

Abstract
Astrocytes in the CNS respond to tissue damage by becoming reactive. They migrate, undergo hypertrophy, and form a glial scar that inhibits axon regeneration. Therefore, limiting astrocytic responses represents a potential therapeutic strategy to improve functional recovery. It was recently shown that the epidermal growth factor (EGF) receptor is upregulated in astrocytes after injury and promotes their transformation into reactive astrocytes. Furthermore, EGF receptor inhibitors were shown to enhance axon regeneration in the injured optic nerve and promote recovery after spinal cord injury. However, the signaling pathways involved were not elucidated. Here we show that in cultures of adult spinal cord astrocytes EGF activates the mTOR pathway, a key regulator of astrocyte physiology. This occurs through Akt-mediated phosphorylation of the GTPase-activating protein Tuberin, which inhibits Tuberin's ability to inactivate the small GTPase Rheb. Indeed, we found that Rheb is required for EGF-dependent mTOR activation in spinal cord astrocytes, whereas the Ras–MAP kinase pathway does not appear to be involved. Moreover, astrocyte growth and EGF-dependent chemoattraction were inhibited by the mTOR-selective drug rapamycin. We also detected elevated levels of activated EGF receptor and mTOR signaling in reactive astrocytes in vivo in an ischemic model of spinal cord injury. Furthermore, increased Rheb expression likely contributes to mTOR activation in the injured spinal cord. Interestingly, injured rats treated with rapamycin showed reduced signs of reactive gliosis, suggesting that rapamycin could be used to harness astrocytic responses in the damaged nervous system to promote an environment more permissive to axon regeneration.
Keywords: Akt, Tuberin, cell migration, rapamycin, ischemia, spinal cord injury
Introduction
Injuries to the CNS not only cause damage to neuronal circuits but also induce dramatic changes in the glial cells (David and Lacroix, 2003; Silver and Miller, 2004; Yiu and He, 2006; Busch and Silver, 2007). Following spinal cord injury, astrocytes upregulate expression of intermediate filament proteins, such as vimentin, nestin, and glial fibrillary acidic protein, as well as proteoglycans and other molecules that are inhibitory to axon growth. Although some proliferation occurs following injury, the most dramatic astrocyte responses involve migration toward the site of injury and hypertrophy. Over time, reactive astrocytes deposit extracellular matrix molecules and form a glial scar. The glial scar plays an important role in the repair process because it contributes to the reestablishment of homeostasis by insulating the injury site and restoring the integrity of the blood–brain barrier (Faulkner et al., 2004; Silver and Miller, 2004). However, the glial scar also represents a physical and biochemical barrier that greatly contributes to the failure of damaged axons to regenerate and reestablish functional connections, thus hindering functional recovery (David and Lacroix, 2003; Faulkner et al., 2004; Silver and Miller, 2004).
Growth factors, such as EGF and TGFα, have been implicated in the transformation of quiescent astrocytes into reactive astrocytes, and it has been recently reported that EGF receptor expression and activation are upregulated in astrocytes after injuries to the CNS (L. Liu et al., 2006). However, the signaling pathways stimulated by EGF that lead to astrocyte activation have not been fully characterized. The serine/threonine kinase mTOR (mammalian target of rapamycin) is a key regulator of cell size and proliferation downstream of growth factor receptors, in addition to its role in mediating cell responses to nutrients (Sabatini, 2006; Shaw and Cantley, 2006; Chiang and Abraham, 2007). Various mTOR upstream regulators have been reported to play an important role in astrocytes. For example, inactivation of a negative regulator of the pathway, the tumor suppressor Pten, promotes astrocyte hypertrophy and proliferation, which can lead to the formation of benign glial tumors called hamartomas as well as malignant glial tumors (Li et al., 1997; Fraser et al., 2004; Wullschleger et al., 2006). Inactivation of another tumor suppressor, Tuberin, also leads to glial cell hypertrophy and the formation of glial hamartomas (Wullschleger et al., 2006). Tuberin is a GTPase-activating protein for Rheb, a Ras family GTPase that activates mTOR by disrupting the inhibitory interaction of the FK506-binding protein FKBP38 (Bai et al., 2007). Akt, Tuberin, and Rheb activate mTOR when it is part of a protein complex called mTORC1 (mTOR complex 1), which is exquisitely sensitive to the drug rapamycin (Bain et al., 2007; Bhaskar and Hay, 2007; Chiang and Abraham, 2007). Interestingly, rapamycin acts by promoting the inhibitory interaction with mTOR of another FK506-binding protein, FKBP12. mTOR also exists as part of a second protein complex, called mTORC2, which is regulated differently and is insensitive to acute inhibition by rapamycin. Our data suggest that a pathway involving mTORC1 plays an important role in spinal cord astrocytes downstream of the EGF receptor.
Materials and Methods
Antibodies and other reagents.
Antibodies against the phosphorylated EGF receptor (Tyr845), p70 S6 kinase, phosphorylated p70 S6 kinase (Thr389), Erk1/Erk2, phosphorylated Erk1/Erk2 (Thr202/Tyr204), Akt, phosphorylated Akt (Ser473), phosphorylated Tuberin (Thr1462), phosphorylated S6 (Ser235/Ser236), and cleaved caspase 3 and the Rheb antibody for immunofluorescence microscopy were from Cell Signaling; the anti-Rheb antibody for immunoblotting was from AbNova; the EGF receptor and Tuberin antibodies were from Santa Cruz Biotechnology; the GFAP, GLT-1, and nestin antibodies were from Millipore Bioscience Research Reagents; the paxillin antibody was from BD Transduction Laboratories; the S100β antibody was from AbCam; the GAPDH antibody was from Ambion; the vimentin antibody was from Zymed/Invitrogen; the 3G10 antibody (David et al., 1992) was from Seikagaku Kogyo. Laminin was from BD Transduction Laboratories; MTT, EGF, DMSO, heparinase III, and phosphatase inhibitor cocktails 1 and 2 were from Sigma; rapamycin was from LC Laboratories; and PD98059, LY294002, and FTI-277 were from EMD Biosciences. Horseradish peroxidase-conjugated secondary antibodies for immunoblotting were from GE Healthcare, and Alexa Fluor-conjugated secondary antibodies for immunofluorescence were from Invitrogen. Rhodamine-phalloidin was from Invitrogen.
Astrocyte cultures.
Astrocyte cultures were prepared from spinal cords of adult male Sprague Dawley rats (P65–P70, weighing 300–350 g) using a method previously described (Tawfik et al., 2006) with some modifications. The spinal cords were ejected from the vertebral column using a saline-filled syringe. The tissue was chemically dissociated with 0.25% trypsin-EDTA for 10 min followed by mechanical trituration in DMEM (Invitrogen). After centrifugation at 1200 rpm for 5 min, the cells were suspended in DMEM containing 10% fetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin and plated in a flask coated with poly-l-lysine (Sigma). The cultures were maintained in a humidified atmosphere of 95% air/5% CO2 at 37°C for 10 d, with changes of the culture medium at days 4 and 7. Approximately on day 10 and 11, oligodendrocytes and microglial cells growing on top of the confluent astrocyte layer were removed by shaking at 200 rpm for 2 h at 37°C and replacing the culture medium. The next day, the cells were trypsinized and replated in six-well plates (40,000 cells/well). The cultures were used for experiments when confluent (typically within 4–6 d).
