308 by PDK1. In accordance with the hypothesis, Asn 53 was found to be very close to the PH-kinase interface in the inactive PKB conformer (see the last section). The binding of the phosphoinositides induced a change in conformation of the PKB PH domain that was discovered by comparison of the structures of the bound and unbound PH domains.
Change in conformation of PKB PH domain upon binding to PtdIns(3,4,5) P3
Significant changes in the conformation of the phosphoinositides-binding pocket was detected in PKBα PH domain unbound (Apo form) resolved at 1.65 Å [44] that was further confirmed by circular dichroism. Such changes had not yet been depicted in other PtdIns (3,4,5) P3 binding PH domains. In particular, a significant shift in the position of variable loop 3 (VL3) of 7.4 Å was detected. Interestingly, a hydrophobic residue at the top of VL3 (Trp 80) was found to be solvent exposed in the Apo form. This finding indicated that Trp 80 might be interacting with another domain of PKB in the context of the full-length protein or with another protein altogether. The critical function of the Trp 80 in the structure of full-length PKB was determined later using molecular dynamics and will be extensively discussed on the last section. The binding to Ins (1,3,4,5) P4 induced also the re-ordering of the variable loop 2 (VL2) from a flexible structure to an α helix upon a 7.6-Å shift, creating a patch of solvent exposed acidic residues (Asp 44, Asp 46, Glu 49 and Glu 40) whose purpose has not yet been elucidated. Interestingly, (Glu 40 Lys) mutation was shown to induce an increased PKB basal activity [11]. Two possible explanations were proposed. The first one was that it might increase the affinity for the membrane. The second suggested that since the residue was not involved in the binding to PtdIns (3,4,5) P3, the mutation might be destabilising the interface between PH and kinase domain allowing an easier access of Thr 308 by PDK1. The dynamic model of PKB PH–kinase interaction presented in the last section [16] showed that Glu 40 is positioned at the surface of the PH domain far away from the PH–kinase interface and would be close to the membrane when bound to PtdIns (3,4,5) P3. Therefore, the effect of the mutation seems likely to be due to an increase in the affinity of the PH domain for the membrane.
The NMR study on PKBβ PH domain (residues 1–111) interacting with Ins (1,3,4,5) P4, [8] confirmed that the overall structure of the PH domain was similar to that of PKBα PH domain in the crystal structure. However, major differences appeared in the variable loops VL2 and VL3. Although VL3 was shown well defined in the crystal structure, most of the residues were not visible in the NMR study. Since the amino acid sequence of the loop in the two isoforms was almost identical, the result could only be explained by the differences in the dynamics of VL3. This result was in accordance with a high flexibility of the loop that might have been stabilised in the crystal structure by the packing. VL2 structure in the same manner was not very well defined, due to its intrinsic flexibility. It is of interest to note that the sequence between the two isoforms of PKB in this region is very different. Whereas VL2 forms a helix upon binding to Ins (1,3,4,5) P4 in PKBα PH crystal structure, it is very unlikely that this would happen for PKBβ PH domain due to its high content in prolines in the equivalent region.
In conclusion, the PKB PH domain interacts with the phosphoinositides at the plasma membrane by interaction with the phosphates in position D3 and D4 of the inositol ring. This binding induces a change in the conformation of the PH domain. The unexpected positioning of specific residues on the isolated PH domain was also indicative of its possible interaction with another protein or another domain of PKB prior to stimulation. However, the lack of information on the relative positioning of PKB domains prevented the drawing of any conclusions regarding the role of each domain in the regulation of PKB activity. Therefore, dynamic information was necessary to comprehend fully PKB activation mechanism.
A change in the conformation of full-length PKB upon plasma membrane translocation was proposed to be responsible for the activation of the kinase. Therefore, the subcellular localisation, the activation status, the binding to activators as well as the changes in conformation of PKB in real time and in intact cell were studied using in situ resonance transfer (FRET) imaging.
Imaging PKB dynamics in intact cells by FRET
Variations in the fluorescence lifetime or the fluorescence intensity ratio of fluorescent reporters allowed the monitoring of multiple dynamic events by live cell imaging. A series of genetically encoded fluorescent biosensors for PKB activation, phosphorylation, conformational change or lipid binding were made (for review, see [63]). They consisted either of full-length PKB, of the assembly of modular blocks from PKB or other protein domains or specific peptide sequences fused at the N- and C-terminal to fluorescent proteins appropriate as a FRET pair. Moreover, the inter-molecular interaction of PKB with its direct activator, the Thr 308 kinase PDK1, was investigated.
Monitoring PKB conformational changes in live cells
The change in PKB conformation upon binding with 3-phosphoinositides at the plasma membrane was detected in intact cells by FRET using fluorescence lifetime imaging microscopy (FLIM). Initially, the green fluorescent protein (GFP) and the yellow fluorescent protein (YFP) were fused to PKBα (GFP-PKBα-YFP) [15] and then YFP was replaced by monomeric red fluorescent protein (mRFP) to create the more sensitive sensor (GFP-PKBα-mRFP) [14]. An increased FRET efficiency at the plasma membrane upon PDGF stimulation indicated that PKB had changed in conformation upon translocation (Fig. 2). The same group independently determined that the appearance of Ser 473 phosphorylation was concomitant to PKB translocation at the plasma membrane upon PDGF stimulation in intact cells [1]. This readout of PKB activation was measured by classical FRET using GFP-PKB as a donor and a phosphospecific antibody for Ser 473 labelled with the Cy3 dye as an acceptor. The highly FRETing species corresponding to the active conformation of PKB started to detach from the plasma membrane 5 min after translocation and the nucleus was populated with active PKB after 20 min. Accordingly, Kunkel et al. showed that a wave of PKB substrate phosphorylations was detected starting from the plasma membrane followed by the cytoplasm and finally to the nucleus [38]. These data suggested a turnover of PKB activation whereby new molecules of inactive PKB would translocate to the plasma membrane to be activated, whereas previously activated molecules detached, to eventually become dephosphorylated. Therefore, the maintenance of the active conformation would be dependent on the presence of the phosphorylations on Thr 308 and Ser 473. These movements of the protein would continue as long as the 3-phosphoinositides would be present.
