

Abstract
In neurons, Presenilin 1(PS1)/γ-secretase is located at the synapses, bound to N-cadherin. We have previously reported that N-cadherin-mediated cell-cell contact promotes cell-surface expression of PS1/γ-secretase. We postulated that N-cadherin-mediated trafficking of PS1 might impact synaptic PS1-APP interactions and Aβ generation. In the present report, we evaluate the effect of N-cadherin-based contacts on Aβ production. We demonstrate that stable expression of N-cadherin in Chinese Hamster Ovary (CHO) cells, expressing the Swedish mutant of human amyloid precursor protein (APP) leads to enhanced secretion of Aβ in the medium. Moreover, N-cadherin expression decreased Aβ42/40 ratio. The effect of N-cadherin expression on Aβ production was accompanied by the enhanced accessibility of PS1/γ-secretase to APP as well as a conformational change of PS1, as demonstrated by the fluorescence lifetime imaging technique (FLIM). These results indicate that N-cadherin-mediated synaptic adhesion may modulate Aβ secretion as well as the Aβ42/40 ratio via PS1/N-cadherin interactions.
Keywords: presenilin 1, N-cadherin, amyloid β, synapse, Alzheimer’s disease
Amyloid β (Aβ) peptides are the major components of senile plaques, a pathological hallmark of Alzheimer’s disease (AD), and are generated by the intramembranous cleavage of the amyloid precursor protein (APP) C-terminal fragment by Presenilin1 (PS1)/γ-secretase (De Strooper et al, 1998). PS1 is a multitransmembrane protein with a 30-kDa N-terminal fragment (NT), a 20-kDa C-terminal fragment (CT) and a large cytoplasmic loop domain (Thinakaran et al, 1996). Most of the PS1 mutations associated with familial AD (FAD) are known to increase the ratio of Aβ42 to Aβ40 (Aβ42/40 ratio), thereby increasing the more aggregation-prone Aβ42 relative to Aβ40 (Citron et al, 1997), which is considered at present to be an important molecular background of FAD pathogenesis. Using fluorescence lifetime imaging microscopy (FLIM), we have previously demonstrated that FAD-linked mutations in PS1 change the spatial relationship between PS1 NT and CT, increasing proximity of the two epitopes (Berezovska et al, 2005). This effect was contrary to that observed after the treatment with Aβ42-lowering nonsteroidal anti-inflammatory drugs (NSAIDs) which leads to the opposite conformational effect with PS1 NT and CT further apart (Lleo et al, 2004). These findings suggested that conformational change in PS1 due to mutations or to allosteric influences provides a possible structural basis for altered Aβ42/40 ratio.
In neurons, PS1 binds to β-catenin and N-cadherin at the synapse (Georgakopoulos et al, 1999). N-cadherin is essential for forming synaptic contact as well as for specific neuronal function such as synaptic plasticity (Bozdagi et al, 2000; Togashi et al, 2002). Accumulating evidence suggests that Aβ release may be regulated by synaptic activity (Kamenetz et al, 2003; Cirrito et al, 2005; Lesne et al, 2005). However, it remains largely unknown how PS1/γ-secretase-mediated APP cleavage is regulated by synaptic activity. We have recently demonstrated that N-cadherin promotes the cell-surface expression of PS1/γ-secretase via direct interaction with PS1 loop domain (Uemura et al, 2007). This result indicated that N-cadherin may recruit PS1/γ-secretase to synaptic sites. Thus we hypothesize that N-cadherin-based synaptic adhesion may influence Aβ production.
Here, we demonstrate that stable expression of N-cadherin in cadherin-deficient CHO cells expressing human APP Swedish mutant (APPSw) enhances the Aβ levels in the medium, possibly by increasing the accessibility of APP to PS1/γ-secretase. Moreover, N-cadherin expression induces a structural change in PS1, similar to that previously observed to accompany NSAID-induced decrease in Aβ42/40 ratio. These results indicate that N-cadherin-PS1 interactions may modulate Aβ production at the synapse, providing novel insight into AD pathophysiology.
Materials and Methods
Plasmid constructs
The construction of the expression vector encoding human N-cadherin tagged with HA at its C-terminus was described previously (Uemura et al, 2006b). The construction of the plasmid, expressing wtPS1 and the production of deletion mutant of PS1 (Δ340–350PS1), which is unable to interact with N-cadherin was described previously (Uemura et al, 2007). Precise cloning of all reading frame was verified by sequencing. The expression vector of APP-GFP was described elsewhere (Kinoshita et al, 2002). The original PS1-GFP (in the loop) construct was a generous gift from Dr. Kaether (Ludwig-Maximilians University, Germany) and was created byintroducing a Not1-GFP-Not1 between codon 351 and 352 of the cytoplasmic loop of human PS1. The RFP fragment with Not1 restriction sites at 5′ and 3′ ends was generated by PCR and GFP was replaced by RFP.
Cell culture and transfection
Chinese hamster ovary (CHO) cells were maintained in DMEM/F12 (Invitrogen) supplemented with 10% FBS. Transient transfection of wtPS1, PS1 mutant (Δ340–350PS1) and N-cadherin into cells were achieved by lipofection method, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Chinese hamster ovary (CHO) cells, stably expressing Swedish (K670/M671->N/L) mutant human APP695 (APPSw-CHO cells) and CHO cells stably expressing both Swedish mutant APP and human N-cadherin (APPSw/Ncad-CHO cells) were obtained as described elsewhere (Uemura et al, 2007). Primary cultured neurons were obtained from the hippocampus of fetal rats (17–19 days gestation) as described previously (Uemura et al, 2006a). Cultures were incubated in EMEM supplemented with 10% fetal calf serum or 10%horse serum.
