

Abstract
One- and two-photon stimulated emission transitions were investigated by the fluorescence quenching of a sulfonyl-containing fluorene compound, 2,7-bis(4-(phenylsulfonyl)styryl)-9,9-didecyl-9H-fluorene (1), in solution at room temperature using a picosecond pump−probe technique. The nature of stimulated transitions under various fluorescence excitation and quenching conditions was analyzed theoretically, and good agreement with experimental data was demonstrated. Two-photon stimulated transitions S1 → S0 were shown for 1 at λq = 1064 nm, representing the first report of two-photon stimulated emission depletion (STED) in a molecular system. The two-photon stimulated emission cross section, δ2PE(λq), of fluorene 1 was estimated to be ∼240−280 GM, suggesting that this compound may be a good candidate for use in two-photon STED microscopy.
1. Introduction
The nature of one- and two-photon stimulated transition processes in organic molecules is a subject of both scientific and technological interest due to a number of nonlinear optical properties, such as two-photon induced fluorescence (2PF),1,2 nonlinear transmittance,3,4 two-photon absorption (2PA) processes,5−7 and light amplification of stimulated emission (lasing).8,9 Several different types of stimulated transitions play important roles in the mechanisms of electric field−molecular structure interactions and should be considered in a broad variety of nonlinear optical measurements. For example, possible effects of saturation10,11 and stimulated emission(12) should be taken into account in the determination of 2PA cross sections by the 2PF method when sufficiently large pulse energies are used and the excitation wavelength overlaps with the fluorescence spectrum of the fluorophore. In the case of 2PA cross section measurements by the open-aperture Z-scan method,(13) possible excited-state absorption (ESA) and stimulated one-photon emission also need to be analyzed, as they can make significant contributions to the observed data.(14)
Excited-state dynamics and stimulated transitions that occur in organic molecules can be studied by fluorescence-quenching methodology, as described previously by Lakowicz and co-workers.15−17 This methodology allows modification of the molecular orientational distribution in the excited states and creates anisotropic molecular ensembles with specific fluorescence properties.(17) As an example, the fluorescence anisotropy properties of 4-dimethylamino-4′-cyanostilbene and tetraphenylbutadiene in the presence of stimulated light quenching have been described.18,19 In some cases, a noticeable deviation from a quadratic dependence of fluorescence emission on excitation power was observed.11,19 Quite significantly, the use of stimulated emission transitions has provided a means to overcome the diffraction limit in fluorescence microscopy,20,21 resulting in the development of high resolution (∼60-nm) stimulated emission depletion (STED) microscopy.22,23
Herein, the nature of one- and two-photon stimulated emission transitions for a new sulfonyl-containing fluorene, 2,7-bis(4-(phenylsulfonyl)styryl)-9,9-didecyl-9H-fluorene (1, Figure 1a), is investigated in toluene and tetrahydrofuran (THF) at room temperature by a fluorescence quenching methodology based on different fluorescence excitation conditions. One-photon fluorescence excitation followed by one- or two-photon quenching was performed using a picosecond pump−probe technique and two laser beams at different wavelengths. Two-photon induced fluorescence along with concurrent one-photon quenching was detected using a single picosecond laser beam. Fluorene 1 is a particularly attractive molecule for such a study because it is a highly fluorescent, photochemically stable compound that exhibits efficient one- and two-photon stimulated transitions.
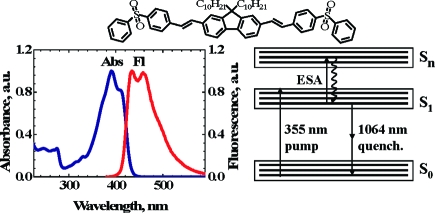
Figure 1.

(Top) Molecular structure of fluorene 1. (a) Normalized absorption (1, 2) and emission (1′, 2′) spectra of 1 in toluene (1, 1′) and THF (2, 2′). (b−d) Electronic molecular model of 1 and corresponding stimulated transitions: (b) one-photon excitation (pump) laser beam at 355 nm, followed by delayed one-photon quenching laser beam at 532 nm; (c) one-photon excitation (355 nm) and delayed two-photon quenching (1064 nm); (d) two-photon excitation and simultaneous one-photon quenching by a single laser beam at 532 nm. ESA indicates possible excited-state absorption processes.