Fig. 2.

Model for GFP and mRFP change in distance upon GFP-PKB-mRFP change in conformation. a The kinase domain of PKB is shown in purple ribbons with GFP and mRFP, respectively, in green and red ribbons. In gold, PKB PH domain is shown in its “PH-in” conformation. The white dashed lines represent the linkages between the PKB domains and GFP or mRFP. b When the PH domain moves to its “PH-out” conformation (orange), GFP moves to a new location. During this movement, the distance between GFP and mRFP becomes smaller (distances shown in white lines are taken between the centres of domains or proteins)
Ananthanarayanan et al. described the role of PKB phosphorylations on PKB conformation [4]. A membrane targeted (Lyn targeting sequence) full-length PKBα flanked with cyan fluorescent protein (CFP; N-terminal) and YFP (C-terminal) was created. This reporter named pm-reporter of Akt action (ReAktion) was used to monitor PKB change in conformation upon stimulation. The authors determined that the mutation of Thr 308 not only to Ala but also to an amino acid that would mimic the phosphorylation (Asp) prevented the detection of a change in conformation. This result with the Asp mutant seemed quite surprising since the mutation Thr 308 Asp is known to activate the kinase, and it was shown by them and others [14] that the phosphorylation of Thr 308 correlated with the change in conformation. It is possible that although the mutation to alanine maintained the protein in an inactive and inactivable conformation preventing the change in conformation to occur, the aspartate mutant may already bear an active conformation that would not detectably (using this FRET read out) change upon stimulation. The phosphorylation status of Ser 473 did not affect the change in conformation upon stimulation. Interestingly, the mutation of Thr 308 to Asp on a non-membrane targeted ReAktion prevented its translocation to the plasma membrane upon activation, indicating that phosphorylation on Thr 308 was likely to play a role in the detachment from the plasma membrane in the presence of 3′-phosphoinositides. The binding of isolated PKB PH with the kinase domain was shown by co-immunoprecipitation. The interaction was increased by stimulation and in particular needed Thr 308 phosphorylation. These results suggested that in addition to the previously published inactive PKB PH–kinase domains interaction monitored by FRET [14], another PH–kinase domain interaction of a different nature seemed to occur in PKB active conformation. Two distinct mode of interaction between the two domains could indeed be envisioned since an increased interaction dependent on Thr 308 phosphorylation was incompatible with the mechanism of interaction presented by the other group. The authors suggested that the increased affinity of the PH domain for the phosphorylated kinase domain could explain the detachment from the plasma membrane despite the high level of PtdIns (3,4,5)P3. In these two studies, the change in conformation of PKB was correlated to its phosphorylation status but the direct detection of PKB activity, described in the next section, was performed independently using also FRET imaging.
Monitoring PKB activation in situ
PKB activation state was examined in situ by looking at the level of phosphorylation of an engineered fluorescent substrate specific for PKB. The biosensors Aktus [53] and later B kinase activity reporter (BKAR) [38] were created for this purpose. The two reporters corresponded to the fusion of a specific PKB substrate sequence with a phosphate-binding domain, flanked by CFP at the N terminus and YFP (citrine) at the C terminus. The variations of FRET were indicative of the phosphorylation status of the reporter and therefore of the activity of PKB. Compared to Aktus, BKAR displayed an increased sensitivity that could detect the activity of endogenous PKB. The reporter could also be reversibly phosphorylated allowing the following of the deactivation signal. The authors showed that BKAR located at the plasma membrane was phosphorylated faster than the reporter located in the cytosol and that a further delay was necessary for phosphorylations to occur in the nucleus. Furthermore, the monitoring of BKAR dephosphorylation by okadaic acid-sensitive phosphatases suggested that the activity of the phosphatases varied depending on the compartments. A more robust phosphatase activity was detected in the cytosol than in the nucleus or the plasma membrane. When targeted to the plasma membrane, the dephosphorylation of the reporter was overcome by PKB-induced phosphorylation. Therefore, using BKAR, Kunkel et al. could monitor a spatiotemporal gradient of PKB activation going from the plasma membrane to the nucleus and follow how the signal could last depending on the localisation in the cell. It is possible that the deactivation of PKB by the dephosphorylation of Thr 308 and Ser 473 linked to okadaic acid-sensitive phosphatases could also be affected in the similar way as the substrates to terminate the signal.
More recently, a bioluminescent reporter for PKB activity (bioluminescence Akt reporter) was designed as an alternative to the FRET systems and was used in vivo in mice [64]. It worked in the same manner as Aktus and BKAR, but the CFP/YFP pair was replaced by the N- and C-terminal domains (N-Luc and C-Luc) of the firefly luciferase reporter molecule. Zhang et al. could measure quantitatively and dynamically the activity of PKB in tumours xenographs upon different treatments. The study of the turnover of the 3′-phosphoinositides at the plasma membrane and the dynamic of interaction with PDK1 allowed to correlate the activity and conformation of PKB to upstream activation signals.
Sensing the 3′-phosphoinositides in live cells
The spatiotemporal regulation of the 3′-phosphoinositides activators of PKB was investigated by using a specific 3′-phosphoinositides sensor called InPAkt [5]. InPAkt was comprised of a PKB PH domain fused with a pseudoligand and flanked with CFP and YFP. In absence of 3′-phosphoinositides, PKB PH would interact with the pseudoligand peptide sequence. Upon activation, the PKB PH domain would detach from the pseudoligand to interact with the 3′-phosphoinositides in a reversible manner (change in FRET). Upon stimulation, InPAkt translocated to the plasma membrane and FRET was detected, indicating a production of 3′-phosphoinositides mainly at the plasma membrane. No FRET was detected in the nucleus using a nuclear-localised sensor, indicating that there may not be detectable amounts of phosphoinositides produced in the nucleus upon stimulation.