Antibodies and Chemical Reagents
Mouse monoclonal anti-N-cadherin C-terminus and anti-β-catenin antibodies are obtained from Transduction Laboratories. Mouse monoclonal anti-β-actin antibody, mouse monoclonal anti-N-cadherin N-terminus antibody (N-cadherin neutralizing antibody, GC-4), rabbit polyclonal anti-nicastrin antibody, rabbit polyclonal anti-APP C-terminus antibody and control normal mouse IgG are from Sigma. Rabbit polyclonal anti-PS1 N-terminal fragment (NTF) and control normal rabbit IgG were from Santa Cruz. Rabbit polyclonal anti-BACE1 antibody was from Calbiochem. Rat monoclonal anti-PS1 NTF antibody was from Chemicon. Alexa Fluor 546 goat anti-mouse IgG, Alexa Fluor 546-phalloidin, and Alexa Fluor 488 goat anti-rabbit IgG, and Cy3-anti-rabbit IgG were obtained from Molecular Probes. Anti-mouse and rabbit horseradish peroxidase-conjugated secondary antibodies are from Amersham Biosciences.
Cell treatment by reagents
For the inhibition of N-cadherin-mediated cell-cell contact, cells were treated with 80μg/ml of N-cadherin-neutralizing antibody (GC-4) in OPTI-MEM for indicated period of time. Control cells were treated with an equal amount of normal mouse IgG.
Western Blot and Immunoprecipitation
Preparation of protein samples, the Western blot and immunoprecipitation analysis were carried out as described elsewhere (Uemura et al, 2007).
Immunostaining
The samples for immunostaining were prepared as described elsewhere (Uemura et al, 2007). After fixation, samples were examined using a laser scanning confocal microscopy, LSM 510 META (Zeiss) or BZ-9000 fluorescent microscopy (KEYENCE).
Measurement of BACE1 activity
β-secretase activity was measured by using β-secretase activity kit (R&D systems). Briefly, 2.5×105 cells of APPSw-CHO cells or APPSw/Ncad-CHO cells were plated in φ3.5cm dish and cultured overnight. Cells were collected and lysed by adding 500μl of 1x cell extraction buffer. Protein concentration of each cell lysate was determined by the Bradford method (Uemura et al, 2003) and equal amount of protein was subjected to the β-secretase activity assay, according to the manufacturer’s instruction.
Fluorescence Lifetime Imaging Microscopy (FLIM) assay
To analyze the PS1 conformation and/or PS1-APP interactions in intact cells expressing or not expressing N-cadherin, the APPSw-CHO or APPSw/Ncad-CHO cells were fixed and double-immunostained with corresponding antibodies labeled with Cy3 and Alexa 488 for the FLIM analysis. To monitor PS1 conformation we used goat anti-PS1 NT and rabbit anti-PS1 CT antibodies from Sigma. For the analysis of PS1-APP interactions we used mouse anti-PS1 antibody raised against amino acids 267–378 in the major TM6–7 loop domain (Chemicon, Temecula, CA) and an antibody to APP CT (Sigma). The fluorescence lifetime of a donor fluorophore (Alexa 488) was measured as described previously (Berezovska et al, 2005). In order to confirm the N-cadherin-mediated cell adhesion effect on Aβ production, we also examined the proximity between APP and PS1 in the presence of N-cadherin-neutralizing antibody (GC-4). For this blocking experiment, we modified the protocol for the FLIM assay since GC-4 is a mouse monoclonal antibody, which might cross-react the immunohistochemical results described above. We did two complementary FLIM experiments: 1) CHO cells stably expressing APPSw and N-cadherin were treated for 6 hrs with 80 ug/ml of either anti-N-cadherin blocking antibody (GC-4) or normal IgG as a control. The cells were fixed and immunostained with antibodies against APP (rabbit anti-APP CT, Sigma) and PS1 (goat anti-PS1 NT, Sigma) for the FLIM analysis. 2) The cells were transfected with C-terminally labeled APP-GFP and PS1-RFP (tagged in the TM6–7 loop region), treated with CG4 or IgG and the FLIM analysis was performed on the living cells.
Measurement of extracellular Aβ
Aβ peptides produced by rat hippocampus primary neurons were measured by using Mouse/Rat Amyloid β (1–40) (N) Assay Kit (IBL). Primary neurons, cultured in φ3.5cm dish were washed once with OPTI-MEM and then incubated in OPTI-MEM for indicated periods of time. After incubation, the culture medium was collected, centrifugated at 600×g for 5min, and the 100μl of the aliquot was used for the extracellular sample. Aβ40 and Aβ42 peptides produced by APPSw-CHO cells or APPSw/Ncad-CHO cells were measured by using Human β Amyloid (1–40) and (1–42) ELIZA Kit (WAKO), respectively. 6×105 of APPSw-CHO cells or APPSw/Ncad-CHO cells cultured in φ3.5cm dish were washed once with OPTI-MEM and then incubated in OPTI-MEM for indicated period of time. After incubation, the culture medium was collected, centrifugated 600×g, 5min, and the 100μl of the aliquot was used for measurement of extracellular Aβ.
Statistical analysis
All values are given in means±s.e. Comparisons were performed using a paired Student’s t-test. For comparison of multiparametric analysis, one-way factorial ANOVA, followed by the post hoc analysis by Fisher’s PLSD was used. P<0.05 was considered to indicate a significant difference. n=4 indicates four independent experiments.