For EGF stimulation, confluent astrocyte cultures were starved for 24 h in 0.1% FBS-containing medium and then stimulated with 100 ng/ml EGF or vehicle for 15 min. In some experiments the cultures were pretreated for 30 min with 100 nm rapamycin, 25 μm PD98059, 50 μm LY294002, or vehicle only as a control before adding EGF. For farnesyltransferase inhibition, the cultures were incubated for 24 h with 10 μm FTI-277. For the growth assays and the Akt phosphorylation time course shown in Figure 3, astrocytes were initially sparse (3000 cells/well in a 96-well plate), and EGF and rapamycin were added every day and the culture medium was replaced every third day.
Figure 3.

mTORC1 is required for the growth of cultured spinal cord astrocytes. A, Fetal bovine serum promotes the growth of astrocytes. Astrocytes were grown for the indicated number of days and their growth was quantified using the MTT assay. The graph shows averages ± SE from three independent experiments, each with triplicate measurements. ***p < 0.001 for the comparison with control-treated astrocytes by two-way ANOVA and Bonferroni's post hoc test. B, C, Rapamycin inhibits astrocyte growth. The graph in B shows averages ± SE from three independent experiments, each with triplicate measurements using an MTT assay. The graph in C shows averages ± SE from triplicate measurements of cell numbers counted with a hemocytometer. **p < 0.01 and ***p < 0.001 for the comparison between serum- and rapamycin-treated astrocytes by two-way ANOVA and Bonferroni's post hoc test. D, Confluency and rapamycin inhibit S6 kinase phosphorylation and enhance Akt phosphorylation. Lysates from untreated and rapamycin-treated cells were probed by immunoblotting with the indicated antibodies. All the lanes are from the same gel, and a lane between untreated and rapamycin-treated samples was digitally removed. E, Rapamycin treatment increases the levels of IRS-2 and does not increase the levels of cleaved caspase 3. Lysates from untreated and rapamycin-treated cells were probed by immunoblotting with the indicated antibodies.
Growth assays.
To measure cell growth using the MTT assay, the culture medium was replaced with 100 μl of 5 mg/ml MTT in serum-free medium. After 4 h the supernatant was aspirated from the wells without disturbing the formazan precipitate, the formazan crystals were dissolved in 100% DMSO and the absorbance was measured at 570 nm with a microplate reader. To measure cell numbers, astrocytes cultured in six-well plates were trypsinized, incubated in suspension with trypan blue to identify dead cells, and counted using a hemocytometer.
Transwell migration assay.
Astrocyte cultures were trypsinized and incubated in suspension in serum-free medium with or without 100 nm rapamycin for 15 min. Fifty thousand cells were added to the top compartment of each Transwell (8 μm filter pore diameter, Corning) precoated with 10 μg/ml laminin and containing serum-free medium in both upper and lower compartments. To stimulate chemotactic migration, 20 ng/ml EGF was added in the lower compartment. For rapamycin treatment, 100 nm rapamycin was present in the upper and lower compartments. The astrocytes were allowed to migrate for 4 h, and then the filters were washed with PBS and the cells from the top surface of the filters were removed with a cotton swab. The filters were then fixed with 4% formaldehyde and cell nuclei were stained with DAPI (Invitrogen). The nuclei of cells that had migrated to the bottom surface of the filter were photographed under a fluorescence microscope (6 fields/filter using a 10× objective) and counted with NIH ImageJ. The average number of cells in three replicate wells was determined for each condition in each of three independent experiments.
Lentivirus packaging and infection.
For lentivirus packaging, 6 × 106 293FT cells (Invitrogen) were transfected using Lipofectamine 2000 (Invitrogen) with a mixture of 6 μg of pLKO.1-Rheb shRNA plasmid or control pLKO.1-GFP shRNA (OpenBiosystems), 2 μg of envelope-encoding pMD2.G plasmid, and 2 μg of the packaging vector psPAX2 plasmid (Addgene). The culture supernatant containing the packaged virus was harvested 48 h after transfection and titered using C6 rat glioma cells. For viral infection, confluent plates of astrocytes were incubated for 24 h with 200 μl of virus (2 × 105 transducing units/ml) and 6 μg/ml polybrene as transduction enhancer. After infection, the astrocytes were grown in medium containing 0.5 μg/ml puromycin, which was replaced every third day, to eliminate noninfected cells. After 10 d, the astrocytes were grown to confluency in puromycin-free medium.
Real-time PCR.
All the reagents used were from Applied Biosystems. The TaqMan Probes and primers for Rheb (assay ID Rn00566263_m1) were “assay-on-demand” gene expression products. Cultured astrocytes were washed with PBS and total RNA was extracted using RNA-STAT-60 and reverse transcribed using random hexanucleotide primers. TaqMan PCR amplification reactions were performed in a volume of 25 μl prepared using the TaqMan Universal Master mix and containing 50 ng of cDNA in MicroAmp Optical Plates with MicroAmp Optical Caps. Incubations at 50°C for 2 min and at 95°C for 10 min were performed to activate the AmpliTaq polymerase, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Hypoxanthine-guanine phosphoribosyltransferase was used as a loading control for each sample, and the standard curve method was used for quantification (Boyle et al., 2003).
Spinal cord ischemia.
Spinal cord ischemia was induced in anesthetized male Sprague Dawley rats according to a previously described protocol (Taira and Marsala, 1996; Hefferan et al., 2007). This protocol involves inflating a balloon catheter placed in the descending thoracic aorta at the level of the left subclavian artery to occlude aortic blood flow for 10 min while also maintaining systemic hypotension. After 3 weeks, rats were anesthetized with pentobarbital and phenytoin and transcardially perfused with 200 ml of heparinized saline solution followed by 250 ml of 4% formaldehyde in PBS to obtain lumbar spinal cord tissue for immunohistochemical analysis. The initial acute inflammatory phase involving massive infiltration of macrophages and activated microglia has subsided after the first week, and 3 weeks after injury reactive gliosis is well developed (Marsala et al., 2004).
To examine the effects of rapamycin on the development of reactive gliosis in the injured spinal cord, 9 rats were injected intraperitoneally with 1 mg/kg rapamycin in 5% polyethylene glycol 400 and 5% Tween 80 daily for 8 d starting 2 d after injury while 5 rats were similarly injected with vehicle control. The rats were killed 1 d after the last administration of rapamycin. All experiments were performed according to protocols approved by the Institutional Animal Care Committee of the University of California, San Diego.
Immunoblotting.
Astrocyte cultures and lumbar spinal cords were lysed in 0.5% Triton X-100, 3% SDS, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 1 mm EDTA-containing protease inhibitors, NaF, and phosphatase inhibitor cocktails 1 and 2. Samples were analyzed by SDS-PAGE followed by immunoblotting. Membranes were incubated with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies and chemiluminescence reagents. Membranes were stripped using the ReBlot Western Blot Recycling Kit (Millipore) before reprobing with a different antibody.
Immunolabeling.