By using BKAR (see above section), it seemed that a wave of PKB activation started from the membrane then appeared in the cytosol to finally reach the nucleus [38]. Therefore, the authors were interested in examining the production of 3′-phosphoinositides upon time whilst concomitantly monitoring PKB activation (with BKAR). Interestingly, as long as the stimulation was maintained, the phosphoinositides levels remained high (measured up to 40 min). This suggested that PKB could detach from the plasma membrane even in presence of high levels of PtdIns (3,4,5) P3.
Recently, Lasserre et al. reported using a new fluorescence correlation spectroscopy (FCS) approach that the recruitment of PKB PH domain at the plasma membrane was promoted by the accumulation of PtdIns (3,4,5) P3 to dynamic membrane nano-domains. The authors hypothesised that the partitioning of PKB to these nano-domains might be necessary for its efficient phosphorylation due to the close proximity of co-recruited upstream activators like PDK1, TORC2 or DNA-PK.
In the study of the dynamic relationship of PKB and PDK1 monitored by FRET (see next section) the two partners were shown to be not only co-localising but actually interacting at the plasma membrane [40].
Dynamics of PKB and PDK1 interaction in intact cells
Since the direct in vitro or in vivo interaction of PKB with its activator PDK1 had not been observed, the dynamic relationship between PKB and PDK1 was examined in intact cells using FRET by two-photon FLIM [14]. The authors found that RFP-PKB and GFP-PDK1 interacted in the cytoplasm prior to stimulation. Furthermore, the stimulation of the cells induced the translocation of a pool of this complex towards the plasma membrane. Whereas the plasma membrane interaction of PKB and PDK1 was shown to be dependent on the production of 3′-phophoinositides (LY294002 treatment), the cytoplasmic interaction appeared to be independent. In accordance, the mutation of PKB or PDK1 PH domains (PKB Arg 25 Cys and PDK1 Arg 472–474 Leu) to prevent their binding to PtdIns (3,4,5) P3 precluded the interaction at the plasma membrane but not in the cytoplasm. Since PDK1 had been known to dock with the phosphorylated HM of some AGC kinases, the role of PDK1 PIF pocket and phosphate-binding pocket as well as the role of PKB phosphorylated HM was investigated for the binding of PKB. The PIF and phosphate-binding pocket defective mutants (PDK1 Leu 155 Glu and PDK1 Arg 131 Ala, respectively) and the PKB Thr 308 Ala/Ser 473 Ala mutant that could not be phosphorylated on the HM were generated. All three mutants retained their capacity to interact with a wild-type partner in the cytoplasm and at the plasma membrane upon stimulation, indicating that the interaction of PKB and PDK1 was independent of the PIF and the phosphate-binding pockets and did not require phosphorylation of Thr 308 or Ser 473. In line with this observation, it has also been shown by using knock-in mutants that the PIF and phosphate-binding pockets of PDK1 were not implicated in the activation of PKB [21, 22]. Thus, the interaction in the cytoplasm occurs via mechanisms distinct from that of other AGC kinases.
Therefore, by compiling the results of these dynamic studies, PDK1 and PKB would be in constant equilibrium between an associated and a dissociated form in the cytoplasm prior to stimulation (Fig. 3). Upon stimulation, the equilibrium would be shifted towards the plasma membrane due to the binding of their PH domain with the locally produced 3′-phosphoinositides. The change in conformation of PKB upon interaction with the 3′-phosphoinositides would allow the access of the Thr 308 to associated PDK1 and trigger the phosphorylation and activation of PKB. The detachment of activated PKB from the plasma membrane would lead to the phosphorylation of the substrates in the cytoplasm and then the nucleus. The duration of the signal would depend on the rate of dephosphorylation of the substrates and of the turnover of the phosphoinositides.
Fig. 3.

Schematic model of the interaction of PKB with PDK1. Prior to stimulation, PKB and PDK1 form a complex in dynamic equilibrium in the cytoplasm. In the PKB inactive conformation, PKB PH and kinase domains interact, noted as “PH-in”. Upon PDGF stimulation, the PKB/PDK1 complex is recruited to the plasma membrane due to interaction with phosphoinositides (in orange). The equilibrium of PKB/PDK1 interaction is shifted towards the associated form. The interaction of PKB PH domain with the lipids induces a change in conformation of PKB, noted as “PH-out”. In this conformation, Thr 308 becomes accessible to PDK1 (the phosphorylation sites are represented by yellow circles). After phosphorylation of Ser 473 and Thr 308 and loading of ATP, PKB dissociates from the plasma membrane in its active conformation to return to the cytoplasm and the nucleus. PKB re-adopts an inactive conformation upon dephosphorylation of its Thr 308 and Ser 473 sites
In order to understand the exact nature of the mechanisms involved in PKB activation dynamics, structural information was essential. The topology of the 3-D structure of the protein related to its activation states was investigated using mass spectrometry and molecular dynamics.
Inter-domain interactions in vitro by cross-linking and tandem mass spectrometry
The 3-D structure of recombinant full-length PKB was [31] first investigated in solution by using chemical cross-linking and tandem mass spectrometry. The cross-linking revealed distinct spatial arrangements of individual domains at different steps of PKB activation: in resting and membrane bound states, when phosphorylated, and upon substrate binding. Information on the interaction distance between the domains were looked into using a range of lysine specific bi-functional cross linkers of varying spacer arm lengths including bis-sulfosuccinimidyl suberate (BS3), disuccinimidyl suberate (DSS) and disuccinimidyl glutarate. Tandem mass spectrometry with static nano-electro spray ionisation (ESI) and high performance liquid chromatography/nano-ESI combined with 18O labelling of the tryptic digests was used to confirm the through space interactions of the domains and to allow quantitative comparison of the peaks from the various states of PKB.
Two cross-linkings emerged on the inactive recombinant PKB (non-phosphorylated on Thr 308 and Ser 473) using the long arms hydrophobic cross-linker DSS or hydrophilic cross-linker BS3. These results indicated that the modified lysines were accessible on the surface of the protein. The first represented the cross-linking of the PH domain (Lys 30) with the kinase domain (Lys 389). The second represented the cross-linking of the kinase domain (Lys 284) with the C-terminal part (Lys 426). When using a shorter arm reagent (DSG), the cross-linking of the kinase with the C terminus, but not of the PH with the kinase, could be detected, suggesting a shorter distance between the kinase and C terminus than between the PH and kinase domains.