2. Theoretical Description
The integral of the fluorescence emission, IFL, excited and quenched in solution by laser pulses, reflects the type and efficiency of different stimulated optical transitions that might occur in a medium.19,24 The dependence of IFL on excitation power provides insight into the nature of stimulated optical processes(11) while affording important information on certain aspects of potential applications (such as the amplification parameters for stimulated emission(25) and potential spatial resolution in fluorescence microscopy(21)). In this article, a simple three-level model of 1 (Figure 1b−d) was utilized for an analytical description of the observed fluorescence emission under different fluorescence excitation and quenching conditions. For simplicity, only singlet electronic states, S0, S1, and Sn (the ground state and first and higher excited states, respectively) were taken into account. Laser pulses were assumed to be Gaussian in space [transverse beam radius, r0 (HW1/e2M)] and time [pulse duration, τ (HW1/eM)] with peak irradiance (in cm−2 s−1) of I0 = λP2EP/π3/2hcr02τ, where λP, EP, h, and c are the excitation wavelength, the pulse energy, Planck’s constant, and the velocity of light in vacuum, respectively.(14)
Two different sets of fluorescence excitation and quenching conditions were analyzed. In the first case, two beams were employed (pump−probe technique) with different excitation and quenching wavelengths: one-photon fluorescence excitation and, after some delay, one- or two-photon fluorescence quenching (Figure 1b,c). The second case corresponded to a single laser beam for two-photon fluorescence excitation and simultaneous (during the same laser pulse) one-photon fluorescence quenching (Figure 1d). It was assumed that (i) the laser beams excited a nearly cylindrical volume inside the cuvette containing an isotropic molecular solution; (ii) linear and nonlinear transmittance of the solution was ∼100%, i.e., the beam irradiance was constant in the longitudinal direction; (iii) the population of the first excited state was much greater than that of higher excited states and much less than that of the ground state, i.e., N1 ≫ Nn and N1 ≪ N0 ≈ N, where N0, N1, and Nn are the populations of S0, S1, and Sn, respectively, per unit volume and N is the total concentration; (iv) the molecular fluorescence lifetime was much longer than the pulse duration (τF ≫ τ); (v) the fluorescence quantum yield (η) was constant within the entire absorption range, i.e., possible ESA processes did not lead to losses in IFL due to radiationless transitions Sn → S0; and (vi) the laser repetition rate was much less than the inverse of the fluorescence lifetime (f ≪ 1/τF), and no accumulative effects occurred in the medium. All of these assumptions are reasonable given the carefully controlled sets of experimental conditions employed.
2.1. One-Photon Excitation and One-Photon Fluorescence Quenching
In the case of one-photon excitation and the assumptions above, each laser pulse creates a population, N1(r,t), that can be obtained from the equations
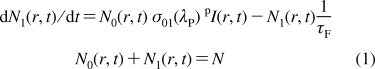 |
where r, t, and λP are the transverse coordinate, time, and excitation wavelength, respectively; σ01(λP) is the one-photon absorption cross section at λP; pI(r,t) = pI0 exp(−t2/τ2 − 2r2/pr02) is the pulse irradiance; and the superscript p corresponds to the pump beam. According to assumptions iii and iv and the dominant role of the stimulated transition [σ01(λP) pI(r,t) ≫ 1/τF], the population N1(r,t) reaches its largest value, Np1(r), toward the end of the excitation pulse, and this maximum value can be expressed as
The value of Np1(r) determines the total number of induced fluorescence photons per excitation pulse, IF0, that can be registered by the detection system as
where φ, η, and L are the collection efficiency of the registration system, the fluorescence quantum yield of the molecule, and the length of the cuvette, respectively. The second one-photon quenching laser pulse at wavelength λq decreases the value of Np1(r) after time delay τd (2τ < τd ≪ τF). The quenching laser pulse overlaps in space with the pumping volume and propagates in the same direction. The decrease in Np1(r) is determined by the stimulated one-photon transitions S1 → S0 with corresponding cross section σ10(λq) (Figure 1b). This leads to a decrease in IF0, which is recorded perpendicular to the pumping and quenching beams. Thus, the value of Nq1(r) after one pulse (quenching) can be obtained as
where the superscript q corresponds to quenching. In this case, the total number of experimentally registered fluorescence photons is
The degree of fluorescence quenching can be obtained from eqs 3 and 5 as
where pr0, qr0, and qEP are the pump and quenching beam radii (HW1/e2M) and the pulse energy of the quenching beam, respectively. According to eq 6, the value of 1 − IF/IF0 is directly proportional to the quenching pulse energy in the case of one-photon quenching.