Cultured astrocytes were fixed in 4% formaldehyde in PBS for 15 min, permeabilized in PHEM buffer (120 mm PIPES, 50 mm HEPES, 20 mm EGTA, and 8 mm magnesium) with 0.1% Triton X-100 for 5 min, blocked for 1 h in PHEM buffer with 5% goat serum, incubated for 1 h with primary antibodies in PHEM buffer with 5% goat serum, and incubated with Alexa Fluor-conjugated secondary antibodies (1:300) in PHEM buffer with 5% goat serum. For detecting the 3G10 epitope, after fixation the astrocytes were incubated at 37°C with 0.005 U/μl heparinase III for 3 h. After washing with PHEM buffer, the cells were blocked for 20 min in PHEM buffer with 5% goat serum, incubated for 3 h with the 3G10 antibody in PHEM buffer with 5% goat serum, and incubated with Alexa Fluor-conjugated secondary antibody (1:300) in PHEM buffer with 5% goat serum.
Spinal cords were dissected and postfixed in 4% formaldehyde in PBS overnight at 4°C and then cryoprotected in 30% sucrose PBS and frozen. Transverse spinal cord sections were permeabilized with PBS, 0.2% Triton X-100 for 10 min, blocked for 1 h in PBS, 5% goat serum, 0.2% Triton X-100, and then incubated overnight with primary antibodies in PBS, 0.2% Triton X-100, 5% goat serum, followed by an 1 h incubation with Alexa Fluor-conjugated secondary antibodies (1:250) in the same solution. Images were captured using a fluorescence microscope or a multiphoton laser point scanning confocal microscopy system (Radiance 2100/AGR-3Q, Bio-Rad Laboratories) equipped with Lasersharp 2000 software (Bio-Rad Laboratories). Some images were captured using a Leica SP2 confocal microscope. Confocal images for all groups with the same labeling in Figure 7 and supplemental Figure 5 (available at www.jneurosci.org as supplemental material) were captured using identical settings and constructed from three optical layers spaced 1 μm. The images used for image analysis were captured as described below.
Figure 7.
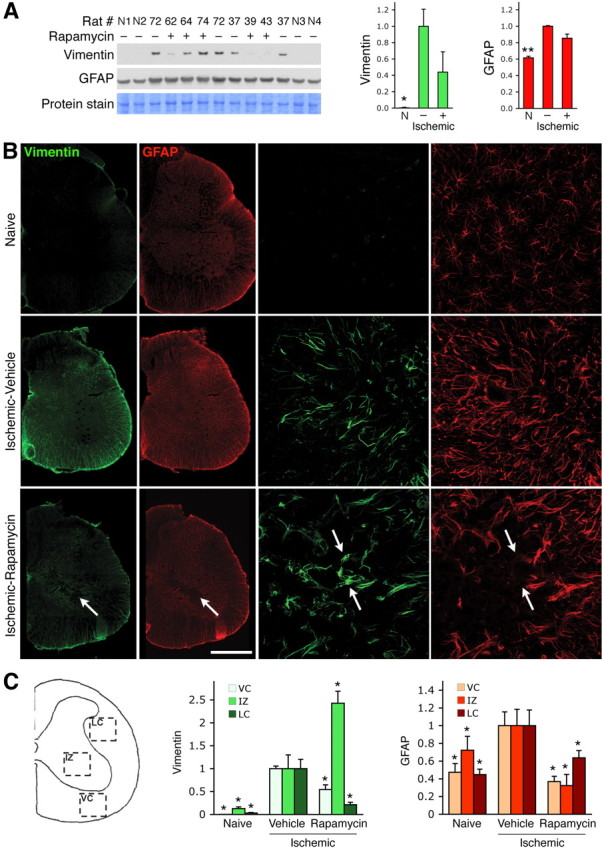
Rapamycin administration decreases reactive gliosis in the injured spinal cord. A, Lysates from the lumbar spinal cord of uninjured naive rats (N1 through N4), and rats treated with vehicle control (−) or rapamycin (+) after spinal cord ischemia, were probed by immunoblotting with antibodies to vimentin and GFAP. The levels of vimentin and GFAP determined from the optical density of the bands were normalized for protein loading based on two Coomassie Blue-stained protein bands (the ∼75 kDa protein band shown in the figure and the ∼18 kDa band shown in supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The histograms show average vimentin and GFAP levels relative to the levels in the injured, vehicle control-treated rats. *p < 0.05 and **p < 0.01 by one-way ANOVA followed by Dunnett's post hoc test for the comparison with vehicle-treated injured spinal cord. B, Vimentin and GFAP immunoreactivity in the spinal cords of naive and vehicle control-treated (Ischemic-Vehicle) or rapamycin-treated (Ischemic-Rapamycin) spinal cords. Reduction in overall vimentin and GFAP immunoreactivity in the rapamycin-treated compared with vehicle-treated spinal cords suggests decreased reactive gliosis. The arrows in the bottom left panels indicate the epicenter of injury in the rapamycin-treated spinal cord. The right panels, showing higher magnification views of the intermediate zone, reveal that there are few GFAP-positive cells in the injury epicenter of the rapamycin-treated spinal cord; examples of vimentin-positive but GFAP-negative cells in the injury epicenter are indicated by arrows. The scale bar represents 500 μm in the left panels and 70 μm in the right panels. C, The scheme on the left illustrates the spinal cord regions used for quantification: VC, ventral column; IZ, intermediate zone; LC, lateral column. The histograms show average vimentin and GFAP levels in the ventral column, intermediate zone, and lateral column, normalized to the levels in the injured, vehicle-treated spinal cords. *p < 0.05 by one-way ANOVA followed by Dunnett's post hoc test for the comparison with vehicle-treated injured spinal cord.
Image analysis.
Double immunohistochemical labeling for GFAP and vimentin was performed using the same antibody solution for all free-floating sections (30 μm thick) and processed as described above. Four sections (from L4 and L5 spinal cord segments) were analyzed from each animal, including three naive, three vehicle-treated, and four rapamycin-treated animals. Fluorescent images were captured using a Leica DMLB microscope with a Zeiss Axiocam MRm monochrome camera. Using identical camera settings, images were obtained from the lateral and ventral columns and the intermediate zones on both sides of each section. Using the pixel histogram generated for each original image by Image-Pro Plus (v.6.2.0.424; Media Cybernetics), the product of the pixel number and pixel intensity value (0–255) was computed and summed for the entire image. This provided a composite measure of changes in both immunoreactive area and fluorescence intensity. Any postprocessing was done with Adobe CS3 (Adobe Systems) with equal changes to any images being compared.
Results
EGF activates the mTORC1 pathway in cultured spinal cord astrocytes
To examine EGF-dependent signaling pathways that may induce changes in astrocytes following injury, we used cultured astrocytes isolated from adult rat spinal cord. Greater than 95% of the cells in these cultures express the astrocytic marker glial fibrillary acidic protein (GFAP) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material, and data not shown). Furthermore, the cultures express nestin, vimentin (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and the EGF receptor (Fig. 1A), which are normally present at very low levels in astrocytes of the adult nervous system but are known to be upregulated in reactive astrocytes at sites of injury (Frisén et al., 1995; Ridet et al., 1997; Pekny et al., 1999; Menet et al., 2003; B. Liu et al., 2006). This is consistent with the notion that astrocytes acquire some of the characteristics of reactive astrocytes in response to stimuli in the cell culture environment (Eddleston and Mucke, 1993; B. Liu et al., 2006). We also detected widespread expression of the calcium-binding protein S100β and heparan sulfate proteoglycans, which have been reported to be present at substantial levels in astrocytes of the adult nervous system and to be further upregulated in reactive astrocytes (Properzi and Fawcett, 2004; do Carmo Cunha et al., 2007; Properzi et al., 2008). Immunoblotting and immunocytochemistry experiments also verified increased EGF receptor tyrosine phosphorylation in response to EGF stimulation of the cultured astrocytes after serum starvation (Fig. 1A; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To determine whether the mTORC1 pathway was activated following EGF stimulation, we used antibodies that detect phosphorylation of p70 S6 kinase on threonine 389, a site well known to be selectively phosphorylated by mTORC1 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and widely used to monitor mTORC1 activation (Sabatini, 2006; Bhaskar and Hay, 2007). EGF stimulation substantially increased the levels of phosphorylated pS6 kinase, indicating activation of mTORC1 in spinal cord astrocytes (Fig. 1A).