Results
N-cadherin expression enhances Aβ secretion and reduces Aβ42/40 ratio
The purpose of our study is to define the effect of a synaptic adhesion molecule, N-cadherin, on Aβ production by using biochemical (Western blot and ELISA) and fluorescence resonance energy transfer (FRET)-based FLIM assay. First, we determined whether stable expression of N-cadherin could enhance the production of Aβ. To test this, CHO cells stably expressing Swedish (K670N/M671L) mutant human APP695 (APPSw-CHO cells) and CHO cells stably expressing both APPSw and human N-cadherin (APPSw/Ncad-CHO cells) were established. The expression levels of BACE1 and γ-secretase components were similar between APPSw-CHO and APPSw/Ncad-CHO cells (Figure 1A). BACE1 activity was not significantly different between these cell lines (Figure 1B). Immunocytochemical analysis using anti-N-cadherin and anti-PS1 antibodies revealed co-localization of these proteins at the sites of cell-cell contact and at the cell surface (Figure 1C–F, see also (Uemura et al, 2007)), indicating that PS1/γ-secretase was recruited to the cell-surface upon formation of N-cadherin-based cell-cell contact.
Figure 1. Characterization of APPSw/Ncad-CHO cells.

(A) APPSw-CHO cells and APPSw/Ncad-CHO cells were analyzed by Western blot. N-cadherin was expressed only in APPSw/Ncad-CHO cells. The expression levels of APP, BACE1, nicastrin, PS1 NT were similar in both cell lines. The bottom lane indicates the β-actin level, which was used as a loading control.
(B) APPSw-CHO cells and APPSw/Ncad-CHO cells were lysed and β-secretase activity in the lysate was measured. No significant difference was found between these cell lines (p=0.15, n=4).
(C–F) APPSw/Ncad-CHO cells were immunostained with with rabbit polyclonal anti-PS1 NT (C) and mouse monoclonal anti-N-cadherin antibodies (D). Merged image is shown in (D). Merged image with nuclear DAPI staining is shown in (F). The fixed samples were analyzed by BZ-9000 fluorescent microscopy (KEYENCE). N-cadherin (D) and PS1 (C) immunoreactivities are co-localized at the cell-cell contact sites (arrowheads). Scale bar: 20μm
Next, we compared the levels of Aβ40 and Aβ42 in the medium between APPSw-CHO and APPSw/Ncad-CHO cells. Both Aβ40 (Figure 2A) and Aβ42 (Figure 2B) levels were increased by stable expression of N-cadherin. Interestingly, the Aβ42/40 ratio was significantly reduced in N-cadherin expressing cells (Figure 2C). We established four independent clones of APPSw/Ncad-CHO cells, all of which produced significantly higher amounts of extracellular Aβ40, compared to the original APPSw-CHO cells (Figure 2D). Moreover, in order to confirm that enhanced Aβ secretion in APPSw/Ncad-CHO cells is specifically caused by the expression of N-cadherin, we used well-characterized N-cadherin-neutralizing antibody (GC-4) (De Wever et al, 2004) to inhibit N-cadherin-mediated contacts. The N-cadherin-neutralizing antibody inhibited the release of Aβ40 from APPSw/Ncad-CHO cells (Figure 2E, white columns), whereas it had no effect on APPSw-CHO cells (Figure 2E, black columns). The level of Aβ40 secreted from APPSw/Ncad-CHO cells after N-cadherin-neutralizing antibody treatment was similar to that of the original APPSw-CHO cells, indicating that the enhanced extracellular release of Aβ from these cells was specifically caused by the N-cadherin expression. Next, to confirm the effect of N-cadherin expression on the metabolism of wild-type APP, we established CHO cell line, which expresses wild-type APP with (APPWt/Ncad-CHO cells) or without (APPWt-CHO cells) N-cadherin (Supplementary Figure 1A). Both stable and transient expression of N-cadherin reduced Aβ42/40 ratio in the background of wild-type APP expression. Thus, these results strongly suggest that N-cadherin influences wild-type as well as mutant APP metabolism (Supplementary Figure 1B–E).
Figure 2. N-cadherin expression enhances extracellular Aβlevels and reduces Aβ42/40 ratio.
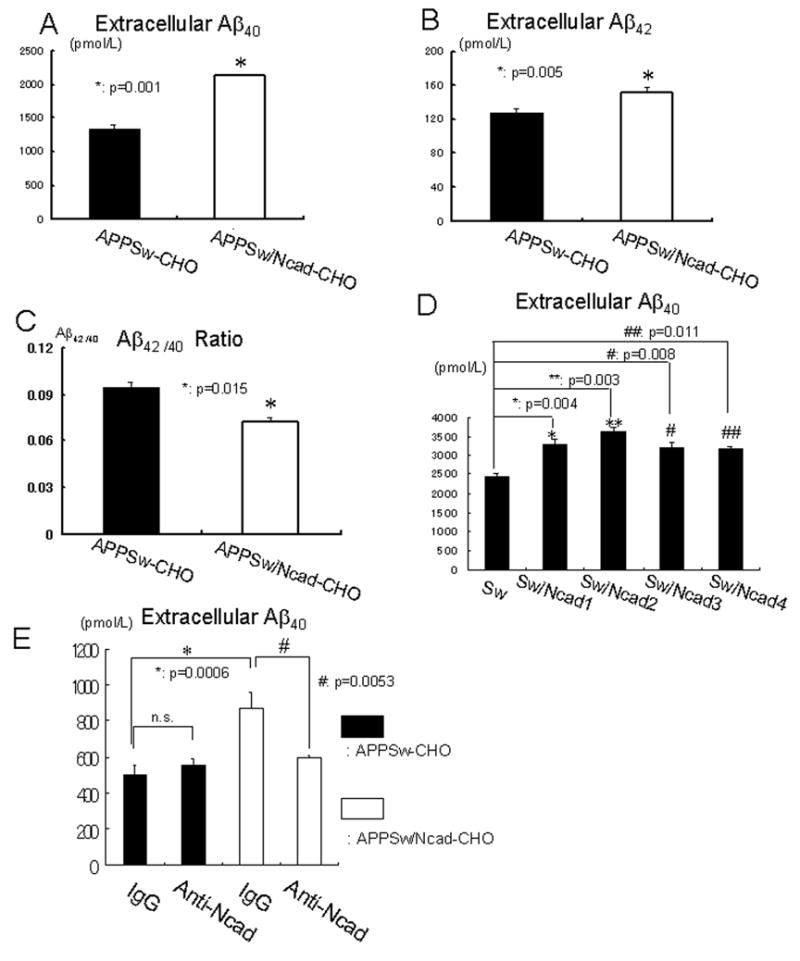
(A) APPSw-CHO cells or APPSw/Ncad-CHO cells were incubated in OPTI-MEM for 12 hours. The amount of extracellular Aβ40 was significantly elevated in APPSw/Ncad-CHO cells, compared to APPSw-CHO cells (n=8, p=0.001).
