

Abstract
Motivation: Statistical phylogenetics is computationally intensive, resulting in considerable attention meted on techniques for parallelization. Codon-based models allow for independent rates of synonymous and replacement substitutions and have the potential to more adequately model the process of protein-coding sequence evolution with a resulting increase in phylogenetic accuracy. Unfortunately, due to the high number of codon states, computational burden has largely thwarted phylogenetic reconstruction under codon models, particularly at the genomic-scale. Here, we describe novel algorithms and methods for evaluating phylogenies under arbitrary molecular evolutionary models on graphics processing units (GPUs), making use of the large number of processing cores to efficiently parallelize calculations even for large state-size models.
Results: We implement the approach in an existing Bayesian framework and apply the algorithms to estimating the phylogeny of 62 complete mitochondrial genomes of carnivores under a 60-state codon model. We see a near 90-fold speed increase over an optimized CPU-based computation and a >140-fold increase over the currently available implementation, making this the first practical use of codon models for phylogenetic inference over whole mitochondrial or microorganism genomes.
Availability and implementation: Source code provided in BEAGLE: Broad-platform Evolutionary Analysis General Likelihood Evaluator, a cross-platform/processor library for phylogenetic likelihood computation (http://beagle-lib.googlecode.com/). We employ a BEAGLE-implementation using the Bayesian phylogenetics framework BEAST (http://beast.bio.ed.ac.uk/).
Contact: msuchard@ucla.edu; a.rambaut@ed.ac.uk
1 INTRODUCTION
The startling, recent advances in sequencing technology are fueling a concomitant increase in the scale and ambition of phylogenetic analyses. However, this enthusiasm belies a fundamental limitation in statistical phylogenetics: as the number of sequences increases, the size of parameter space—specifically the number of possible phylogenetic histories—explodes. To make matters worse, under any modestly realistic model of sequence evolution, considerable computational effort is required to evaluate each history. Troubling, this effort also skyrockets with the number of sequences and complexity of the sequence characters from nucleotides, through amino acids to codons. Although statistically efficient techniques such as Markov chain Monte Carlo (MCMC) help limit computation effort by concentrating evaluations on only those histories that make a significant contribution to the posterior probability density, improved MCMC methods alone cannot mitigate the non-linear nature of the increase in computational burden. Fortunately, the power of computers available to biologists, famously, has also been growing exponentially with remarkable consistency (Moore, 1998) over a similar time scale to the advances in sequencing technology. One aspect of this is the ubiquitous availability of multi-processor and multi-core computers, inviting novel parallel algorithms to make efficient use of these machines.
The last decade has seen a rapid adoption of parallel computing for molecular phylogenetics (Altekar et al., 2004; Feng et al., 2003, 2007; Keane et al., 2005; Minh et al., 2005; Moret et al., 2002; Schmidt et al., 2002; Stamatakis et al., 2005). Concentrating largely on advances for clusters of networked computers, researchers mix and match from a number of parallelization approaches. The first distributes separate histories across multiple computers or CPU cores for independent evaluation (Keane et al., 2005; Moret et al., 2002; Schmidt et al., 2002). The second partitions the sequence data into conditionally independent blocks for distribution across the cluster (Feng et al., 2003; Stamatakis et al., 2005). The third simultaneously runs multiple MCMC samplers in a synchronized fashion (Altekar et al., 2004; Feng et al., 2003). Recently using a data partitioning approach, Feng et al. (2007) even demonstrate success on a tera-flop cluster, achieving almost linear speedup with the number of nodes employed. However, cluster-based approaches carry with them non-negligible computational over-head in the communication between parallel processes and, critically, linear speedup in the number of CPU processing cores leads to considerable financial costs to purchase hardware or rent super-computer time.
There exists, however, a much less expensive resource available in many desktop computers, the graphics processing unit (GPU), largely unexploited for computational statistics in biology (Charalambous et al., 2005; Manavski and Valle, 2008). GPUs are dedicated numerical processors designed for rendering 3D computer graphics. In essence, they consist of hundreds of processor cores on a single chip that can be programmed to apply the same numerical operations simultaneously to each element of large data arrays under a single instruction, multiple data (SIMD) paradigm. Because the same operations, called kernels, function simultaneously, GPUs can achieve extremely high arithmetic intensity if one can transfer the input data and output results onto and off of the processors quickly. An extension to common programming languages [CUDA: (NVIDIA, 2008)] opens up the GPU to general purpose computing, and the computational power of these units has increased to the stage where they can process data-intensive problems many orders of magnitude faster than conventional CPUs.