2.2. One-Photon Excitation and Two-Photon Fluorescence Quenching
For one-photon excitation and two-photon fluorescence quenching, the same population, Np1(r), created by one-photon excitation (eq 4) can be quenched by an IR pulse at the wavelength 2λq with the same duration τ and propagating in the same direction with time delay τd (Figure 1c). Any ESA processes at 2λq do not affect the integrated fluorescence intensity (assumption v), so two-photon stimulated transitions S1 → S0 [with corresponding two-photon emission cross section δ2PE(2λq)] should be the only reason for a decrease in IF0. It is interesting to note that this fluorescence quenching methodology allows one to elucidate two-photon stimulated emission transitions, distinguishing them from possible one-photon ESA effects. In this case, the value of the S1 population after the quenching pulse, N2Pq1(r), can be expressed as
and the total number of registered fluorescence photons is given by
Taking into account eqs 3 and 8, the degree of fluorescence quenching can be represented as
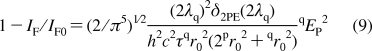 |
From eq 9, for the case of two-photon quenching, the value of 1 − IF/IF0 is directly proportional to the square of quenching pulse energy, and the slope of the dependence 1 − IF/IF0 = f(qEP2) allows one to determine the two-photon emission cross section, δ2PE(2λq).
2.3. Two-Photon Excitation and One-Photon Fluorescence Quenching with a Single Laser Beam
A single laser beam can be used for two-photon excitation and simultaneous one-photon quenching of the induced fluorescence emission (Figure 1d). In this case, eq 1 should be expressed as
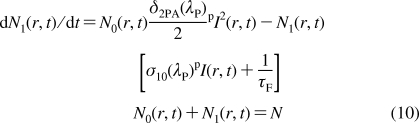 |
where σ10(λP) is the one-photon stimulated emission cross section of the transition S1 → S0. For sufficiently low pulse irradiance, that is, pI(r,t) ≪ {1/[τFσ10(λP)], 1/[τFδ2PA(λP)]1/2}, solution of eq 10 gives the value of Nq1(r), and the corresponding registered integrated fluorescence emission can be obtained as
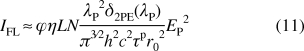 |
From eq 11, one finds IFL ∼ EP2, in the range of relatively low excitation irradiance. In general, the numerical solution of eq 10 affords a nearly linear dependence as IFL ∼ EP; for intermediate excitation irradiance, 1/[τFσ10(λP)] ≪ pI(r,t) ≪ σ10(λP)/δ2PA(λP); and IFL is nearly constant for pI(r,t) ≫ {σ10(λP)/δ2PA(λP), 1/[τFδ2PA(λP)]1/2}.
3. Experimental Section
3.1. Synthesis of Fluorene 1
The synthesis of sulfonyl-containing fluorene 1 is shown in Scheme 1. Precursors 2, 3, 5, and 6 were prepared according to the literature.(26) Compound 4 was obtained from the oxidation of 3, using a modified literature procedure.(27) Horner−Wadsworth−Emmons coupling was performed as described below, affording 1 in 38% yield. All glassware was flame-dried and cooled in a desiccator over CaCl2. All reactions were carried out under N2 atmosphere. All solvents and commercially available reagents were used without further purification unless otherwise indicated.
Scheme 1. Synthesis of Bis(Sulfonyl)-Containing Fluorene Derivative 1.