When inactive PKB was incubated with liposomes containing PE/PC/PS/PIP3 (50/18/30/2) in order to mimic the plasma membrane, the two inter-domain interactions were lost. These results indicated that the domains had moved further apart than by the spatial constraint of 24 Å allowed by the DSS cross-linker.
In the active double-phosphorylated Thr 308 and Ser 473 PKB, the interaction between the PH and the kinase domain reappeared whereas surprisingly, the interaction with the kinase and the C terminus did not. It was only when the substrate GSK3 peptide and ATP analogue were bound to the active PKB (phosphorylated on Thr 308 and Ser 473) that both cross-linkings, PH–kinase and kinase-C terminus were reformed.
Together, these data confirmed that different conformational states of PKB existed at various stages of PKB activation in vitro. The data were consistent with and complementary to structural studies and biochemistry assays. Furthermore, it provided new insights into the 3-D structure of PKB and in particular into the spatial proximity of the PKB domains in various states of activation that could not been achieved by crystallography or FRET alone. However, the length of the cross-linking could not provide information on the precise positioning of the domains. Indeed, a linker of 20–24 Å of length might not differentiate between close but distinct interactions. It is in fact very likely (see section on “Probing PKB 3-D structure by molecular modelling and dynamic simulations”) that the PH–kinase interaction monitored by cross-linking in the inactive state of PKB might not be of the same nature as the interaction monitored in the cross-linked activated state of PKB (phosphorylated Thr 308 and Ser 473). Therefore, the use of molecular modelling and dynamics associated to biochemical analysis became essential to comprehend how the domain interactions define PKB function at a molecular level.
Probing PKB 3-D structure by molecular modelling and dynamic simulations
Allosteric regulation of PKB by PH and HM regulatory domains
In vitro and cellular evidence had suggested a specific role for the PKB PH domain in regulating the accessibility of the activation loop residue of the kinase domain to PDK1 [51]. By using molecular modelling, a detailed representation of PKBα PH and kinase domains interaction was presented. The docking was performed based on hydrophobicity in water [39] and hydrophobic patches were located on the kinase and on the PH domains. The electrostatic potential maps and the residue maps were compared and a strong complementarity was observed between the two domains. Interestingly, the residue Trp 80 at the extremity of the VL3 of the PH domain that was previously found solvent exposed on the crystal structure of isolated PKBα PH domain [44] was inserted inside a deep cleft in the kinase domain around residues Lys 297, Glu 298 and Glu 314. To confirm the interaction model in situ, the authors used the fluorescent PKB reporter GFP-PKB-mRFP to monitor changes in the distance or the orientation of the domains by FRET. The mutation of two polar residues Gln 79 and Thr 82 at the interface of the two domains was predicted to disrupt their interaction and hence the FRET detected. Therefore, the mutations of these residues to Glu were performed to create the mutant reporter GFP-PKB(79 Glu–82 Glu)-mRFP. The change in the FRET efficiency of mutated GFP-PKB(79 Glu–82 Glu)-mRFP compared to the wild-type reporter confirmed that the mutation of these residues at the interface of the domains perturbed their interaction, therefore validating the model.
These results suggested that PKB PH domain would have a dual function on PKB activation: inhibitory and stimulatory. In basal conditions, PKB would be maintained in an inactive conformation by the interaction of its PH and kinase domains (“PH-in”). The “PH-in” conformation would be responsible for preventing the phosphorylation of Thr 308 by the associated PDK1. Upon stimulation, PKB PH-domain interaction with phosphoinositides and its concomitant change in conformation to “PH-out” at the plasma membrane would allow co-recruited PDK1 to phosphorylate Thr 308 upon stimulation.
The following use of molecular dynamics [16] led to specific information about the PH–kinase interface and its regulation. The unexpected formation of a PH-induced cavity (Fig. 4) in the same region as the previously described PKB hydrophobic pocket (binding site for PKB HM domain) [61] was observed. The refined dynamic model also showed that through this PH-induced cavity, the Trp 80 residue was accessible from outside whilst PKB remained in its inactive conformer. In order to probe the dynamic model, an allosteric inhibitor of PKB (AKT inhibitor VIII) was used [9, 10, 41, 65]. Its action was proposed to specifically target the “PH-in” conformer of PKB in a manner dependent on the PH domain and the Trp 80 [29]. The inhibition of the in vivo change in conformation of the GFP-PKB-mRFP conformational probe dependent on the presence of Trp 80 upon inhibitor treatment confirmed this hypothesis. By using molecular dynamic simulations, it was suggested that AKT inhibitor VIII would lock PKB in the inactive conformation by interacting with Trp 80 through the PH-induced cavity. The use of AKT inhibitor VIII allowed to correlating the size of the cavity with the potency of the inhibitor. Contrary to PKBα, PKBγ had been shown to have a reduced sensitivity towards the inhibitor and concomitantly the molecular dynamics showed that a PH domain-induced cavity was not formed in PKBγ. Additionally, by creating a chimera of the PH domain of PKBα with the kinase domain of PKBγ, a partial re-opening of a cavity was observed and a concomitant enhanced sensitivity for the inhibitor compared to PKBγ detected. In summary, AKT inhibitor VIII was successfully used to show that a PH domain-induced cavity was present in the inactive PKB “PH-in” conformer.
Fig. 4.