Here, we describe novel algorithms to make efficient use of the particular architecture of GPUs for the calculation of the likelihood of molecular sequence data under continuous-time Markov chain (CTMC) models of evolution. Our algorithms build upon Silberstein et al. (2008), who demonstrate efficient on-chip memory caching for sum–product calculations on the GPU. Our approach is fundamentally different from previous phylogenetic parallelization, including an exploit of GPUs (Charalambous et al., 2005), as we exploit optimized caching, SIMD and the extremely low overhead in spawning GPU threads to distribute individual summations over the unobserved CTMC states within the sum–product algorithm. With this significantly higher degree of parallelization, we demonstrate near 90-fold increases in evaluation speed for models of codon evolution. Codon substitution models (Goldman and Yang, 1994; Muse and Gaut, 1994) decouple the rates of substitution between amino acids and those between nucleotides that do not alter the protein sequence. Such models have the potential to provide a more accurate description of the evolution of protein-coding nucleotide sequences (Shapiro et al., 2006) and thus more accurate phylogenetic analyses (Ren et al., 2005), but to date have largely only been practically employed to make inferences conditioned on a single phylogenetic history.
2 METHODS
To harness the hundreds of processing cores available on GPUs in phylogenetics, we introduce many-core algorithms to compute the likelihood of n aligned molecular sequences D given a phylogenetic tree τ with n tips and a CTMC model that characterizes sequence evolution along τ and allows for rate variation along the alignment. To describe these novel algorithms, we first review standard approaches to the data likelihood, Pr (D), computation (Felsenstein, 1981; Lange, 1997). We then highlight two time-consuming steps in these approaches: (i) computing the probabilities of observing two specific sequences at either end of each branch in τ and (ii) integrating over all possible unobserved sequences at the internal nodes of τ. Finally, we demonstrate how massive parallelization of these steps generates substantial speedup in computing Pr (D).
2.1 Computing the marginalized data likelihood
The data D=(D1,…, D1) comprise C alignment columns, where column data Dc=(Dc1,…, Dcn) for c = 1,…, C contain one homologous sequence character from each of the n taxa. Each character exists in one of S possible states that we arbitrarily label {1,…, S}. Relating these taxa, τ is an acyclic graph grown from n external nodes of degree 1, n − 2 internal nodes of degree 3 and one root node of degree 2. Connecting these nodes are 2n − 2 edges or branches with their associated lengths T =(t1,…,t2n−2).
To keep track of these nodes and branches during computation, we require several additional labelings. We label the root and internal nodes with integers {1,…, n − 1} starting from the root and label the tips of τ arbitrarily with integers {1,…, n}. We let ℐ identify the set of internal branches and ℰ identify the set of terminal branches of τ. For each branch b∈ℐ, we denote the internal node labels of the parent and child of branch b by ψ(b) and ϕ(b), respectively. We use the same notation for each terminal branch b except ψ(b) is an internal node index, while ϕ(b) is a tip index.
Following standard practice since Felsenstein (1981) we assume that column data Dc are conditionally independent and identically distributed. Thus, it suffices to compute the likelihood for only each unique Dc and reweigh appropriately using standard data compression techniques. To ease exposition in this section, we assume all C columns are unique. Let s=(s1,…,sC) where sc =(sc1,…, sc,n−1) denote the unobserved internal node sequence states. To incorporate rate variation, we use the very popular discretized models (Yang, 1994) that modulate the CTMC for each column independently through a finite number of rates r∈{1,…, R}. Then, the joint likelihood of the unobserved internal node data and the observed data at the tips of τ given the rates r=(r1,…, rC) becomes
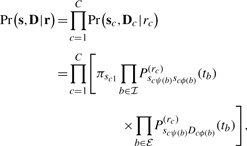 |
(1) |
where π=(π1,…, πS) are the prior probabilities of the unobserved character in each column at the root node and P(r)sj(t) for s, j∈{1,…, S} are the finite-time transition probabilities of character j existing at the end of a branch with length t given character s at the start under rate r. These probabilities derive from the rate-modulated CTMC and we discuss their calculation in a later section. Often, π also derives from the CTMC.