Preparation of (4-(Bromomethyl)phenyl)(phenyl) Sulfone (4)
The synthesis of (4-(bromomethyl)phenyl)(phenyl) sulfone (4) was performed according to a modified procedure from Hsiao et al.(27) Hydrogen peroxide (30%, 0.100 g, 0.80 mmol) was added to a solution of (4-(bromomethyl)phenyl)(phenyl) sulfide (3, 0.141 g, 0.51 mmol) in glacial acetic acid (0.200 g). The mixture was mildly heated until the exothermic reaction was initiated, and an additional 0.100 g of 30% hydrogen peroxide was added. The mixture was then taken to 100 °C and monitored by thin layer chromatography (TLC) every 15 min. Upon completion of the reaction (2 h), the mixture was cooled to room temperature, and the acetic acid was evaporated in vacuo. The crude product was dissolved in Et2O, washed twice with saturated NaHCO3 (aqueous), washed twice with saturated NaCl (aqueous), dried over anhydrous MgSO4, and filtered. After concentration of the filtrate in vacuo, the resulting product was recrystallized (1:1 acetone/water) to obtain 0.155 g (98%) of a colorless solid; mp 135−136 °C (lit. 132 °C).(28)1H NMR (300 MHz, CDCl3): 7.94 (m, 4H, Ar—H), 7.92 (m, 5H, Ar—H), 4.45 (s, 2H, —CH2—).13C NMR (75 MHz, CDCl3) 143.3 (C), 141.6 (C), 141.4 (C), 133.6 (CH), 130.1 (CH), 129.6 (CH), 128.4 (CH), 127.9 (CH), 31.8 (CH2).
Preparation of (4,4′-(1E,1′E)-2,2′-(9,9-Didecyl-9H-fluorene-2,7-diyl)bis(ethene-2,1-diyl)bis(4,1-phenylene))bis(phenylsulfone) (1)
In a two-neck, 250-mL round-bottom flask, 0.245 g (0.79 mmol) of (4-(bromomethyl)phenyl)(phenyl) sulfone (4) was dissolved in 6 mL of dry, freshly distilled P(OEt)3. The mixture was taken to reflux and monitored by TLC until complete conversion of the starting material was observed (5 h). Unreacted P(OEt)3 was evaporated by vacuum distillation, affording a viscous, pale yellow oil that was used for the Horner−Wadsworth−Emmons reaction without further purification. The phosphonate was dissolved in 10 mL of dry dimethylformamide (DMF), and 0.228 g of NaH was added portionwise to the solution. The mixture was stirred at room temperature for 1 h. A solution of 2,7-diformylflourene, 6, (0.198 g, 0.39 mmol) in 5 mL of dry DMF was added dropwise to the solution, which was then monitored by TLC until no 6 was observed (24 h). Once the reaction had reached completion, the solution was added dropwise to 150 mL of ice-chilled water. The resulting yellow oil was extracted with CH2Cl2 (twice), washed (three times) with water, and dried over MgSO4. The crude product was purified by column chromatography on silica gel (under N2) using a 1:1 mixture of ethyl acetate/hexane as the eluent. A yellow solid was obtained (0.142 g, 38% yield for two steps); mp 150.9−151.5 °C. 1H NMR (300 MHz, CDCl3): 7.96 (m, 8H), 7.63 (m, 16H), 7.28 (d, 2H), 7.12 (d, 16.5 Hz, 2H), 1.99 (m, 4H), 1.10 (m, 28H), 0.77 (t, 0.6.3 Hz, 6H), 0.63 (m, 0.4H). 13C NMR (75 MHz, CDCl3): 151.5 (C), 142.3 (C), 141.5 (C), 139.4 (C), 135.3 (C), 132.9 (C), 132.8 (CH), 131.2 (CH), 129.1 (CH), 128.0 (CH), 127.3 (CH), 126.7 (C), 126.7 (CH), 121.0 (C), 120.2 (C). 108.1 (C), 55.0 (C), 31.8 (C), 29.9 (C), 29.5 (C), 29.2 (C), 23.7 (C), 22.6 (C), 14.0 (C). HRMS for (C61H70O4S2): theoretical [M + H]+, 931.4788; [M + NH4+]+, 948.5054; [M + Na]+, 953.4608. Found: [M + H]+, 931.4769; [M + NH4+]+, 948.5057; [M + Na]+, 953.4604.