(B) APPSw-CHO cells or APPSw/Ncad-CHO cells were incubated in OPTI-MEM for 12 hours. After incubation, culture medium was collected and the amount of extracellular Aβ42 was measured. Extracellular Aβ42 was significantly elevated in APPSw/Ncad-CHO cells, compared to APPSw-CHO cells (n=8, p=0.005).
(C) The Aβ42/40 ratio in the medium was significantly decreased in APPSw/Ncad-CHO cells, compared to APPSw-CHO cells (n=8, p=0.015).
(D) APPSw-CHO (Sw) cells or four independent stable cell lines of APPSw/Ncad-CHO cells (SwNcad1–4) were incubated in OPTI-MEM for 24 hours. After incubation, the amount of extracellular Aβ40 was measured. Secreted extracellular Aβ40 was significantly elevated in every APPSw/Ncad-CHO stable cell line (SwNcad1–4), compared to that in APPSw-CHO cells (Sw) (n=4).
(E) APPSw-CHO cells or APPSw/Ncad-CHO cells were incubated in fresh OPTI-MEM containing either control IgG or N-cadherin-neutralizing antibody for 6 hours. After incubation, the amount of extracellular Aβ40 was measured. N-cadherin-neutralizing antibody significantly reduced the extracellular Aβ40 release into the medium in APPSw/Ncad-CHO cells (p=0.03, n=4). Conversely, N-cadherin-neutralizing antibody had no effect on the extracellular Aβ40 release into the medium in APPSw -CHO cells (p=0.3, n=4).
N-cadherin expression increases the accessibility of PS1/γ-secretase to its substrate APP
We have previously demonstrated by the FLIM assay that close association of PS1 and APP preferentially occurs in the distal subcellular compartments (Berezovska et al, 2003). In addition, we have shown that N-cadherin/PS1 interaction changes subcellular distribution of the PS1/γ-secretase, thereby enhancing its expression at the cell-surface (Uemura et al, 2007). Thus, we postulated that enhanced secretion of Aβ in N-cadherin expressing cells may be attributed to better accessibility of APP to PS1/γ-secretase. To test this hypothesis we used an established FLIM assay to monitor APP-PS1 interactions (Berezovska et al, 2003). PS1 was immunostained with an anti-PS1 loop region antibody labeled with Alexa 488 (FRET donor) and the APP CT was immunostained with a Cy3-labeled antibody (FRET acceptor). The fluorescence lifetime of the Alexa 488 donor fluorophore shortens in close vicinity (<10 nm) of a FRET acceptor fluorophore. The degree of the lifetime shortening is a quantitative measure of proximity. The donor fluorophore lifetime can be color-coded and displayed on a pixel-by-pixel basis through the entire image of the cell: if APP and PS1 molecules are closer together, the donor fluorescence lifetime will be shorter, and the color will be closer to red. The FLIM analysis showed that Alexa 488 lifetime was significantly shortened in APPSw/Ncad-CHO cells, compared to that in APPSw-CHO cells, indicating that PS1 and APP came into closer proximity (or increased percentage of molecules are in close proximity to one another) in the presence of N-cadherin (Table 1). Pseudocolor FLIM image showed more red pixels per cell (i.e. more interacting molecules per cell) in APPSw/Ncad-CHO cells (Figure 3B), compared to that in APPSw-CHO cells (Figure 3A). This indicates that N-cadherin expression may increase the accessibility of PS1/γ-secretase to its substrate APP. In order to examine the effect of N-cadherin-mediated cell adhesion on APP/PS1 interaction, we also examined the proximity of APP and PS1 in the presence of N-cadherin-neutralizing antibody (GC-4). We performed two complementary FLIM experiments; one with immunohistochemistry using goat anti-PS-NT antibody and rabbit anti-APP-CT antibody (Table 2-1) and the other using live cells expressing APP-GFP and PS1-RFP (Table 2--2), in the presence of either GC-4 or normal mouse IgG as a control. In both blocking experiments, we observed significantly longer donor fluorophore lifetime in GC4 treated cells, comparing to that in IgG treated cells, indicating that N-cadherin-based cell-cell adhesion specifically modulates the accessibility of APP to PS1/γ-secretase. To confirm these results biochemically, we transfected N-cadherin into HEK293 cells and analyzed whether N-cadherin expression enhances the APP-PS1 interaction by immunoprecipitation. As expected, APP-PS1 interaction was increased in N-cadherin expressing cells (Supplement Figure 2), indicating that N-cadherin expression brings APP and PS1/γ-secretase in closer proximity.
TABLE 1.
FRET between PS1 loop and APP CT in CHOSw compared to NcadCHOSw cells
| cell line | FRET donor (Alexa 488) | FRET acceptor (Cy3) | Alexa 488 lifetime in ps (mean ± S.E.) | p value (compared with CHOSw PS1 loop APP CT) |
|---|---|---|---|---|
| APPSw-CHO (n = 10) | PS1 loop | None (negative control) | 1932+/−7 | p < 0.0001 |
| APPSw-CHO (n = 10) | PS1 loop | APP CT | 1790 ± 22 | |
| APPSw/Ncad-CHO (n = 12) | PS1 loop | APP CT | 1644 ± 25 | p < 0.0001 |
Figure 3. N-cadherin expression in CHO cells increases PS1-APP interactions and induces conformational change of PS1/γ-secretase.