Unpacking Equation (1), if we guess the internal node states and rate for each column, the likelihood reduces simply to the product of character transition probabilities over all of the branches in τ and across all columns. However, s and r are never observed and, hence, we sit with a further time-consuming integration. Letting Pr (r) represent the prior probability mass function over rates, we recover the observed data likelihood by summing over all possible internal node states and column rates,
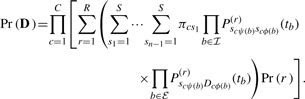 |
(2) |
2.1.1 Traditional computation
Judiciously distributing the sum over s within the product over branches in Equation (2) reduces its computational complexity from exponential to polynomial in n and forms a sum–product algorithm, also known as Felsenstein's Peeling algorithm in phylogenetics (Felsenstein, 1981). This approach invites a post-order traversal of the internal nodes in τ and the computation of a recursive function at each node that depends only on its immediate children; to the best of our knowledge, all likelihood-based phylogenetic software exploits this recursion.
Let Fu={Furcs} be an R×C×S matrix of forward, often called partial or fractional, likelihoods at node u. Element Fus is the probability of the observed data at only the tips that descend from node u, given that the state of u is s. If u is a tip, then we initialize partial likelihoods via equation Fus =1{s=Du}. In case of missing or ambiguous data, Du denotes the subset of possible character states, and forward likelihoods are set to Fus =1{s∈Du}. During post-order, or upward, traversal of τ, we compute forward likelihoods for each internal node u using the recursion
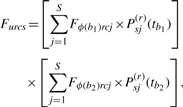 |
(3) |

where b1 and b2 are indices of the branches descending from node u and ϕ(b1) and ϕ(b2) are the corresponding children of u.
Given the final recursive computations F1rcs1 at the root, we recover the data likelihood through a final summation over the root state s1 and column rate r across all columns,
| (4) |
2.1.2 Many-core computation
Computing Equation (3) for all (r, c, s) is generally O(RCS2) but is at best order O(RCS) when both children are tips and have unambiguous data. In either case, this high computational cost invites parallelization. For each node u in the recursion, we propose distributing these computations across many-core processors such that each (r, c, s)-entry executes in its own short-lived thread. This fine scale of parallelization differs substantially from previous approaches that partition the columns into conditionally independent blocks and distribute the blocks across separate processing cores and is possible because GPUs share common memory and sport negligible costs to spawn and destroy threads, potentiating significant speedup improvement.
Algorithm 1 outlines our implementation of Equation (3). For each (r, c, s)-thread, the algorithm dedicates only a small portion of its code to actually computing Furcs. Most of the work involves efficiently reading and caching the large vectors of child forward likelihoods {Fϕ(b1)rcj} and {Fϕ(b2)rcj} for j=1,…, S and even larger finite-time transition probability matrices {P(r)sj(tb1} and {P(r)sj(tb2} for s, j=1,…, S to maximize computational throughput.
To maximize data throughput with global memory, the GPU hardware combines, or ‘coalesces’, memory read/writes of 16 consecutive threads into a single wide memory transaction. If one cannot coalesce global memory access, then separate memory transactions occur for each thread, resulting in high latency. Thus, our algorithm attempts to read/write only multiples-of-16 values at a time. One approach we take embeds models in which S is not a multiple of 16 into a larger space by zero-padding extra entries in the forward likelihoods and transition probabilities. For example, codon models have a number of stop-codons (depending on the exact genetic code being employed), which are not considered valid states within the CTMC. This marginally reduces S <64; however, S is no longer a multiple of 16, so we treat the stop-codons as zero-probability states, yielding the full S =64. For nucleotide models, we simply process four (r, c, s)-entries, instead of one, in a single thread.
On-board the GPU processing units themselves sits up to 16 KB of memory shared between all threads grouped into a thread-block with a maximum of 512 threads per block. Shared memory performance is between 100- to 150-fold faster than even ‘coalesced’ global memory transactions. However, 16 KB is very small, about 5400 single-precision values. To optimize performance, we attempt to cooperatively prefetch the largest possible chunks of data sitting in global memory using coalesced transactions and cache these values in shared memory, in an order that maximizes their re-use across the threads in a block. Efficient caching requires carefully partitioning the R × C × S threads into blocks.
Two considerations direct our thread-block construction. First, within a rate class and column, Furcs for all states s depend on the same S forward likelihoods for both child nodes that we wish to load from global memory once. Second, within a rate class, Furcs for each column c depends on the same finite-time transition probabilities. Consequentially, we construct R ×⌈C/CBS⌉ thread-blocks, where ⌈·⌉ is the ceiling function and column-block size (CBS) is a design constant, controlling the number of columns processed in a block. Each block shares S × CBS threads that correspond to all states for CBS columns. Processing multiple columns allows for the reuse of cached finite-time transition probabilities. To this end, we choose CBS as large as possible such that S × CBS≤512. For codon models, CBS =8.