3.2. Optical Measurements
All measurements were performed in spectroscopic-grade toluene and THF at room temperature. The absorption spectra were obtained with an Agilent 8453 UV−visible spectrophotometer in 10-mm-path-length quartz cuvettes for concentrations, C, of ∼(1−2) × 10−5 M. The steady-state fluorescence spectra were recorded with a PTI QuantaMaster spectrofluorimeter in 10-mm spectrofluorometric quartz cuvettes for dilute solutions (C ≈ 10−6 M). All spectra were corrected for the spectral responsivity of the PTI detection system. The fluorescence quantum yields of 1 were determined relative to 9,10-diphenylanthracene in cyclohexane (η = 0.95).(29) Fluorescence lifetimes were measured with a PicoHarp300 time-correlated single-photon-counting system and 76 MHz femtosecond excitation (MIRA 900, Coherent).
Optical stimulated transitions in 1 were investigated using a picosecond Nd:YAG laser (PL 2143 B Ekspla) operating with exit wavelengths of 1064 nm (base), 532 nm (second harmonic), and 355 nm (third harmonic); a pulse duration of τ ≈ 21 ps (HW1/eM); nearly Gaussian time and space pulse intensity distributions; and a 10 Hz repetition rate. The experimental setup is illustrated in Figure 2. Two separate laser beams were used (pump−probe method(25)) for one-photon excitation and one- or two-photon induced quenching. Excitation laser pulses at λP = 355 nm with EP ≤ 12 μJ were focused into a 1-mm quartz cuvette (concentration C ≈ 5 × 10−5 M) with a waist of radius pr0 ≈ 0.15 mm. Fluorescence emission was observed perpendicular to the excitation beam. This fluorescence was partially quenched by another laser pulse at λq = 532 nm (one-photon quenching, qEP ≤ 50 μJ), propagating in the same direction, fully overlapped in space with the excitation volume and delayed for τd ≈ 80 ps relative to the excitation pulse. The amount of spontaneously emitted fluorescence photons during delay time τd was negligible given the condition τd ≪ τF. After the quenching pulse, the energy of the rest of the collected fluorescence photons was measured with a silicon photodetector. For two-photon induced quenching, the laser beam at λq = 1064 nm (qEP ≤ 200 μJ) was used at the same excitation conditions as for one-photon quenching: λP = 355 nm, EP ≤ 12 μJ, pr0 ≈ 0.15 mm, τd ≈ 80 ps, and full spatial overlap.
Figure 2.

Experimental setup: (1) picosecond laser, (2) beam splitters, (3) polarizers, (4) wave plate λ/2, (5) 100% reflection mirror, (6) neutral density filters, (7) calibrated silicon photodetectors, (8) time delay line, (9) focusing lens (25 cm), (10) 1- or 10-mm quartz cuvettes with investigated solutions, (11) optical collection system, (12) set of neutral and interferometric filters, (13) fiber-optic spectrometer. Optical elements 3 and 4 were used for ease of changing the pulse energies for the wavelengths used.
A single laser beam at λP = 532 nm was used in the case of two-photon excitation and simultaneous one-photon induced quenching of the fluorescence emission of 1. According to Figure 1a, the wavelength λP = 532 nm is within the fluorescence emission spectral range of 1 and the losses of pulse energy EP, determined by the ground-state one-photon absorption, are negligible. In this case, the laser beam with EP ≤ 200 μJ was focused in a 10-mm quartz cuvette (concentration C ≈ 3 × 10−4 M) to a waist of radius pr0 ≈ 0.3 mm. It should be mentioned that, in all measurements, the polarization of the laser beams was vertical, and the quality of the filtered scattered excitation and quenching light was controlled by a fiber-optic spectrometer USB2000 (Ocean Optics Inc.). No photochemical or other accumulative effects were observed.