Figure 1.

EGF stimulation of astrocytes from adult spinal cord activates the mTOR pathway. A, EGF promotes phosphorylation of the EGF receptor and S6 kinase. Astrocytes were stimulated for 15 min with EGF (+) or vehicle (−), and lysates were probed by immunoblotting with antibodies to phosphorylated EGF receptor (p-EGFR) and phosphorylated S6 kinase (p-S6K) and reprobed with antibodies to the EGF receptor and S6 kinase. B, The Erk MAP kinase pathway is not involved in EGF-dependent S6 kinase phosphorylation. Astrocytes treated with the PD98059 inhibitor (+) or vehicle (−) were stimulated with EGF, and lysates were probed by immunoblotting with antibodies to phosphorylated S6 kinase (p-S6K) or phosphorylated Erk1 and Erk2 MAP kinases (p-Erk) and reprobed with antibodies detecting the nonphosphorylated forms of the proteins. The levels of phosphorylated S6 kinase determined from the optical density of the bands were normalized to total S6 kinase protein levels. The histogram shows average levels of phosphorylated S6 kinase relative to the level after EGF treatment. The error bars represent the SE from three independent experiments. PD98059 did not cause significant differences in S6 phosphorylation as determined by one-way ANOVA and Bonferroni's post hoc test. C, The PI3 kinase–Akt pathway mediates EGF-dependent Tuberin phosphorylation. Astrocytes incubated with the LY294002 PI3 kinase inhibitor (+) or vehicle (−) were stimulated with EGF, and lysates were probed by immunoblotting with the indicated antibodies. The levels of phosphorylated proteins were quantified as described in B for S6 kinase. The histogram shows average levels of phosphorylated Tuberin and phosphorylated Akt relative to the levels after EGF treatment. The error bars represent the SE from three independent experiments. ***p < 0.001 for the comparison between untreated and LY294002-treated by one-way ANOVA and Bonferroni's post hoc test. D, The mTORC1-selective inhibitor rapamycin blocks S6 kinase phosphorylation but not Akt phosphorylation. Astrocytes incubated with rapamycin (+) or vehicle (−) were stimulated with EGF, and the lysates were probed by immunoblotting with the indicated antibodies.
Studies with other cell types suggest that several signaling connections may lead to mTORC1 activation following EGF stimulation (Sabatini, 2006; Shaw and Cantley, 2006; Chiang and Abraham, 2007). For example, the Erk1/Erk2 MAP kinases and their downstream target p90 Rsk can phosphorylate Tuberin and inhibit its activity, leading to downstream Rheb and mTORC1 activation (Bhaskar and Hay, 2007; Wu et al., 2007). EGF treatment promoted phosphorylation and activation of Erk1/Erk2 in cultured astrocytes (Fig. 1B), as expected because the Ras–MAP kinase pathway is a major pathway known to be activated by the EGF receptor (Citri and Yarden, 2006). However, treatment with an inhibitor of the Erk pathway, PD98059, did not significantly reduce S6 kinase phosphorylation (Fig. 1B). Thus, activation of mTOR in spinal cord astrocytes stimulated with EGF does not depend on Erk1/Erk2 activation.
EGF stimulation also caused phosphorylation of Akt at serine 473 (Fig. 1C; supplemental Fig. 3, available at www.jneurosci.org as supplemental material), which is one of the sites involved in Akt activation (Bhaskar and Hay, 2007; Chiang and Abraham, 2007). Akt in turn can activate mTORC1 by phosphorylating Tuberin at serine 939 and threonine 1462, leading to its inactivation (Wu et al., 2007). Tuberin T1462 phosphorylation was indeed increased following EGF stimulation and the PI3 kinase inhibitor, LY294002, abolished not only Akt but also Tuberin phosphorylation, suggesting that Tuberin phosphorylation occurs downstream of PI3 kinase and Akt (Fig. 1C; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Although LY294002 can also target mTOR (Bain et al., 2007), inhibition of mTORC1 with rapamycin did not reduce Akt phosphorylation (Fig. 1D). Rather, we observed an upregulation of Akt phosphorylation similar to that previously described in several other cell types treated with rapamycin (Wullschleger et al., 2006; Bhaskar and Hay, 2007). However, rapamycin abolished S6 kinase phosphorylation (Fig. 1D), confirming that this phosphorylation depends on mTORC1 because rapamycin is a very selective mTOR inhibitor (Bain et al., 2007; Chiang and Abraham, 2007). Together, these data suggest that EGF activates mTORC1 in spinal cord astrocytes through Akt-mediated Tuberin inactivation (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). However, Akt has also been reported to activate mTOR through different signaling mechanisms that are independent of Tuberin and Rheb (Shaw and Cantley, 2006; Bhaskar and Hay, 2007; Chiang and Abraham, 2007).
To assess the involvement of Rheb in EGF-dependent mTORC1 activation, we used the farnesyltransferase inhibitor FTI-277 because Rheb cannot activate mTORC1 if its membrane localization is impaired by the absence of the carboxy-terminal farnesyl group (Castro et al., 2003). Treatment of cultured astrocytes with FTI-277 significantly inhibited the EGF-dependent increase in S6 kinase phosphorylation (Fig. 2A), suggesting that Rheb participates in mTORC1 activation. We also knocked down Rheb mRNA and protein expression by using an shRNA delivered by lentiviral infection (Fig. 2B,C). Rheb knockdown inhibited S6 kinase phosphorylation both in the absence and in the presence of EGF (Fig. 2D). This indicates that Rheb is necessary for mTORC1 activation by EGF (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and possibly other stimuli, in astrocytes from the adult spinal cord.
Figure 2.