The final complication of Algorithm 1 arises when all S2 transition probabilities do not fit in shared memory. Instead, the threads cooperatively prefetch columns of the matrix in peeling-block size (PBS) length chunks. We choose PBS as large as possible without overflowing shared memory; for codon models in single precision, PBS=8. To enable coalesced global memory reads of the matrix columns, we exploit a column-wise flattened representation of the finite-time transition matrices; this differs from the standard row-wise representation in modern computing.
2.2 Computing the finite-time transition probabilities
Integral to the data likelihood are the finite-time transition probabilities 𝒫(r)sj(t) that characterize how state s mutates to state j along a branch of length t at rate r. Common practice in likelihood-based phylogenetics models the evolution of molecular characters as an irreducible, reversible S-state CTMC with infinitesimal generator Λ = Λ(θ). Unknown generator parameters θ govern the behavior of the chain. For nucleotide characters, popular parameterizations include the Hasegawa et al. (1985, HKY85) and Lanave et al. (1984, GTR) models. Due to their computational complexity, codon models remain less explored. Goldman and Yang (1994) and Yang et al. (2000, M0) introduce a S =61-state model with parameters θ =(κ, ω, π), where κ measures the transition:transversion rate ratio, ω controls the relative rate of non-synonymous to synonymous substitutions and π models the stationary distribution of characters and is often fixed to empirical estimates.
For many commonly used CTMCs and notably for all codon models, closed-form expression for the finite-time transition probabilities given θ do not exist and researchers exploit numerical eigendecomposition of Λ to recover the probabilities through
| (5) |
where μr is the rate multiplier for rate class r and E is the matrix of eigenvectors of Λ with corresponding eigenvalues λ1,…, λS. These eigenvalues and E are implicit functions of θ. Generally, one thinks of matrix diagonalization as a rate-limiting step (Schadt et al., 1998) in phylogenetic reconstruction, as diagonalization proceeds at O(S3); however, a typical MCMC sampler attempts to update branch lengths and rate-multipliers several orders of magnitude more often than the sampler updates θ. Consequentially, computation effort in recomputing Equation (5) for each rate class r and branch length t in τ far outweighs diagonalization of Λ.
2.2.1 Many-core computation
While time-consuming diagonalization occurs infrequently, constructing finite-time transition probabilities from a diagonalization is a major bottleneck and parallelization affords advantages in these calculations as well. Specifically, updating infinitesimal parameters θ and one rediagonalization leads to recalculating all probabilities for each rate class r along all 2n−2 branches in τ. Also, updating an internal node height in τ leads to recalculating the transition probabilities for all R for the two or three branches incident to the affected internal node.
Each of these updates starts with same eigenvector matrices and multiplies these against differently scaled and exponentiated eigenvalues. After precalculating the scaling factors, we perform these matrix multiplications in parallel. Our algorithm is a natural extension to usual GPU-based matrix multiplication kernels for arbitrary matrices A and B, see e.g. Choi et al. (1994) and Lee et al. (1997). Since A and B are often too large to fetch completely onto the GPU shared memory, these kernels take a block strategy. Each block of threads performs a loop. In each loop step, the threads form the product Asub Bsub of smaller submatrices that do fit within the shared memory of the block. The threads cooperatively prefetch all entries in Asub and Bsub. Then, each thread computes one entry of the product; this depends on many of the prefetched values, and so the kernel maximizes shared memory reuse. The threads tabulate results as the loop moves the submatrices across A and down B. We modify this algorithm in two ways. First, the threads within a block prefetch the appropriate submatrix diagonal entries of Drt and form the three-matrix product; and second, we greatly extend the number of blocks to perform evaluation for all rate classes and branch lengths simultaneously. Figure 1 depicts this blocking strategy when multiplying EDrtE−1. The figure assumes two blocks span S; in general, we set the block dimension to 16 to ensure coalesced global memory transactions. Finally, greatly speeding memory retrieval in the peeling algorithm, we store each resultant matrix P(r)(t) in a column-wise, transposed representation in global memory.
Fig. 1.

Parallel thread block design for computing the finite-time transition probabilities P(r)(t)=EDrt E−1 for all R rate classes along all 2n−2 branches simultaneously. Example assumes that two prefetch blocks (shaded) span the matrices.