4. Results and Discussion
The linear absorption and steady-state fluorescence spectra of 1 in toluene and THF at room temperature are presented in Figure 1a. The absorption spectra were nearly independent of solvent properties, whereas the fluorescence spectra exhibited a slight solvatochromic dependence. Fluorene 1 has nearly identical extinction coefficients (molar absorptivity) in toluene and THF [εmaxabs ≈ (92−95) × 10−3 M−1 cm−1] and a single exponential fluorescence decay with lifetimes of τF ≈ 0.85 ns (toluene) and τF ≈ 0.74 ns (THF). The fluorescence quantum yields of 1 were η ≈ 1 and independent of the excitation wavelength in all solvents, as shown in Figure 3a. This suggests no participation of direct radiationless transitions Sn → S0. Therefore, the fluorescence intensity of 1 was not quenched by an ESA mechanism. A comprehensive analysis of the linear spectral properties of 1, including excitation anisotropy spectra, is reported separately.(30)
Figure 3.

(a) Spectral dependence of the fluorescence quantum yield η (1) and normalized absorption spectrum of 1 in toluene. (b) Dependence of integral fluorescence emission in toluene. (c,d) Dependences 1 − IF/IF0 = f(qEP) for one-photon fluorescence quenching at λq = 532 nm in (c) toluene and (d) THF. (e,f) Two-photon fluorescence quenching at λq = 1064 nm in (e) toluene and (f) THF.
One-photon induced fluorescence of 1, and its delayed one- and two-photon quenching, can be analyzed from the data presented in Figure 3b−f. The dependences of the integrated fluorescence emission IFL on the excitation pulse energy EP at λP = 355 nm (Figure 3b) revealed a linear one-photon population of the fluorescent level S1 over the energy range used. A linear dependence IFL = f(EP) confirmed the validity of assumptions ii, iii, and v. The one-photon quenching wavelength λq = 532 nm was chosen to be within the fluorescence emission spectral range of 1, and the quenching pulse was delayed to τd ≈ 80 ps > 2τ to avoid overlap with the excitation pulse. The dependences of 1 − IF/IF0 on the quenching pulse energy qEP for one-photon excitation at 355 nm and one-photon quenching at 532 nm are presented in Figure 3c,d. From these data, the values of 1 − IF/IF0 were found to exhibit a linear dependence on qEP in both solvents, in agreement with eq 6. The values of the one-photon stimulated transition cross sections, σ10(λq), were estimated from the slopes of these dependences. For example, σ10(532 nm) ≈ 2.5 × 10−17 cm2 in toluene, which is close to the corresponding value of σ10(532 nm) ≈ 3 × 10−17 cm2 calculated from the known steady-state absorption εabs(λ) and fluorescence IFL(λ) spectra.
The values of 1 − IF/IF0 exhibited a quadratic dependence on the quenching pulse energy qEP (see Figure 3e,f) in the case of two-photon fluorescence quenching at λq = 1064 nm, which is in good agreement with eq 9. It should be mentioned that λq = 1064 nm is outside the fluorescence emission spectral range of 1 and, thus, one-photon stimulated transitions at this wavelength are negligible. Possible ESA processes at λq = 1064 nm from the S1 electronic state do not affect the fluorescence intensity, as discussed previously. As a result, the quadratic dependences 1 − IF/IF0 = f(qEP) reveal a pure two-photon stimulated quenching of the fluorescence. The values of the corresponding cross sections were estimated from the slopes of the dependences in Figure 3e,f: δ2PE(1064 nm) ≈ 240 and 280 GM for 1 in toluene and THF, respectively.
The nature of the stimulated transitions under two-photon fluorescence excitation and simultaneous one-photon fluorescence quenching at 532 nm was analyzed from the dependences presented in Figure 4. The quenching wavelength is located sufficiently far from the linear absorption bands and can be absorbed only through the 2PA mechanism. The dependences of the integrated fluorescence emission IFL of 1 on excitation pulse energy EP exhibited nearly quadratic character at low excitation irradiance (EP ≤ 15 μJ), as shown in the insets of Figure 4. These results are in good agreement with eq 11, describing a pure 2PA mechanism when the stimulated transitions S1 → S0 are neglected. As can be seen in Figure 4, the dependence IFL = f(EP) becomes close to linear in the intermediate range of excitation irradiance (EP > 15 μJ), providing evidence for the contribution of one-photon quenching at 532 nm. Further increase in the pulse energy to EP > 200 μJ, along with an expected saturation effect, was not realized because of laser damage of the cuvette. For comparison, the same dependence IFL = f(EP) was obtained for p-terphenyl in toluene; see Figure 5. The fluorescence spectrum of this compound (curve 2 in Figure 5a) did not overlap with λP = 532 nm. Thus, one-photon stimulated quenching is negligible. As can be seen in Figure 4b, the dependence IFL = f(EP) was close to quadratic over the entire energy range. These results for p-terphenyl in toluene at λP = 532 nm were anticipated, reflecting the two-photon excitation origin of the induced fluorescence.