PKBα PH domain creates a cavity in the kinase domain. a Structure of the PKBα PH domain (gold) and reconstructed kinase domain (blue) in complex (PKBα “PH-in” conformer) after short dynamic runs and minimisation in water and physiological salt concentration. The secondary structure of the kinase domain is represented as a white ribbon. The PH domain residue Trp 80 (red) is visible through an open cavity in the kinase domain. b Vertical cross section of the dynamic structure shown in a at the level of the PH-induced cavity. The distance between the Trp 80 residue in red and the protein surface is about 8–8.5 Å (red arrow). The cavity is filled with water molecules shown as white pearls
The group of Yang et al. had shown that a channel existed in the structure of the isolated kinase domain of PKA that appeared to contain conserved residues amongst all AGC kinases [62]. Furthermore, the hydrophobic motif of PKA was interacting with the channel. In PKB, the positioning of the HM in a hydrophobic groove in the N-lobe of the isolated active PKBβ kinase domain had been described in the crystal structure 1O6K [61]. However, the position of the HM in inactive full-length PKB had not been investigated. A dynamic model showed that the HM passed right across the PH-induced cavity. Specifically, the sequence Phe469-X-X-Phe472-Ser of the HM was kinked above the cavity, positioning Phe 469 and Phe 472 at a binding distance from Trp 80 (Fig. 5). The interaction between Phe 469/472 and Trp 80 suggested a role for the HM motif in the regulation of PKB prior to its translocation and Ser 473 phosphorylation. Furthermore, the C-terminal HM did not seem to be implicated in AKT inhibitor VIII binding but could potentially overlap at a common binding site.
Fig. 5.
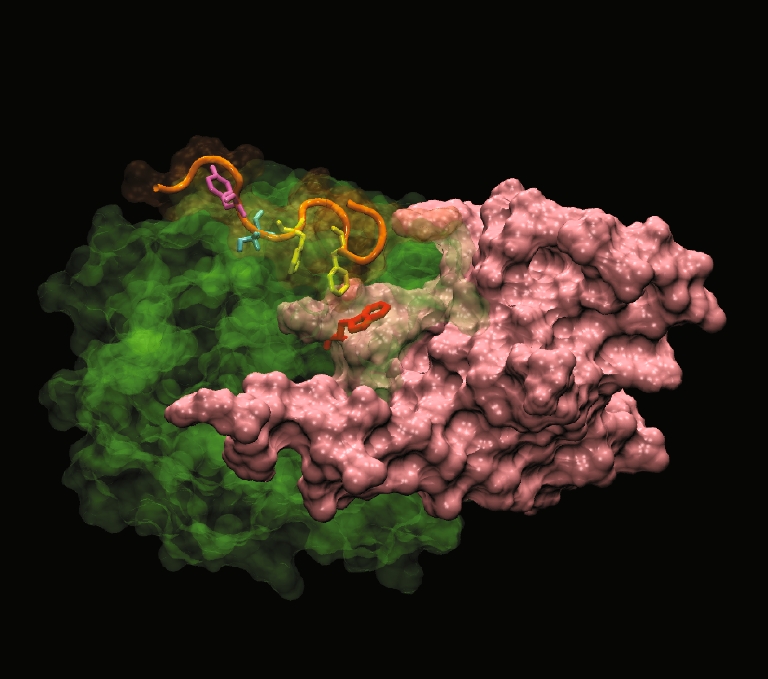
The C-terminal hydrophobic motif of PKBα in the “PH-in” conformer is positioned at the apex of the cavity PKBα C-terminal hydrophobic motif HM (orange) interacting with PKBα kinase domain (green) in the “PH-in” conformer. The PH domain is in pink and the PH domain residue Trp 80 is in red. The model shows that two phenyalanines (in yellow) from the sequence F469XXF472S of HM are found positioned right above the PH-induced cavity in PKBα kinase domain. Ser 473 is represented in cyan and Tyr 474 in magenta
Taken together, these results show that prior to stimulation, the presence of the PH domain associated to the kinase domain of PKB (inactive conformation) induces the formation of a cavity in the kinase domain [16]. Interestingly, the change in conformation of PKB [14] allows the separation of the domains leading to the closing of the cavity and the formation of a hydrophobic groove that is conserved in other AGC kinases [62]. The interaction of the HM with the kinase domain occurs in the same region. In the inactive conformation of PKB, the HM would bind to the Trp 80 through the cavity. In the active conformer of PKB, it is buried inside a channel in the hydrophobic groove [62]. Therefore, in the same manner as for the PH domain, it is likely that the HM would play a dual role in the regulation of PKB activity. It would help on one hand in maintaining the inactive conformation of PKB by locking the conformation and on the other hand would activate allosterically the kinase by re-arranging the catalytic site.
Role of phosphorylated Thr 450 in PKB regulation by the HM regulatory domain
The role of the residue Thr 450 (numbering for PKBα) commonly known as the turn motif [59] was investigated in the regulation of PKB activation by the HM [30]. This constitutively phosphorylated residue [2] is conserved in many AGC kinases and is required for full PKB activation in addition to Thr 308 and Ser 473. Recently, it was shown that TORC2 was responsible for the phosphorylation of Thr 450 in addition to Ser 473 [33]. The authors suggested that the term of turn motif was not appropriate for the residue Thr 450 since it did not correspond to the turn motif residue of PKA. Instead, the term of Z (for zipper) motif was proposed and will be used in this review. Since in the crystal structure of ΔPH-PKBβ [62] a portion of the C-terminal directly following the kinase domain could not be resolve, information was not obtained for the position or the function of the Z motif. By using molecular modelling, a phospho Ser/Thr binding site was identified in the kinase domain of PKBβ where the highly conserved basic residues Lys 160, Lys 165, Arg, 184 and Arg 224 interacted with phosphorylated Thr 451. These results suggested that the constitutive phosphorylation of the Z motif promoted the zipper-like binding of the C-terminal domain to the kinase domain potentially promoting the correct positioning of the HM. Accordingly, the mutation of the Thr 450 (PKBα) or of the four basic residues induced a decrease in PKB activity. Furthermore, the binding of the Z motif prevented Thr 450 dephosphorylation, indicating that in vivo Thr 450 being constitutively phosphorylated the Z motif might be always interacting. In conclusion, the binding of the Z motif into the kinase domain would be an anchoring point increasing the local concentration of HM in the immediate vicinity of the hydrophobic groove leading to its correct positioning.