Rheb is required for EGF-dependent S6 kinase phosphorylation in spinal cord astrocytes. A, The FTI-277 farnesyltransferase inhibitor decreases phosphorylation of S6 kinase. Astrocytes incubated with FTI-277 were stimulated with EGF and lysates were probed by immunoblotting with antibodies to phosphorylated S6 kinase (p-S6K) or Rheb, and reprobed with antibodies to S6 kinase. Note that nonfarnesylated Rheb has a different molecular weight, so Rheb immunoreactivity appears weaker because the protein is no longer concentrated in a single band. The levels of phosphorylated S6 kinase determined from the optical density of the bands were normalized to total S6 kinase protein levels. The histogram shows average levels of phosphorylated S6 kinase relative to the level after EGF treatment. The error bars represent the SE from three independent experiments, each with duplicate measurements. **p < 0.01 for the comparison between untreated and FTI-277-treated astrocytes by one-way ANOVA and Bonferroni's post hoc test. B, C, Lentiviral delivery of Rheb shRNA decreases Rheb mRNA and protein levels in spinal cord astrocytes. Astrocytes were infected with a lentivirus encoding Rheb shRNA or a control (GFP) shRNA. In B, Rheb mRNA was quantified by quantitative real-time PCR, and the histogram shows the averages ± SE from three independent experiments, each with duplicate measurements. ***p < 0.001 for the comparison with cells treated with control shRNA by one-way ANOVA and Bonferroni's post hoc test. In C, astrocytes were incubated with rapamycin (+) or vehicle (−) and stimulated with EGF. The levels of Rheb protein were determined from the optical density of the bands and normalized to the levels of β-tubulin in the lysates. The histogram shows average levels of Rheb relative to the level in unstimulated control cells. The error bars represent the SE from four independent experiments, each with duplicate measurements. ***p < 0.001 for the comparison of control shRNA- versus Rheb shRNA-treated samples by two-way ANOVA. D, Rheb knockdown decreases S6 kinase phosphorylation. Astrocytes infected with lentivirus encoding Rheb shRNA or a control shRNA were incubated with rapamycin (+) or vehicle (−) and stimulated with EGF. The histogram shows the average levels of phosphorylated S6 kinase normalized to total S6 kinase and relative to the level in EGF-stimulated control astrocytes. The error bars represent the SE from four independent experiments, each with duplicate samples. ***p < 0.001 for the comparison of control shRNA versus Rheb shRNA by one-way ANOVA and Bonferroni's post hoc test.
The mTORC1 inhibitor rapamycin negatively affects the growth and EGF-dependent migration of spinal cord astrocytes
Because rapamycin has been shown to inhibit the growth of many cell types, indicating a critical role of mTORC1 in cell proliferation (Sabatini, 2006; Shaw and Cantley, 2006; Chiang and Abraham, 2007), we examined the effects of EGF and rapamycin on the growth of spinal cord astrocytes over a period of several days by using the MTT cell viability assay. Stimulation with EGF in the absence of serum was not sufficient to promote the growth of astrocytes from adult spinal cord (Fig. 3A). On the other hand, astrocyte growth induced by serum was almost completely inhibited by rapamycin (Fig. 3B,C), concomitant with inhibition of S6 kinase phosphorylation (Fig. 3D). Phosphorylation of S6 kinase also decreased in the absence of rapamycin, once the cells became confluent and stopped growing (days 4–6). Concomitant with the decrease in S6 kinase phosphorylation induced by either confluency or rapamycin, we observed an increase in Akt phosphorylation on S473 (Fig. 3D). Elevated Akt phosphorylation in rapamycin-treated cells was accompanied by an increase in the levels of the scaffolding protein insulin receptor substrate-2 (IRS-2), which is an upstream regulator of Akt (Fig. 3E). The increase in phosphorylated Akt suggests that survival signals may be upregulated (Manning and Cantley, 2007) and therefore that the effects of rapamycin probably do not involve increased cell death. Indeed, we did not detect cleaved caspase 3 in the rapamycin-treated astrocytes while low levels were detectable in the untreated, confluent astrocytes (Fig. 3E).
Astrocytes are known to migrate toward sites of spinal cord injury (Faulkner et al., 2004). Immunolabeling for filamentous actin and paxillin, a component of focal adhesions, showed that the cultured astrocytes were well spread and had many actin stress fibers and focal adhesions (Fig. 4A), consistent with a high adhesion to the substrate and a low level of cell motility. EGF treatment for 15 min decreased cell spreading, focal adhesions, and stress fibers while causing the formation of long processes, as previously described and consistent with the ability of EGF to promote astrocyte motility (B. Liu et al., 2006). Rapamycin treatment prevented the EGF-dependent morphological changes in spinal cord astrocytes (Fig. 4A) as well as the chemotactic effects of EGF in Transwell migration assays (Fig. 4B), suggesting a role for mTORC1 in the regulation of spinal cord astrocyte migration toward EGF. Akt phosphorylation on S473 was increased in cultured spinal cord astrocytes treated with rapamycin for 4 h, corresponding to the duration of the Transwell migration assay (Fig. 4C). These results implicate mTORC1 activated downstream of Tuberin and Rheb in the regulation of astrocyte migration and indicate that rapamycin could be used to inhibit not only astrocyte growth but also chemotactic migration.
Figure 4.

mTORC1 promotes EGF-dependent cytoskeletal reorganization and migration of spinal cord astrocytes. A, EGF induces morphological changes in astrocytes. Astrocytes treated with rapamycin were stimulated for 15 min with EGF, fixed, and stained for paxillin (green), F-actin (red), and DNA (blue). Scale bar, 20 μm. B, Rapamycin inhibits astrocyte chemotactic migration toward EGF. Astrocytes were seeded on Transwell filters coated with laminin and allowed to migrate through the filters toward EGF for 4 h. The histogram shows the average number of cells that migrated to the lower side of the filters in the different conditions relative to control. Error bars indicate the SE from three independent experiments, each with triplicate samples. C, Lysates from astrocytes treated for 4 h with rapamycin and untreated controls were probed by immunoblotting with antibodies to phosphorylated Akt (p-Akt) and phosphorylated S6 kinase (p-S6 kinase) and reprobed with antibodies to Akt and S6 kinase.
The mTOR pathway is activated in the rat spinal cord after ischemic injury
To determine whether activation of mTORC1 also occurs in reactive astrocytes in vivo, we used a model in which spinal cord injury is induced by transient occlusion of the descending thoracic aorta combined with systemic hypotension, leading to transient lumbar spinal cord ischemia (Taira and Marsala, 1996; Hefferan et al., 2007). We examined spinal cord frozen sections by immunohistochemistry 3 weeks after reperfusion, while control spinal cords were obtained from rats that underwent a sham operation. We detected prominent upregulation of markers of activated astrocytes, such as vimentin, glial fibrillary acidic protein, the glial glutamate transporter GLT-1, and the NG2 chondroitin sulfate proteoglycan, in the white matter of the injured spinal cord (Fig. 5A and data not shown). Furthermore, we observed increased labeling with a phosphospecific antibody that recognizes the activated EGF receptor and possibly also activated ErbB2 (Fig. 5B), similar to the increase in activated EGF receptor previously reported in a spinal cord contusion model and other CNS injury models (B. Liu et al., 2006; Erschbamer et al., 2007; Liu and Neufeld, 2007). The substantial colocalization of phosphorylated EGF receptor with the glial glutamate transporter GLT-1 and the partial colocalization with the glial intermediate filament protein vimentin (Fig. 5B), which is similar to the colocalization with vimentin observed in cultured astrocytes (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggest that the activated EGF receptor is present in reactive astrocytes and may regulate their responses to the ischemic injury.
Figure 5.