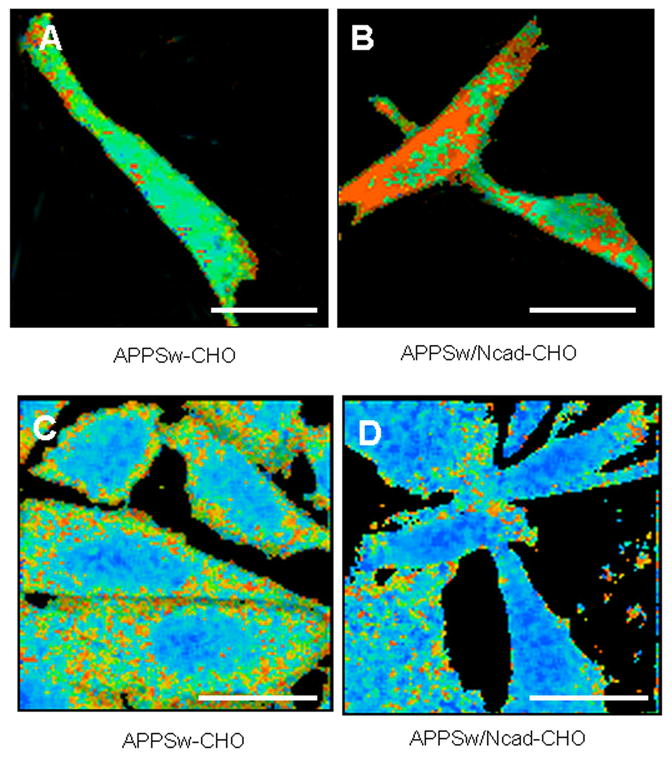
(A and B) For the FLIM assay, PS1 is stained at its loop region with Alexa 488 (FRET donor) and APP is stained at its CT with Cy3(FRET acceptor). The fluorescence lifetime of Alexa 488 is displayed as a pseudocolor image: if PS1 and APP molecules are closer together, the donor fluorescence lifetime will be shorter, and the color will be closer to red. Alexa488 lifetime was significantly shortened in APPSw/Ncad-CHO cells (B), compared to that in APPSw-CHO cells (A), indicating that PS1 and APP came into closer proximity in the presence of N-cadherin. Scale bar: 10μm
(C and D) APPSw-CHO (C) or APPSw/Ncad-CHO (D) cells were immunostained with antibodies against PS1 NT (Alexa 488) and CT (Cy3). The proximity between PS1 NT and CT was evaluated by measuring lifetime of the Alexa 488 donor fluorophore (PS1 NT Alexa 488) in the FLIM assay. The fluorescence lifetime of Alexa 488 is displayed as a pseudocolor image. Red pixels indicate close proximity between PS1 N- and C-termini. Alexa 488 lifetime in APPSw/Ncad-CHO (D) cells was significantly increased, compared to that in APPSw-CHO cells (C), indicating that N-cadherin “opened” PS1 conformation with NT and CT further apart. Scale bar: 10μm
TABLE 2.
| TABLE 2-1. FRET between PS1 N-terminus and APP CT in NcadCHOSw cells treated with N-cadherin neutralizing GC-4 antibody or IgG as a control | ||||
|---|---|---|---|---|
| cell line | FRET donor (Alexa 488) | FRET acceptor (Cy3) | Alexa 488 lifetime in ps (mean ± S.E.) | p value (compared with control IgG) |
| NcadCHOSw (Negative Control) | PS1 NT | None | 1903±28 | |
| NcadCHOSw with control IgG (n = 21) | PS1 NT | APP CT | 1677 ± 22 | |
| NcadCHOSw with GC-4 (n = 17) | PS1 NT | APP CT | 1789 ± 100 | p < 0.01 |
| TABLE 2-2. FRET between PS1 loop and APP CT in living NcadCHOSw cells treated with GC-4 or IgG | ||||
|---|---|---|---|---|
| cell line | FRET donor | FRET acceptor | Alexa 488 lifetime in ps (mean ± S.E.) | p value (compared with control IgG) |
| NcadCHOSw (Negative Control) | APP-GFP | None | 2076±62 | |
| NcadCHOSw with control IgG (n = 21) | APP-GFP | PS1-RFP(loop) | 1646 ± 223 | |
| NcadCHOSw with GC-4 (n = 17) | APP-GFP | PS1-RFP(loop) | 1859 ± 74 | p < 0.01 |
N-cadherin expression induces the conformational change of PS1
Whereas total Aβ was increased in N-cadherin expressing cells, the Aβ42/40 ratio was reduced (Figure. 2C). We and others have demonstrated previously that Aβ42/40 ratio correlates with PS1 conformation in intact cells: familial Alzheimer’s disease mutations in PS1 that elevate Aβ42/40 ratio decreased (Berezovska et al, 2005), while Aβ42–lowering NSAIDs (Lleo et al, 2004) or structural changes in γ-secretase component, Pen2 (Isoo et al, 2007) increased, PS1 NT-CT proximity. Therefore, we investigated whether change in Aβ42/40 ratio observed in cells with tighter cell-cell adhesion mediated by N-cadherin is due to a conformational change in PS1/γ-secretase. The proximity between PS1 NT and CT in fixed and detergent permeabilized cells was evaluated by measuring lifetime of the Alexa 488 donor fluorophore (PS1 NT Alexa 488) in the absence (negative control) and presence of the Cy3 acceptor on the PS1 CT. As expected, the Alexa 488 donor fluorophore lifetime shortened when the PS1 CT was labeled with the Cy3 acceptor (Table 3), consistent with the close proximity between the PS1 NT and CT in APPSw-CHO cells. In contrast, Alexa 488 lifetime in APPSw/Ncad-CHO cells was significantly longer (1821+−14 psec), compared to that in APPSw-CHO cells, indicating that N-cadherin “opened” the PS1 conformation with NT and CT being further apart (Table 2, Figure 3C, D). Thus, these results are in agreement with the previous findings that more “open” PS1 conformation correlates with generation of the shorter Aβ species (Lleo et al, 2004), and therefore decreased Aβ42/40 ratio in APPSw/Ncad-CHO cells may be attributed to the change in conformation of the PS1/γ-secretase due to N-cadherin overexpression.