Figure 4.

Dependences of the integrated fluorescence intensity of 1 in (a) toluene and (b) THF on excitation pulse energy EP at λP = 532 nm. Insets reveal the initial parts of the corresponding dependences.
Figure 5.

(a) Normalized absorption (1) and fluorescence (2) spectra of p-terphenyl in toluene. (b) Dependence IFL = f(EP) at λP = 532 nm for p-terphenyl in toluene. Inset represents a quadratic dependence.
5. Conclusion
A comprehensive investigation of stimulated transitions in sulfonyl-containing fluorene 1 in toluene and THF was accomplished with a picosecond fluorescence quenching method at room temperature. The fluorescence quenching processes exhibited good agreement with theoretical predictions and can be used to study the nature of stimulated transitions and to determine the corresponding one- and two-photon quenching cross sections. Pure two-photon stimulated transitions S1 → S0 with δ2PE(λq) ≈ 240−280 GM were shown for 1 at λq = 1064 nm, representing the first report of two-photon STED in a molecular system. A complex, nonquadratic dependence of two-photon induced fluorescence intensity on excitation pulse energy was obtained at λP = 532 nm, providing compelling evidence for the contribution of one-photon induced quenching upon two-photon excitation. The determination of the spectral dependences and absolute values of one- and two-photon stimulated emission cross sections is not only fundamentally important but might help develop the tenets for a number of nonlinear optical applications that can exploit STED. In particular, the two-photon stimulated emission processes can be harnessed to further increase the spatial resolution of fluorescence microscopy imaging. Based on the results of this study, sulfonyl-containing fluorene derivative 1, exhibiting a high fluorescence quantum yield and photochemical stability, is a promising fluorescence probe for one- and two-photon STED microscopy, a subject of further investigation in our laboratory.
Acknowledgments
We acknowledge the Civilian Research and Development Foundation (UKB2-2923-KV-07), the Ministry of Education and Science of Ukraine (Grant M/49-2008), and the National Science Foundation (ECS-0524533, ECS-0621715, and CHE-0832622) for support of this work.
References
- Bykova E. E.; Zemlyanov A. A.; Geints Y. E. Proc. SPIE 2006, 6522, 65220Y/1–65220Y/7. [Google Scholar]
- Shao P.; Li Z.; Qin J.; Gong H.; Ding S.; Wang Q. Aust. J. Chem. 2006, 59, 49–52. [Google Scholar]
- Correa D. S.; Goncalves V. C.; Balogh D. T.; Mendonca C. R.; De Boni L. Polymer 2006, 47, 7436–7440. [Google Scholar]
- De Boni L.; Gaffo L.; Misoguti L.; Mendonca C. R. Chem. Phys. Lett. 2006, 419, 417–420. [Google Scholar]
- Webster S.; Fu J.; Padilha L. A.; Przhonska O. V.; Hagan D. J.; Van Stryland E. W.; Bondar M. V.; Slominsky Y. L.; Kachkovski A. D. Chem. Phys. 2008, 348, 143–151. [Google Scholar]
- Gu B.; Ji W.; Patil P. S.; Dharmaprakash S. M. J. Appl. Phys. 2008, 103, 103511/1–103511/6. [Google Scholar]