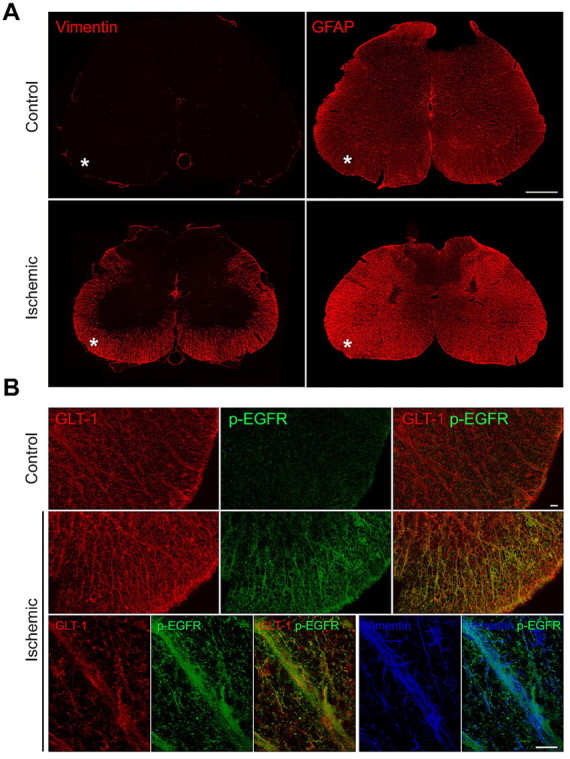
The EGF receptor is activated in vivo in the injured spinal cord. A, The increased vimentin and glial fibrillary acidic protein (GFAP) immunoreactivity reveals the presence of reactive astrocytes in the white matter of the lumbar spinal cord (asterisks) following an ischemic injury. Scale bar, 500 μm. B, Immunolabeling for phosphorylated EGF receptor (green) is increased in the white matter of the spinal cord after ischemia and shows substantial colocalization with the astrocytic glutamate transporter GLT-1 (red) as well as partial colocalization with the cytoskeletal protein vimentin (blue) in triple-labeled sections. Scale bars, 20 μm.
To determine whether the mTOR pathway is activated in reactive spinal cord astrocytes in vivo, we immunolabeled spinal cord sections for phosphorylated ribosomal protein S6, which is a substrate of S6 kinase, because the antibodies to phosphorylated S6 kinase did not work well for immunohistochemistry. Prominently elevated levels of phosphorylated S6 ribosomal protein were detected in the white matter of injured spinal cords compared with controls (Fig. 6A). The substantial colocalization with vimentin staining (Fig. 6A), which is reminiscent of that observed in cultured astrocytes (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), confirmed upregulation of S6 phosphorylation in reactive astrocytes. We then examined whether Rheb was present in the injured spinal cord and may therefore play an in vivo role in regulating mTORC1 activation downstream of the activated EGF receptor in reactive as trocytes, as suggested by our in vitro results. We detected Rheb immunoreactivity in control spinal cords, predominantly in the gray matter (Fig. 6B). Interestingly, Rheb expression was prominently upregulated in the white matter of the injured spinal cord and showed extensive colocalization with vimentin (Fig. 6B). Given that Rheb overexpression typically results in higher levels of activated Rheb (Im et al., 2002), this suggests an additional Rheb-dependent mechanism, besides Tuberin inhibition downstream of the EGF receptor, that may contribute to in vivo activation of mTORC1 in reactive astrocytes of the injured spinal cord.
Figure 6.

Rheb is upregulated and mTORC1 is activated in vivo in the injured spinal cord. A, Immunolabeling for phosphorylated S6 ribosomal protein (green) is dramatically upregulated in the vimentin-positive astrocytes (red) within the white matter of injured spinal cord. Scale bar, 20 μm. B, Immunolabeling for Rheb (green) is also dramatically upregulated in the vimentin-positive astrocytes (red) in the white matter of the injured spinal cord after ischemia (asterisks). Scale bars, 500 μm (top) and 20 μm (bottom).
Rapamycin treatment reduces reactive gliosis in the injured spinal cord
To examine the effects of inhibiting the mTORC1 pathway in astrocytes of the injured spinal cord, rapamycin was injected intraperitoneally for 8 d starting 2 d after transient spinal cord ischemia. By immunoblotting we did not detect vimentin expression in lysates of naive, uninjured spinal cord, whereas substantial levels of GFAP were present, as expected (Fig. 7A; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). We observed significant increases of both vimentin and GFAP in lysates from the injured lumbar spinal cord compared with naive controls, consistent with the development of reactive gliosis. We observed a trend toward a decrease in the overall levels of vimentin and GFAP in the injured spinal cord of rats treated with rapamycin compared with vehicle-treated controls, although there was substantial variability between animals (Fig. 7A; supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
To more accurately measure changes in vimentin and GFAP levels in different regions of the injured spinal cord, we quantified the immunofluorescence signal from labeled spinal cord sections in a different cohort of animals (Fig. 7B; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Quantification of immunofluorescence from the ventral column, intermediate zone, and lateral column showed that reactive gliosis, revealed by increased levels of both vimentin and GFAP immunoreactivity, was significantly reduced in rats treated with rapamycin compared with vehicle-treated control rats (Fig. 7C). The only exception were the levels of vimentin in the intermediate zone, which were increased. This was consistent with the presence of vimentin-positive cells in the injury epicenter (in the intermediate zone), in contrast to the greatly reduced infiltration of GFAP-positive astrocytes in the injury epicenter after rapamycin treatment (Fig. 7B; supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Together with the observed effects of rapamycin on cultured spinal cord astrocytes, these results suggest that rapamycin administration inhibits the development, proliferation, and/or migration of reactive astrocytes in the injured nervous system and may therefore have beneficial effects by reducing reactive gliosis.
Discussion
In this study we show that activation of the EGF receptor in astrocytes triggers a downstream signaling pathway that involves Akt, Tuberin, Rheb, and mTOR. This pathway was elucidated using cultured astrocytes isolated from the adult spinal cord and appears to also be functional in reactive astrocytes in vivo, in a well characterized model of spinal cord injury in which transient ischemia causes neuronal loss, paraplegia, and reactive gliosis (Taira and Marsala, 1996; Hefferan et al., 2007). Immunohistochemistry showed that in reactive astrocytes of the injured spinal cord there was an increase not only in the levels of activated EGF receptor but also in the expression of the mTORC1 activator Rheb. Thus, upregulation of Rheb expression may be another mechanism contributing to mTORC1 activation in vivo (Im et al., 2002). It will be interesting to determine whether increased Rheb expression is a common feature of nervous system injuries characterized by reactive gliosis.
Adult spinal cord astrocytes treated with rapamycin did not grow, confirming that astrocytes require mTORC1 activity to grow in culture, as has been shown for many other cell types (Sabatini, 2006; Shaw and Cantley, 2006; Chiang and Abraham, 2007). However, EGF was not sufficient to stimulate astrocyte growth in the absence of other serum components, suggesting that additional signaling pathways besides activation of mTORC1 are important for growth regulation in these cells. On the other hand, EGF was sufficient to induce changes in the actin cytoskeleton and to stimulate chemotactic migration. These effects of EGF appear to also be mediated by mTORC1 because they were acutely inhibited by rapamycin, which due to its unusual mechanism of action is a very specific mTORC1 inhibitor (Bain et al., 2007). A role for mTORC1 in cell migration has been reported in only a few cell types, such as fibroblasts and trophoblast cells, where growth factor-induced migration can be inhibited by a short exposure to rapamycin, similarly to what we observed in astrocytes (Berven et al., 2004; Qian et al., 2004; Qiu et al., 2004). In contrast, mTOR has a widespread role in the regulation of cytoskeletal remodeling and cell migration when it is part of the mTORC2 complex (Jacinto et al., 2004; Sarbassov et al., 2004). Although mTOR in mTORC2 is insensitive to rapamycin, prolonged exposure to rapamycin can deplete the levels of mTOR available to form mTORC2 complexes (Sarbassov et al., 2006). However, such indirect inhibition of mTORC2 is not likely to be responsible for the effects of rapamycin on EGF-dependent changes in cytoskeletal organization and migration in adult spinal cord astrocytes due to the relatively short time course of rapamycin exposure in our migration experiments. Furthermore, rapamycin did not decrease Akt phosphorylation at S473, which is the site phosphorylated by mTORC2 (Sarbassov et al., 2005).