Conclusion and outlook
Much effort has been put into understanding how inter- or intra-molecular interactions controlled the activity of kinases (for review, see [28, 50]). In the case of PKB, whose activity has been shown to play a fundamental role in many biological processes, the need for extensive safety levels has been established in order to prevent unwanted activation of the kinase. In fact, four regulatory levels have been implemented that must be cleared in a precise order before full activation. If one step is missing, the kinase cannot be activated. The compilation of the information from the crystal structures, molecular modelling and dynamics, FRET/FLIM analysis, classical biochemistry as well as mass spectrometry has allowed the elaboration of a refined model for the activation of PKB.
Maintenance of PKB inactive conformer
In all kinases, a tight auto-inhibition mechanism is critical for the maintenance of the inactive state since the close proximity of an ATP molecule to an open binding site is sufficient to allow its fast entry into the pocket (Michel Laguerre, personal communication). Therefore, a series of allosteric controls are implemented to prevent the activation of the kinases. These controls occur through conformational rearrangements of the protein, due to dimerisation, interaction with scaffolds, interaction with other proteins or subunits, phosphorylations or intra-molecular bindings (for review, see [55]). One of the most common mechanisms is the obstruction of the ATP-binding site by the activation loop in the inactive state of the kinase. In PKB, the activation loop residue Thr 308 (non-phosphorylated) makes a hydrogen bond with Glu 234 located on the other side of the active site. The phosphorylation of Thr 308 is therefore necessary to break this bond and allow the movement of the activation loop [61]. The way of maintaining an inactive kinase can be unique for different kinases [55]. For PKB, the inter-domain interactions of the regulatory PH and C-terminal domains are critical. Mechanisms of inter-domain inhibition have been described in AGC kinases, specifically for the members the most closely related to PKB, the kinases PKA and PKC. In PKA, the inter-molecular interaction of the regulatory subunits RIα with the kinase cleft of the catalytic subunits C in absence of cyclic adenosine 3′,5′-monophosphate (cAMP) was demonstrated by crystallography [36]. In the case of PKC, a series of biochemical analyses led to the discovery of a pseudosubstrate peptide sequence that leads to autoinhibition [17, 46]. The mechanism of PKB regulation reviewed here shows the first example of the inhibition of a kinase activity by a PH domain.
The intra-molecular interaction of PKB PH and kinase domains was shown to prevent the accessibility of the Thr 308 to the associated upstream activator kinase PDK1 [14]. In addition to the steric hindrance induced by the position of the PH domain, the loop containing the Trp 80 occupies exactly the position that the activation loop would adopt following Thr 308 phosphorylation during PKB activation. Therefore, a two level regulation prevents here the re-organisation of the active site.
The creation of a water-filled cavity in the PKB kinase domain by interaction of the PH domain initiates still another level of regulation that involves the C-terminal part. The Phe 469 and Phe 472 of the C-terminal HM embedded in the kinase domain N-lobe interact with the PH domain residue Trp 80 through the water-filled cavity [16]. This interaction could be responsible for locking the whole complex in an inactive and inactivable conformer. It is interesting to note that the C-terminal has a dual role in the regulation of PKB. On one hand, it interacts at the level of the hydrophobic pocket of the kinase after activation, inducing the re-organisation of the active site residues and on the other hand regulates the stability of the inactive conformer prior to stimulation. Therefore, the role of the C-terminal HM motif in the regulation of PKB activity is more complex than previously anticipated.
Activation of PKB
The activation process of PKB exhibits four different levels of regulation, the analogy being that of two “doors” sequentially unlocked then opened. The generation of PtdIns (3,4,5)P3 at the plasma membrane upon stimulation induces the translocation of PKB towards the cell surface. The phosphorylation of Ser 473 by a kinase located at the plasma membrane (or whose activity would overcome dephosphorylation at this location) would result in a displacement of the C-terminal HM sufficient to disrupt the interaction of Phe 469 and Phe 472 with Trp 80. The PH domain still bound to the kinase domain (“PH-in”) but would be now free to move: “door 1” is unlocked [16]. Accordingly, the results obtained by mass spectrometry indicate that the movement is large enough to prevent the cross-linking of the C terminus with the kinase domain [31].
The change in the conformation of the PKB PH domain due to its binding to PtdIns (3,4,5)P3 at the plasma membrane [44] would likely lead to the expulsion of the PH domain from the kinase domain: “PH-out” conformation. Accordingly, in a related mode of inhibition, the regulatory subunit RIα of PKA undergoes a major conformational change when associated with cAMP that is proposed to free the kinase [36]. Therefore, the “PH-out” conformation allows the activation loop to be exposed to phosphorylation: “door 1” is opened [14].
The interaction of the HM with the kinase domain hydrophobic groove of PKB leads to the re-alignment of the catalytic residues by re-ordering the αC helix. The αC helix has been shown to play a central role in the allosteric activation of other kinases like cyclin-dependent kinase 2, activated by the binding of cyclin A in an inter-molecular interaction [34]. However, it is not known whether the re-alignment of the catalytic site by interaction of the phosphorylated HM is necessary for the phosphorylation of Thr 308. Mass spectrometry data suggested that the interaction of the HM phosphorylated on Ser 473 would only occur when ATP and substrate are bound to the protein [31]. In addition, X-ray crystallography studies only describe the interaction of the HM with the hydrophobic groove of PKB kinase domain in complex with ATP and GSK3 peptide. It is probable that the presence of the PH domain plays an important role in the positioning of the HM and the re-ordering of the catalytic site in activated PKB. However, it could not be investigated in the crystal structure of isolated domains or detailed by mass spectrometry.
The phosphorylation of Thr 308 by associated PDK1 [14] induces a large change in conformation of the activation loop freeing the ATP-binding site [61]: “door 2” is unlocked. The total amount of negative charges due to the phosphate on the Thr 308 and the three phosphates from the ATP could compensate exactly the four phosphates of PtdIns (3,4,5) P3 head group and trigger the detachment of PKB from the plasma membrane. Activated cytosolic PKB can therefore bind its substrates: “door 2” is opened.
It is possible that active cytosolic PKB PH domain may re-engage to a new interaction with the kinase domain [31] although through a different mode of interaction than the one described in the “PH-in” conformation [14, 16]. This new binding might also be necessary for the actual detachment from the membrane [4].