- Allain C.; Schmidt F.; Lartia R.; Bordeau G.; Fiorini-Debuisschert C.; Charra F.; Tauc P.; Teulade-Fichou M.-P. ChemBioChem 2007, 8, 424–433. [DOI] [PubMed] [Google Scholar]
- Lattante S.; Barbarella G.; Favaretto L.; Gigli G.; Cingolani R.; Anni M. Appl. Phys. Lett. 2006, 89, 051111/1–051111/3. [Google Scholar]
- Kobayashi T.; Savatier J.-B.; Jordan G.; Blau W. J.; Suzuki Y.; Kaino T. Proc. SPIE 2004, 5351, 210–216. [Google Scholar]
- Ansari A.; Szabo A. Biophys. J. 1993, 64, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. H.; Tai O. Y.-H.; Wang Y.; Tsai T.-H.; Chang N.-C. J. Chem. Phys. 2005, 122, 084509/1–084509/7. [DOI] [PubMed] [Google Scholar]
- Galanin M. D.; Kirsanov B. P.; Chizhikova Z. A. Sov. Phys. JETP Lett. 1969, 9, 502–507. [Google Scholar]
- Sheik-Bahae M.; Said A. A.; Wei T. H.; Hagan D. J.; Van Stryland E. W. IEEE Quantum Electron. 1990, 26, 760–769. [Google Scholar]
- Belfield K. D.; Bondar M. V.; Hernandez F. E.; Przhonska O. V.; Yao S. J. Phys. Chem. B 2007, 111, 12723–12729. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.; Gryczynski I.. Fluorescence Quenching by Stimulated Emission. In Nonlinear and Two-Photon-Induced Fluorescence; Lakowicz J. R., Ed.; Topics in Fluorescence Spectroscopy; Plenum Press: New York, 1997; Vol. 5, Chapter 8, pp 305−360. [Google Scholar]
- Lakowicz J. R.; Gryczynski I.; Kusba J.; Bogdanov V. Photochem. Photobiol. 1994, 60, 546–562. [DOI] [PubMed] [Google Scholar]
- Kusba J.; Lakowicz J. R. J. Chem. Phys. 1999, 111, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryczynski I.; Kusba J.; Gryczynski Z.; Malak H.; Lakowicz J. R. J. Fluoresc. 1998, 8, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryczynski I.; Bogdanov V.; Lakowicz J. R. J. Fluoresc. 1993, 3, 85–92. [DOI] [PubMed] [Google Scholar]
- Hell S. W.; Wichmann J. Opt. Lett. 1994, 19, 780–782. [DOI] [PubMed] [Google Scholar]
- Klar T. A.; Jakobs S.; Dyba M.; Egner A.; Hell S. W. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V.; Seeger J.; Salditt T.; Hell S. W. J. Phys. B 2005, 38, S695–S705. [Google Scholar]
- Westphal V.; Rizzoli S. O.; Lauterbach M. A.; Kamin D.; Jahn R.; Hell S. W. Science 2008, 320, 246–249. [DOI] [PubMed] [Google Scholar]
- Voropai E. S.; Saechnikov V. A. Zh. Prikl. Spektrosk. 1989, 51, 382–402. [Google Scholar]
- Belfield K. D.; Bondar M. V.; Hernandez F. E.; Morales A. R.; Przhonska O. V.; Schafer K. J. Appl. Opt. 2004, 43, 6339–6343. [DOI] [PubMed] [Google Scholar]
- Yanez C. O.; Andrade C. D.; Belfield K. D. Chem. Commun. 2009, 827–829. [DOI] [PubMed] [Google Scholar]
- Hsiao C. N.; Shechter H. Tetrahedron Lett. 1982, 23, 1963–1966. [Google Scholar]
- Castro Pineiro J. L.; Cooper L. C.; Gilligan M.; Humphries A. C.; Hunt P. A.; Ladduwahetty T.; MacLeod A. M.; Merchant K. J.; Van Niel M. B.; Wilson K. PCT International Patent Application 2006097766, 2006.
- Lakowicz J. R.Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, 1999. [Google Scholar]
- Belfield K. D.; Bondar M. V.; Yanez C. O.; Hernandez F. E.; Przhonska O. V.. J. Mater. Chem. , published online Mar 31, 2009, http://dx.doi.org/10.1039/b820950b. [Google Scholar]