On the contrary, treatment of the astrocytes with rapamycin rapidly and persistently increased Akt phosphorylation at S473. Previous studies have shown that rapamycin can promote Akt phosphorylation in some cell types through inhibition of a negative feedback loop involving S6 kinase-mediated phosphorylation and degradation of insulin receptor substrates (O'Reilly et al., 2006; Wullschleger et al., 2006; Bhaskar and Hay, 2007). By increasing insulin receptor substrate levels, rapamycin inhibition of mTORC1 activity can increase PI3 kinase and, therefore, Akt activation. A similar mechanism appears to account for the inverse regulation of S6 kinase and Akt phosphorylation that we observed in adult spinal cord astrocytes because we observed increased levels of insulin receptor substrate-s (IRS-2) protein in rapamycin-treated astrocytes.
The rapamycin-dependent increase in Akt phosphorylation may play a role in the inhibitory effects of rapamycin on astrocyte migration. The roles of Akt in cell survival and metabolism are well known, while its role in cell migration is less characterized (Manning and Cantley, 2007). Interestingly, recent studies have shown that Akt1 and Akt2 can have different effects on migration depending on the cell type (Stambolic and Woodgett, 2006). For example, activation of Akt1 but not Akt2 decreases EGF-dependent breast epithelial cell migration by inhibiting Erk kinases (Irie et al., 2005) and hyperactivation of Akt1 in breast cancer cells inhibits motility and invasion through a pathway that involves Tuberin phosphorylation and degradation, leading to decreased Rho activation (H. Liu et al., 2006). Furthermore, activation of Akt2 but not Akt1 has been shown to decrease fibroblast migration, likely by inhibiting Rac and Pak1 (Zhou et al., 2006) while phosphorylation and inhibition of GSK-3 by Akt family members can have negative effects on cell migration (Kobayashi et al., 2006). Downregulation of S6 kinase activity may also play a role in the inhibitory action of rapamycin on the cytoskeletal and chemotactic effects of EGF. In addition to its role in the regulation of protein synthesis, S6 kinase has been shown to associate with the actin cytoskeleton and has been implicated in the regulation of cell morphology and migration (Berven et al., 2004; Qian et al., 2004).
An important implication of our findings is that blocking the mTORC1 pathway with rapamycin, which is already in use as an immunosuppressive and antiproliferative drug (Bain et al., 2007; Chiang and Abraham, 2007), could be beneficial in the treatment of spinal cord injury by reducing astrocyte proliferation and migration. Indeed, treating rats with rapamycin after ischemic spinal cord injury lowered the levels of vimentin and GFAP, two intermediate filament proteins that are characteristically upregulated in reactive astrocytes. Furthermore, rapamycin administration decreased infiltration of GFAP-positive astrocytes in the epicenter of the injury, consistent with decreased astrocyte proliferation and/or migration.
It will be interesting to examine whether prolonged rapamycin treatment can also downregulate expression of proteins responsible for astrocyte hypertrophy and reactive gliosis, whose expression may depend on EGF receptor and mTORC1 activity (Junier, 2000; Ess et al., 2004; Fraser et al., 2004; Uhlmann et al., 2004; Smith and Strunz, 2005; B. Liu et al., 2006; Liu and Neufeld, 2007). Moreover, the increased Akt activation seen after rapamycin treatment should enhance astrocyte survival (Manning and Cantley, 2007), which could also be beneficial in conjunction with inhibiting excessive astrocyte growth and motility. While moderate astrocyte gliosis would be desirable to reduce tissue damage and neuronal cell death following injury (Faulkner et al., 2004), excessive gliosis may be limited with the use of rapamycin.
It is encouraging that in vitro rapamycin inhibits astrocyte growth and migration (as we show here) and reduces the size of hypertrophic astrocytes in which mTORC1 is hyperactivated (Uhlmann et al., 2004). Nevertheless, it will be important to carefully examine the in vivo effects of rapamycin on functional recovery after different types of nervous system injury, because of the complexities of the mTORC1 pathway and its importance in regulating the physiology of different cell types in the damaged nervous system (Sabatini, 2006; Shaw and Cantley, 2006; Chiang and Abraham, 2007). In clinical trials for the treatment of tuberous sclerosis-associated astrocytomas and Pten-deficient glioblastomas, rapamycin has already shown some promising growth inhibitory activity toward cells of the glial lineage (Franz et al., 2006; Cloughesy et al., 2008). Other studies with animal models have shown that a brief administration of rapamycin near the time of ischemic injury to the CNS does not have neuroprotective effects (Sharkey and Butcher, 1994; Bochelen et al., 1999; Tachibana et al., 2005). In contrast, a single dose of rapamycin administered 4 h after traumatic brain injury in mice has been shown to reduce inflammatory processes and neuronal loss and to improve functional recovery (Erlich et al., 2007). Interestingly, the mTORC1 pathway has been very recently implicated in axon growth and regeneration (Choi et al., 2008; Park et al., 2008), suggesting that it may be desirable to devise strategies to preferentially target rapamycin to glial cells.
The EGF receptor inhibitors PD168393 and AG1478 promote axon regeneration and functional recovery in preclinical studies using animal models of spinal cord and optic nerve injury through inhibitory effects on reactive astrocytes (B. Liu et al., 2006; Erschbamer et al., 2007) as well as by decreasing neuronal responses to inhibitors of axon outgrowth (Koprivica et al., 2005). Our studies suggest that rapamycin may be useful as an alternative or in combination with EGF receptor inhibitors (Wang et al., 2006) to reduce glial scar formation after spinal cord injury and possibly other injuries to the CNS.
Footnotes
This work was supported by National Institutes of Health Grants DA21654 (C.I.S.), NS051644 (M.M.), and HD025938 (E.B.P.) and by Slovak Academy of Sciences APVV Grant 0314-06 (M.M.). We thank Dr. Trono for the psPAX2 and pMD2.G plasmids, F. Irie and Y. Yamaguchi for help with the detection of heparan sulfate proteoglycans, M. Carmona for help with immunofluorescence images, and L. Olson and G. Chiang for helpful comments on this manuscript.