TABLE 3.
FRET between PS1 NT and CT in CHOSw compared to NcadCHOSw cells
| cell line | FRET donor (Alexa 488) | FRET acceptor (Cy3) | Alexa 488 lifetime in ps (mean ± S.E.) | p value (compared with NcadCHOSw) |
|---|---|---|---|---|
| APPSw-CHO (n = 11) | PS1 NT | None (negative control) | 1897 ± 7 | p < 0.0001 |
| APPSw-CHO (n = 14) | PS1 NT | PS1 CT | 1524 ± 46 | p = 0.0002 |
| APPSw/Ncad-CHO (n = 14) | PS1 NT | PS1 CT | 1821 ± 14 |
PS1/N-cadherin interaction affects both Aβ production and Aβ42/40 ratio
Since N-cadherin interacts with the cytoplasmic loop of PS1 CTF (Georgakopoulos et al, 1999), we next determined whether the PS1/N-cadherin interaction affects Aβ production and/or Aβ42/40 ratio. To test this, either wtPS1 or a PS1 mutant lacking the N-cadherin interaction domain (Δ340–350PS1, (Uemura et al, 2007)) was transfected into APPSw/Ncad-CHO cells. Since PS1/γ-secretase acts in a complex including PS1, Nicastrin, Pen-2 and Aph-1 (Takasugi et al, 2003), Δ340–350PS1 competes with endogenous wild-type PS1 to occupy other components of γ-secretase and act in a dominant-negative fashion (Thinakaran et al, 1997). As expected, immunoprecipitation assay revealed that Δ340–350PS1 does not interact with N-cadherin (Figure 4A). We found that the extracellular levels of both Aβ40 (Figure 4B) and Aβ42 (Figure 4C) were decreased after the transient expression of Δ340–350PS1, compared to wtPS1. In addition, Aβ42/40 ratio in the medium was increased in the Δ340–350PS1 transfectants, compared to that in wtPS1 (Figure 4D), indicating that the PS1/N-cadherin interaction affects both Aβ production and Aβ42/40 ratio.
Figure 4. Loss of PS1/N-cadherin interaction reduces extracellular Aβ levels and enhances Aβ42/40 ratio.
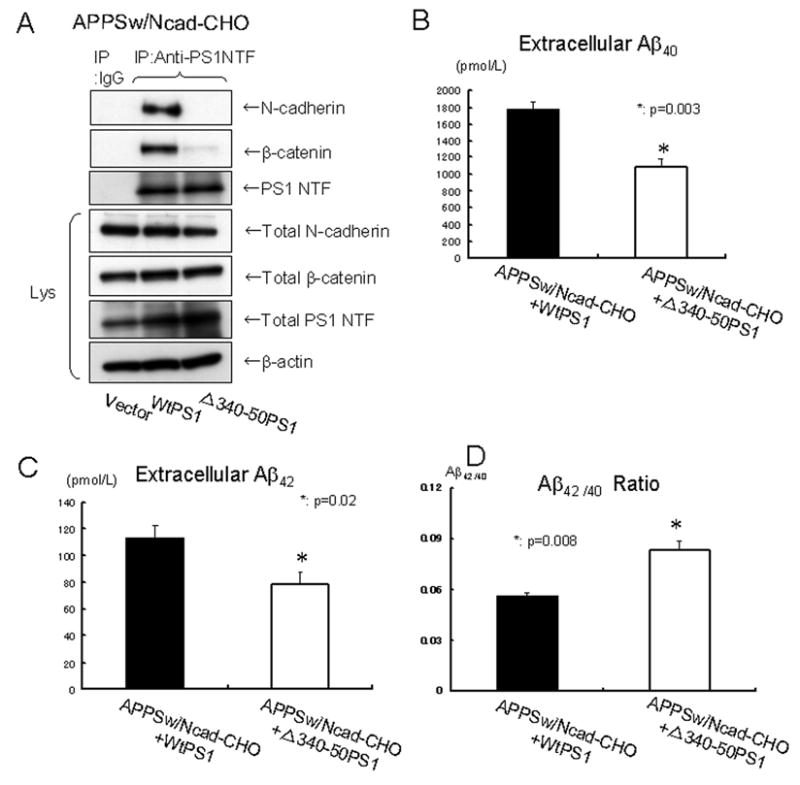
(A) APPSw/Ncad-CHO cells were transfected with either wtPS1 or PS1 mutant lacking PS1/N-cadherin interaction domain (Δ340–350PS1). 24 hours after transfection, cell lysates were immunoprecipitated with rabbit polyclonal anti-PS1 NT antibody or normal rabbit IgG as a control, followed by the Western blot. N-cadherin and β-catenin were efficiently co-immunoprecipitated with wild type PS1. However, very poor N-cadherin and β-catenin signal was detected in the co-immunoprecipitates from APPSw/Ncad-CHO cells transfected with Δ340–350PS1, indicating the lack of PS1/N-cadherin/β-catenin interaction. The expression levels of PS1, N-cadherin andβ-catenin in the cell lysates (Lys) were similar between these cell lines. The bottom lane indicates the β-actin level, used as a loading control.