In order to inactivate PKB, the undoing of all the previous steps in a reverse order would be necessary. The dephosphorylation of Thr 308 and Ser 473 by okadaic-sensitive or insensitive phosphatases and the re-entry of the VL3 (Trp 80) loop in the kinase domain being amongst the most obvious steps. However, a description of these events would be too speculative at present and will require further investigation.
The interaction of the PH domain of PKB with its kinase domain represents a new mechanism for the allosteric control of an AGC kinase that had not been observed thus far. A recent study highlighted the difference in the activation mechanism of the closely related AGC kinases, PKB and PKC, previously thought to be very similar [17]. It will be interesting to investigate whether PDK1, the only other AGC kinase containing a PH domain, possesses a related mode of regulation. The C-terminal regulatory HM is better represented in other AGC kinases and has been shown to have a similar function in the allosteric activation of the kinases. However, in PKB, the HM might also be involved in the regulation of the inactive conformer in cooperation with the PH domain. Its interaction with the Trp 80 residue of the PH domain could secure the protein in the inactive conformation. The phosphorylation of Ser 473 would thus be necessary to unlock the PH-in conformer. Therefore, the sequential phosphorylation of first Ser 473 followed by Thr 308 upon conformation change would trigger the full activation of PKB. A role for phosphorylated Thr 450 of the Z motif (also known as turn motif) in PKB regulation prior to stimulation could also be expected since its interaction with the kinase domain in the “PH-in” conformation is possible. Interestingly, based on the modelling of the PH with the kinase and HM, an unusual topology of the flexible linkers between the PH–kinase and kinase-C-terminal that could not be seen by crystallisation is expected. From the position of the first and last residues visible on the structures, the linkers should cross over one another. The determination of the relative positioning of the linkers is likely to shed light on the molecular mechanism of the coordinated events involving the PH domain and HM in PKB activation.
Abbreviations
- PHLLP
Pleckstrin homology domain leucin-rich repeat protein phosphatase
- TORC2
Target of Rapamycin complex 2
- PDK1
Phosphoinositides-dependent protein kinase 1
- PKB
Protein kinase B
- Aktus and BKAR
B kinase activity reporters
- ReAktion
Reporter of Akt action
- BAR
Bioluminescence Akt reporter
- FRET
Förster resonance energy transfer
- FLIM
Fluorescence lifetime imaging microscopy
- HM
Hydrophobic motif
- GFP
Green fluorescent protein
- YFP
Yellow fluorescent protein
- mRFP
Monomeric red fluorescent protein
- CFP
Cyan fluorescent protein
- PtdIns(3,4,5)P3
InPAkt indicators for phosphoinositides based on Akt
- PH
Pleckstrin homology
- VL2 and VL3
Variable loop 2 and 3
- PRK2
PKC-related kinase 2
- DAPP1
Dual adaptor of phosphotyrosine and 3-phosphoinositides
- GRP1
General receptor for phosphoinositides isoform 1
- BTK
Bruton tyrosine kinase
- PKA
Cyclic AMP dependent protein kinase
- CDK2
Cyclin-dependent kinase 2
Acknowledgements
We would like to thank Dr. Richard Byrne for the critical reading of the manuscript.
References
- 1.Alcor D, Calleja V, Larijani B. Revealing signaling in single cells by single- and two-photon fluorescence lifetime imaging microscopy. Methods Mol Biol. 2009;462:307. doi: 10.1007/978-1-60327-115-8_21. [DOI] [PubMed] [Google Scholar]
- 2.Alessi DR, Andjelkovic M, Caudwell B. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15(23):6541. [PMC free article] [PubMed] [Google Scholar]
- 3.Alessi DR, Deak M, Casamayor A. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7(10):776. doi: 10.1016/S0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 4.Ananthanarayanan B, Fosbrink M, Rahdar M. Live-cell molecular analysis of Akt activation reveals roles for activation loop phosphorylation. J Biol Chem. 2007;282(50):366334. doi: 10.1074/jbc.M706227200. [DOI] [PubMed] [Google Scholar]
- 5.Ananthanarayanan B, Ni Q, Zhang J. Signal propagation from membrane messengers to nuclear effectors revealed by reporters of phosphoinositide dynamics and Akt activity. Proc Natl Acad Sci U S A. 2005;102(42):15081. doi: 10.1073/pnas.0502889102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andjelkovic M, Alessi DR, Meier R. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272(50):31515. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 7.Andjelkovic M, Jakubowicz T, Cron P. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc Natl Acad Sci U S A. 1996;93(12):5699. doi: 10.1073/pnas.93.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auguin D, Barthe P, Auge-Senegas MT. Solution structure and backbone dynamics of the pleckstrin homology domain of the human protein kinase B (PKB/Akt). Interaction with inositol phosphates. J Biomol NMR. 2004;28(2):137. doi: 10.1023/B:JNMR.0000013836.62154.c2. [DOI] [PubMed] [Google Scholar]
- 9.Barnett SF, Bilodeau MT, Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation. Curr Top Med Chem. 2005;5(2):109. doi: 10.2174/1568026053507714. [DOI] [PubMed] [Google Scholar]
- 10.Barnett SF, Defeo-Jones D, Fu S. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385(Pt 2):399. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellacosa A, Chan TO, Ahmed NN. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17(3):313. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 12.Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26(11):657. doi: 10.1016/S0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 13.Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12(2):104. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 14.Calleja V, Alcor D, Laguerre M. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5(4):e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calleja V, Ameer-Beg SM, Vojnovic B. Monitoring conformational changes of proteins in cells by fluorescence lifetime imaging microscopy. Biochem J. 2003;372(Pt 1):33. doi: 10.1042/BJ20030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calleja V, Laguerre M, Parker PJ. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7(1):e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron AJ, Rycker M, Calleja V. Protein kinases, from B to C. Biochem Soc Trans. 2007;35(Pt 5):1013. doi: 10.1042/BST0351013. [DOI] [PubMed] [Google Scholar]
- 18.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 19.Chen R, Kim O, Yang J. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276(34):31858. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- 20.Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008;23(1):1. doi: 10.1177/172460080802300101. [DOI] [PubMed] [Google Scholar]
- 21.Collins BJ, Deak M, Arthur JS. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22(16):4202. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins BJ, Deak M, Murray-Tait V. In vivo role of the phosphate groove of PDK1 defined by knockin mutation. J Cell Sci. 2005;118(Pt 21):5023. doi: 10.1242/jcs.02617. [DOI] [PubMed] [Google Scholar]