References
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berven LA, Willard FS, Crouch MF. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296:183–195. doi: 10.1016/j.yexcr.2003.12.032. [DOI] [PubMed] [Google Scholar]
- Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Bochelen D, Rudin M, Sauter A. Calcineurin inhibitors FK506 and SDZ ASM 981 alleviate the outcome of focal cerebral ischemic/reperfusion injury. J Pharmacol Exp Ther. 1999;288:653–659. [PubMed] [Google Scholar]
- Boyle DL, Rosengren S, Bugbee W, Kavanaugh A, Firestein GS. Quantitative biomarker analysis of synovial gene expression by real-time PCR. Arthritis Res Ther. 2003;5:R352–R360. doi: 10.1186/ar1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Di Nardo A, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5 doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Bai XM, Van der Schueren B, Cassiman JJ, Van den Berghe H. Developmental changes in heparan sulfate expression: in situ detection with mAbs. J Cell Biol. 1992;119:961–975. doi: 10.1083/jcb.119.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S, Lacroix S. Molecular approaches to spinal cord repair. Annu Rev Neurosci. 2003;26:411–440. doi: 10.1146/annurev.neuro.26.043002.094946. [DOI] [PubMed] [Google Scholar]
- do Carmo Cunha J, de Freitas Azevedo Levy B, de Luca BA, de Andrade MS, Gomide VC, Chadi G. Responses of reactive astrocytes containing S100beta protein and fibroblast growth factor-2 in the border and in the adjacent preserved tissue after a contusion injury of the spinal cord in rats: implications for wound repair and neuroregeneration. Wound Repair Regen. 2007;15:134–146. doi: 10.1111/j.1524-475X.2006.00194.x. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Erschbamer M, Pernold K, Olson L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J Neurosci. 2007;27:6428–6435. doi: 10.1523/JNEUROSCI.1037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ess KC, Uhlmann EJ, Li W, Li H, Declue JE, Crino PB, Gutmann DH. Expression profiling in tuberous sclerosis complex (TSC) knockout mouse astrocytes to characterize human TSC brain pathology. Glia. 2004;46:28–40. doi: 10.1002/glia.10324. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Zhu X, Kwon C-H, Uhlmann EJ, Gutmann DH, Baker SJ. Pten loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. doi: 10.1158/0008-5472.CAN-04-2487. [DOI] [PubMed] [Google Scholar]
- Frisén J, Johansson CB, Török C, Risling M, Lendahl U. Rapid, widespread, and longlasting induction of nestin contributes to the generation of glial scar tissue after CNS injury. J Cell Biol. 1995;131:453–464. doi: 10.1083/jcb.131.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferan MP, Kucharova K, Kinjo K, Kakinohana O, Sekerkova G, Nakamura S, Fuchigami T, Tomori Z, Yaksh TL, Kurtz N, Marsala M. Spinal astrocyte glutamate receptor 1 overexpression after ischemic insult facilitates behavioral signs of spasticity and rigidity. J Neurosci. 2007;27:11179–11191. doi: 10.1523/JNEUROSCI.0989-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im E, von Lintig FC, Chen J, Zhuang S, Qui W, Chowdhury S, Worley PF, Boss GR, Pilz RB. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene. 2002;21:6356–6365. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Junier MP. What role(s) for TGFalpha in the central nervous system? Prog Neurobiol. 2000;62:443–473. doi: 10.1016/s0301-0082(00)00017-4. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, Kikuchi A. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol Cell Biol. 2006;26:898–911. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF, He Z. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310:106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liu B, Neufeld AH. Activation of epidermal growth factor receptors in astrocytes: from development to neural injury. J Neurosci Res. 2007;85:3523–3529. doi: 10.1002/jnr.21364. [DOI] [PubMed] [Google Scholar]
- Liu B, Chen H, Johns TG, Neufeld AH. Epidermal growth factor receptor activation: an upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J Neurosci. 2006;26:7532–7540. doi: 10.1523/JNEUROSCI.1004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, Bissell MJ. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103:4134–4139. doi: 10.1073/pnas.0511342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsala M, Kakinohana O, Yaksh TL, Tomori Z, Marsala S, Cizkova D. Spinal implantation of hNT neurons and neuronal precursors: graft survival and functional effects in rats with ischemic spastic paraplegia. Eur J Neurosci. 2004;20:2401–2414. doi: 10.1111/j.1460-9568.2004.03702.x. [DOI] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Giménez y, Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci U S A. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She Q-B, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallén A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisén J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Properzi F, Fawcett JW. Proteoglycans and brain repair. News Physiol Sci. 2004;19:33–38. doi: 10.1152/nips.01449.2003. [DOI] [PubMed] [Google Scholar]
- Properzi F, Lin R, Kwok J, Naidu M, van Kuppevelt TH, Ten Dam GB, Camargo LM, Raha-Chowdhury R, Furukawa Y, Mikami T, Sugahara K, Fawcett JW. Heparan sulphate proteoglycans in glia and in the normal and injured CNS: expression of sulphotransferases and changes in sulphation. Eur J Neurosci. 2008;27:593–604. doi: 10.1111/j.1460-9568.2008.06042.x. [DOI] [PubMed] [Google Scholar]
- Qian Y, Corum L, Meng Q, Blenis J, Zheng JZ, Shi X, Flynn DC, Jiang B-H. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am J Physiol Cell Physiol. 2004;286:C153–C163. doi: 10.1152/ajpcell.00142.2003. [DOI] [PubMed] [Google Scholar]
- Qiu Q, Yang M, Tsang BK, Gruslin A. Both mitogen-activated protein kinase and phosphatidylinositol 3-kinase signalling are required in epidermal growth factor-induced human trophoblast migration. Mol Hum Reprod. 2004;10:677–684. doi: 10.1093/molehr/gah088. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Butcher SP. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature. 1994;371:336–339. doi: 10.1038/371336a0. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006;16:461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Shiiya N, Kunihara T, Wakamatsu Y, Kudo AF, Ooka T, Watanabe S, Yasuda K. Immunophilin ligands FK506 and cyclosporine A improve neurologic and histopathologic outcome after transient spinal cord ischemia in rabbits. J Thorac Cardiovasc Surg. 2005;129:123–128. doi: 10.1016/j.jtcvs.2004.04.047. [DOI] [PubMed] [Google Scholar]
- Taira Y, Marsala M. Effect of proximal arterial perfusion pressure on function, spinal cord blood flow, and histopathologic changes after increasing intervals of aortic occlusion in the rat. Stroke. 1996;27:1850–1858. doi: 10.1161/01.str.27.10.1850. [DOI] [PubMed] [Google Scholar]
- Tawfik VL, Lacroix-Fralish ML, Bercury KK, Nutile-McMenemy N, Harris BT, Deleo JA. Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: heterogeneity of the quiescent phenotype. Glia. 2006;54:193–203. doi: 10.1002/glia.20365. [DOI] [PubMed] [Google Scholar]
- Uhlmann EJ, Li W, Scheidenhelm DK, Gau CL, Tamanoi F, Gutmann DH. Loss of tuberous sclerosis complex 1 (Tsc1) expression results in increased Rheb/S6K pathway signaling important for astrocyte cell size regulation. Glia. 2004;47:180–188. doi: 10.1002/glia.20036. [DOI] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, Mischel PS. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- Wu EHT, Wu KKH, Wong YH. Tuberin: a stimulus-regulated tumor suppressor protein controlled by a diverse array of receptor tyrosine kinases and G protein-coupled receptors. Neurosignals. 2007;15:217–227. doi: 10.1159/000101333. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zhu J, Liu KY, Sabatini BL, Wong STC. Mutual information-based feature selection in studying perturbation of dendritic structure caused by TSC2 inactivation. Neuroinformatics. 2006;4:81–94. doi: 10.1385/NI:4:1:81. [DOI] [PubMed] [Google Scholar]