(B) 6×105 of APPSw/Ncad-CHO cells cultured in φ3.5cm dish were transfected with either wtPS1 or PS1 mutant lacking PS1/N-cadherin interaction domain (Δ340–350PS1). 24 hours after transfection, cells were washed once with OPTI-MEM and incubated in fresh OPTI-MEM for 12 hours. After incubation, the culture medium was collected and the amount of extracellular Aβ40 was measured by ELISA. Extracellular Aβ40 was significantly reduced in the background of Δ340–350PS1 transfection, compared to that in wtPS1 transfected cells (p=0.003, n=4).
(C) The amount of extracellular Aβ42 in the same condition as in (B) was measured by ELISA. Extracellular Aβ42 was significantly reduced in the background of Δ340–350PS1 transfection, compared to that in wtPS1 transfected cells (p=0.02, n=4).
(D) The Aβ42/40 ratio in the conditions (B) and (C) was significantly reduced in in the background of Δ340–350PS1 transfection (p=0.008, n=4).
Discussion
In this report, we demonstrate that introducing N-cadherin into cadherin-deficient CHO cells increased secreted Aβ40 and Aβ42 levels (Figure 2). The expression of N-cadherin in CHO cells elevates cell-surface levels of PS1/γ-secretase ((Uemura et al, 2007), see also Figure 1). Thus, the effect of the cadherin expression on Aβ secretion might be mediated by the change in the subcellular distribution of PS1/γ-secretase. In addition, our FLIM analysis revealed that the N-cadherin expression allowed more PS1 and APP to interact near the cell surface, resulting in a greater amount of fluorophore-labelled epitopes coming into close proximity (Table 1 and see more red pixels in Figure 3B compared to 3A). The FLIM results were confirmed by co-IP experiment (Supplement Figure 2), indicating better accessibility of APP to PS1/γ-secretase in the presence of N-cadherin. These data suggest that subcellular redistribution and better accessibility of PS1/γ-secretase to APP substrate may be the cause of the net increase in total Aβ production in the presence of N-cadherin. The cellular compartment in which APP/PS1 interactions are promoted by N-cadherin was not clarified in the present study. However, since N-cadherin is an important cell adhesion molecule and we have previously demonstrated that N-cadherin promotes cell-surface expression of PS1/γ-secretase ((Uemura et al, 2007), see also Figure 1), the interactions are likely to occur near to the cell surface.
Interestingly, N-cadherin expression not only enhanced Aβ release, but also decreased Aβ42/40 ratio, the latter effect is similar to NSAIDs treatment (Lleo et al, 2004) and opposite to that caused by the FAD-linked PS1 mutations (Berezovska et al, 2005). This effect was associated with the “open” PS1 conformation, driving NT and CT further apart, in N-cadherin expressing cells as revealed by the FLIM assay (Table 2 and Figure 3C, D). Preventing N-cadherin-PS1 interaction either by absence of N-cadherin (Figure 1) or by introducing a PS1 mutant that does not interact with N-cadherin (Figure 4) both have increased Aβ42/40 ratio. In the absence of N-cadherin (APPSw-CHO cells), the Aβ42/40 ratio was around 0.095+−0.006, whereas it was reduced to 0.072+−0.006 in the presence of N-cadherin (APPSw/Ncad-CHO cells) (Figure 2C). The Aβ42/40 ratio under the expression of Δ340–350PS1 was 0.083+−0.009, whereas Aβ42/40 ratio under the expression of wt PS1 was 0.056+−0.004 (Figure 4D). These results indicate that Δ340–350 mutant prevented the decrease in Aβ42/40 ratio induced by N-cadherin expression and restored to baseline Aβ42/40 ratio. Since N-cadherin binds to the cytoplasmic loop of PS1 CT (Georgakopoulos et al, 1999), it is possible that this physical contact causes an allosterical change in PS1 conformation by moving PS1 NT and CT further apart. On the contrary to the N-cadherin expression, the expression of wt PS1 has no effect on Aβ production. Other reports have also demonstrated that single expression of wt PS1 has limited effect on Abeta production in vivo (Citron et al., 1997). We speculate that this apparent contradiction is caused by the lack of other γ-secretase components, when PS1 is expressed alone. γ-secretase is composed of PS1, nicastrin, pen-2 and aph-1 and can remain stable only when these components are available. It was also reported that expression of pen-2 is required for conferring the γ-secretase activity and endoproteolysis of PS1 (Takasugi et al., 2003). Thus, expression of PS1 alone might not have impact on Aβ metabolism significantly.
Thus, expression of N-cadherin modulates Aβ production in two ways: the total amount of Aβ and theAβ42/40 ratio. These are independent readouts of γ-secretase function. According to our experimental data, these changes can be interpreted as reflecting access of N-terminally cleaved APP to functionally active γ-secretase (total amount of Aβ) compared to the exact molecular interaction between PS-1 and the APP substrate (Aβ42/40 ratio). The presence of N-cadherin impacts each of these features, by directing the localization of γ-secretase closer to cell surface membrane as well as a direct allosteric effect on PS-1/γ-secretase conformation.
Accumulating evidence suggests that partial loss of function in PS1/γ-secretase may lead to increased Aβ42/40 ratio as well as to neurodegeneration (Shen and Kelleher RJ, 2007; Wolfe, 2007). In addition, a recent report suggests that Aβ40 mayinhibit amyloid deposition and thus may be physiologically neuroprotective (Kim et al, 2007). In this respect, tight cell-cell contact may enhance the function of PS1/γ-secretase to produce more Aβ40 by inducing its distributional and conformational change.