- 23.Conus NM, Hannan KM, Cristiano BE. Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J Biol Chem. 2002;277(41):38021. doi: 10.1074/jbc.M203387200. [DOI] [PubMed] [Google Scholar]
- 24.Feng J, Park J, Cron P. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279(39):41189. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 25.Ferguson KM, Kavran JM, Sankaran VG. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol Cell. 2000;6(2):373. doi: 10.1016/S1097-2765(00)00037-X. [DOI] [PubMed] [Google Scholar]
- 26.Gambhir A, Hangyas-Mihalyne G, Zaitseva I. Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys J. 2004;86(4):2188. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Gold MG, Barford D, Komander D. Lining the pockets of kinases and phosphatases. Curr Opin Struct Biol. 2006;16(6):693. doi: 10.1016/j.sbi.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Green CJ, Goransson O, Kular GS, et al. Use of Akti and a drug-resistant mutant validates a critical role for PKB/Akt in the insulin-dependent regulation of glucose and system A amino acid uptake. J Biol Chem. 2008;283:27653–27667. doi: 10.1074/jbc.M802623200. [DOI] [PubMed] [Google Scholar]
- 30.Hauge C, Antal TL, Hirschberg D. Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 2007;26(9):2251. doi: 10.1038/sj.emboj.7601682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang BX, Kim HY. Interdomain conformational changes in Akt activation revealed by chemical cross-linking and tandem mass spectrometry. Mol Cell Proteomics. 2006;5(6):1045. doi: 10.1074/mcp.M600026-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Begley M, Morgenstern KA. Crystal structure of an inactive Akt2 kinase domain. Structure. 2003;11(1):21. doi: 10.1016/S0969-2126(02)00937-1. [DOI] [PubMed] [Google Scholar]
- 33.Ikenoue T, Inoki K, Yang Q. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27(14):1919. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeffrey PD, Russo AA, Polyak K. Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature. 1995;376(6538):313. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 35.Kannan N, Haste N, Taylor SS. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A. 2007;104(4):1272. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307(5710):690. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 37.Knighton DR, Zheng JH, Ten Eyck LF. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253(5018):407. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 38.Kunkel MT, Ni Q, Tsien RY. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J Biol Chem. 2005;280(7):5581. doi: 10.1074/jbc.M411534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laguerre M, Saux M, Dubost J-P. MLPP: a program for the calculation of molecular lipophilicity in proteins. Pharm. Sci. 1997;3:217. [Google Scholar]
- 40.Lasserre R, Guo XJ, Conchonaud F. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat Chem Biol. 2008;4(9):538. doi: 10.1038/nchembio.103. [DOI] [PubMed] [Google Scholar]
- 41.Lindsley CW, Zhao Z, Leister WH. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15(3):761. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 42.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase Balpha (PKBalpha) promoted by hyperosmotic stress. EMBO J. 1998;17(24):7294. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milburn CC, Deak M, Kelly SM. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J. 2003;375(Pt 3):531. doi: 10.1042/BJ20031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mora A, Komander D, Aalten DM. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15(2):161. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 46.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370(Pt 2):361. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15(5):661. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 48.Obata T, Yaffe MB, Leparc GG. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275(46):36108. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 49.Parsons DW, Wang TL, Samuels Y. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436(7052):792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 50.Pellicena P, Kuriyan J. Protein–protein interactions in the allosteric regulation of protein kinases. Curr Opin Struct Biol. 2006;16(6):702. doi: 10.1016/j.sbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Sable CL, Filippa N, Filloux C. Involvement of the pleckstrin homology domain in the insulin-stimulated activation of protein kinase B. J Biol Chem. 1998;273(45):29600. doi: 10.1074/jbc.273.45.29600. [DOI] [PubMed] [Google Scholar]
- 52.Sarbassov DD, Guertin DA, Ali SM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005;307(5712):1098. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 53.Sasaki K, Sato M, Umezawa Y. Fluorescent indicators for Akt/protein kinase B and dynamics of Akt activity visualized in living cells. J Biol Chem. 2003;278(33):30945. doi: 10.1074/jbc.M212167200. [DOI] [PubMed] [Google Scholar]
- 54.Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2(10):760. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 55.Shi Z, Resing KA, Ahn NG. Networks for the allosteric control of protein kinases. Curr Opin Struct Biol. 2006;16(6):686. doi: 10.1016/j.sbi.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 56.Stephens L, Anderson K, Stokoe D. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279(5351):710. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 57.Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene. 2005;24(50):7391. doi: 10.1038/sj.onc.1209100. [DOI] [PubMed] [Google Scholar]
- 58.Thomas CC, Deak M, Alessi DR. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol. 2002;12(14):1256. doi: 10.1016/S0960-9822(02)00972-7. [DOI] [PubMed] [Google Scholar]
- 59.Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275(12):8271. doi: 10.1074/jbc.275.12.8271. [DOI] [PubMed] [Google Scholar]
- 60.Tokunaga E, Oki E, Egashira A. Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets. 2008;8(1):27. doi: 10.2174/156800908783497140. [DOI] [PubMed] [Google Scholar]
- 61.Yang J, Cron P, Good VM. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol. 2002;9(12):940. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 62.Yang J, Cron P, Thompson V. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9(6):1227. doi: 10.1016/S1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Allen MD. FRET-based biosensors for protein kinases: illuminating the kinome. Mol Biosyst. 2007;3(11):759. doi: 10.1039/b706628g. [DOI] [PubMed] [Google Scholar]
- 64.Zhang L, Lee KC, Bhojani MS. Molecular imaging of Akt kinase activity. Nat Med. 2007;13(9):1114. doi: 10.1038/nm1608. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Z, Leister WH, Robinson RG. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg Med Chem Lett. 2005;15(4):905. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]