It has recently been shown that neuronal activity modulates the production and secretion of Aβ peptides (Kamenetz et al, 2003; Lesne et al, 2005). In addition, it was demonstrated in vivo that Aβ levels in the brain interstitial fluid are dynamically influenced by synaptic activity (Cirrito et al, 2005). Taken together, Aβ secretion seems to be physiologically regulated in neurons and Aβ itself may have its own physiological function (Pearson and Peers, 2006). On the contrary, converging lines of evidence suggests that natural soluble Aβ oligomers trigger synaptic loss (Spires et al, 2005; Shankar et al, 2007). Therefore, it is plausible that synaptic dissociation caused by Aβ oligomers changes PS1 conformation to produce more Aβ42 thus starting the vicious cycle of Aβ42 generation by modifying the Aβ42/40 ratio. Our present study presents solid evidence that Aβ production and the Aβ42/40 ratio can be modulated by the degree of PS1-N-cadherin interaction, and thus potentially by cell-cell adhesion status. Our current findings, thus, provide a potential link between synaptic contacts and physiological Aβ release, with cadherins being the key player. However, these experiments are carried out by CHO cell lines exogenously expressed with APP or its mutant, which does not allow to conclude about the relevance of the presented mechanism in neurons and remain to be proved in neuron and in vivo settings.
With its potential role in the rearrangement of existing cell-cell contacts (Okamoto et al, 2001; Marambaud et al, 2002; Haas et al, 2005) PS1/γ-secretase may influence synaptic plasticity, which might be affected in AD. Future study in this field could lead to a better understanding of AD synaptic pathophysiology.
Supplementary Material
Acknowledgments
Research described in this article was supported by Philip Morris USA Inc., Philip Morris International (OB, SS) and NIH AG026593 (OB) and by the Ministry of Education, Science, Sportsand Culture (Japan), Grant-in-Aid for 18023021, 18059019 (AK). We thank Dr. Kaether (Ludwig-Maximilians University, Germany) for generously providing original PS1-GFP construct.
Abbreviations List
- PS1
presenilin 1
- Aβ
amyloid β
- APP
amyloid precursor protein
- APPSw
APP Swedish mutant
- FRET
fluorescence resonance energy transfer
- FLIM
fluorescence lifetime imaging technique
- AD
Alzheimer’s disease
- NT
N-terminal fragment
- CT
C-terminal fragment
- FAD
familial AD
- NSAIDS
nonsteroidal anti-inflammatory drugs
- CHO
Chinese Hamster Ovary
References
- Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, Hyman BT. Familial Alzheimer’s disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005;25:3009–3017. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovska O, Ramdya P, Skoch J, Wolfe MS, Bacskai BJ, Hyman BT. Amyloid precursor protein associates with a nicastrin-dependent docking site on the presenilin 1-gamma-secretase complex in cells demonstrated by fluorescence lifetime imaging. J Neurosci. 2003;23:4560–4566. doi: 10.1523/JNEUROSCI.23-11-04560.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Wever O, Westbroek W, Verloes A, Bloemen N, Bracke M, Gespach C, Bruyneel E, Mareel M. Critical role of N-cadherin in myofibroblast invasion and migration in vitro stimulated by colon-cancer-cell-derived TGF-beta or wounding. J Cell Sci. 2004;117:4691–4703. doi: 10.1242/jcs.01322. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos A, Marambaud P, Efthimiopoulos S, Shioi J, Cui W, Li HC, Schutte M, Gordon R, Holstein GR, Martinelli G, Mehta P, Friedrich VL, Jr, Robakis NK. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell. 1999;4:893–902. doi: 10.1016/s1097-2765(00)80219-1. [DOI] [PubMed] [Google Scholar]
- Haas IG, Frank M, Veron N, Kemler R. Presenilin-dependent processing and nuclear function of gamma-protocadherins. J Biol Chem. 2005;280:9313–9319. doi: 10.1074/jbc.M412909200. [DOI] [PubMed] [Google Scholar]
- Isoo N, Sato C, Miyashita H, Shinohara M, Takasugi N, Morohashi Y, Tsuji S, Tomita T, Iwatsubo T. Abeta42 overproduction associated with structural changes in the catalytic pore of gamma-secretase: common effects of Pen-2 N-terminal elongation and fenofibrate. J Biol Chem. 2007;282:12388–12396. doi: 10.1074/jbc.M611549200. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Smith CJ, Berezovska O, Hyman BT. Direct visualization of the gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein: association with Fe65 and translocation to the nucleus. J Neurochem. 2002;82:839–847. doi: 10.1046/j.1471-4159.2002.01016.x. [DOI] [PubMed] [Google Scholar]
- Lesne S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci. 2005;25:9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT. Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med. 2004;10:1065–1066. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, Wong AJ, Saya H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson HA, Peers C. Physiological roles for amyloid beta peptides. J Physiol. 2006;575:5–10. doi: 10.1113/jphysiol.2006.111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3 The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, Sisodia SS. Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem. 1997;272:28415–28422. doi: 10.1074/jbc.272.45.28415. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Bito H, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Activity-dependent regulation of beta-catenin via epsilon-cleavage of N-cadherin. Biochem Biophys Res Commun. 2006a;345:951–958. doi: 10.1016/j.bbrc.2006.04.157. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kihara T, Kuzuya A, Okawa K, Nishimoto T, Ninomiya H, Sugimoto H, Kinoshita A, Shimohama S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci Lett. 2006b;402:278–283. doi: 10.1016/j.neulet.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kitagawa N, Kohno R, Kuzuya A, Kageyama T, Chonabayashi K, Shibasaki H, Shimohama S. Presenilin 1 is involved in maturation and trafficking of N-cadherin to the plasma membrane. J Neurosci Res. 2003;74:184–191. doi: 10.1002/jnr.10753. [DOI] [PubMed] [Google Scholar]
- Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, Kinoshita A. GSK3beta Activity Modifies the Localization and Function of Presenilin 1. J Biol Chem. 2007;282:15823–15832. doi: 10.1074/jbc.M610708200. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:136–140. doi: 10.1038/sj.embor.7400896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.