2.3 Precision
Graphics rendering normally requires only 32-bit (single) precision floating point numbers and thus most GPUs compute at single precision. While the latest generation of GPUs can operate on 64-bit (double) precision numbers, the precision boost comes with a performance cost because the GPU contains far fewer double-precision units. Further, even at double precision, rounding error can still occur while propagating the partial likelihoods up a large tree. In general, we opt for single-precision floats and implement a rescaling procedure when calculating the partial likelihoods to help avoid roundoff. Our approach follows the suggestion of Yang (2000). When an under- or over-flow occurs while computing the likelihood, we record the largest partial likelihood Muc =maxr,s Furcs for each site c. As we peel up the internal nodes, we replace Furcs with Furcs/Muc to help avoid roundoff. We recover the final likelihood for each column at the root through rescaling the resulting calculations by ∏u Muc. In contrast to Yang (2000), we compute Muc and rescale the partial likelihoods in parallel for each site. When S is a power of 2, we further improve performance by performing the maximum-element search over s through a parallel-reduction algorithm. Until another under- or over-flow occurs, we leave Muc fixed.
2.4 Harnessing multiple GPUs
We provide support for multiple GPUs through a simple load balancing scheme in which we divide the data columns amongst approximately equal partitions and distribute one partition to each GPU. This scheme replicates considerable work in calculating the same finite-time transition probabilities on each GPU but the alternative, computing these values once on a master GPU and then distributing, could be much slower as it requires transferring large blocks of data from card to card; this carries extremely high latency. Our approach also facilitates the distribution of different genomic loci to each GPU whilst allowing each to have different models of evolution (for example, combining mitochondrial and nuclear loci requires different genetic codes and thus different transition probability matrices).
3 EXAMPLE
To illustrate the performance gains that GPUs afford in statistical phylogenetics, we explore the evolutionary relationship of mitochondrial genomes from 62 extant carnivores and a pangolin outgroup, compiled from GENBANK. This genomic sequence alignment contains 10 860 nt columns that code for 12 mitochondrial proteins and when translated into a 60-state vertebrate mitochondrial codon model, yields an impressive 3620 columns, of which 3600 are unique. Conducting a phylogenetic analysis on such an extensive dataset using a codon substitution model would previously be considered computationally foolhardy.
Figure 2 displays the tree for these carnivores that we infer from a Bayesian analysis under the M0 codon model (Goldman and Yang, 1994) with a 4-class discrete-Γ model for rate variation (Yang, 1994) and relaxed molecular clock (Drummond et al., 2006). This analysis uses GPU-enabled calculations through MCMC to draw 25 million random samples from the joint posterior of both an unknown phylogeny and unknown parameters characterizing the substitution model. The resulting phylogeny is compatible with the current understanding of the familial relationships of Carnivora and importantly helps resolve relationships within the Arctoidea. This infraorder within the caniform (dog-like) carnivores comprises the Ursidae (bears), Musteloidea (weasles, raccoons, skunks and red panda) and the Pinnipedia (seals and walruses) and the branching order has been debated. Delisle and Strobeck (2005) entertain a nucleotide-based analysis of a smaller dataset of the same 12 genes and show marginal support for either the grouping of Pinnipedia and Ursidae or Musteloidea and Uridae (depending on whether a Bayesian or maximum likelihood approach, respectively, is employed). When run under a conventional GTR nucleotide model, our alignment also yields the Pinnipedia and Ursidae grouping but with less than emphatic support (posterior probability 0.79). However, with the codon model, the same data proffer significant support (posterior probability 0.97) for the Pinnipedia and Musteloidea grouping, a relationship also found for nuclear markers (Flynn et al., 2000; Yu et al., 2004) and ‘supertree’ approaches that attempt to synthesize available phylogenetic knowledge (Bininda-Emonds et al., 1999).
Fig. 2.

Reconstructed codon-based majority clade consensus tree of 62 carnivore mitochondrial protein-coding sequences with the long-tailed pangolin (Manis tetradactyla) as an outgroup. We label clades with posterior probabilities except where they approach 1 and color branches to reflect relative rates of molecular evolution (red/faster, blue/slower) under a relaxed clock.
Table 1 reports the marginal posterior estimates for the tree height in expected substitutions, the transition:transversion rate ratio κ, the non-synonymous:synonymous rate ratio ω, rate variation dispersion α and relaxed clock deviation σ. These estimates demonstrate the high overall rate of synonymous substitution, suggesting that these changes may be saturating faster than amino acid replacement substitutions, an effect not adequately modeled at the nucleotide level. The codon model employed here does allow different rates for synonymous and replacement substitutions and this helps to explain the congruence of the resulting tree with that of nuclear genes that evolve at a slower rate.
Table 1.
Codon substitution model parameter estimates for 62 extant carnivores and the pangolin outgroup
| Posterior | 95% Bayesian | |
|---|---|---|
| mean | credible interval | |
| Tree height | 4.2 | (2.5–5.5) |
| Transition:transversion ratio κ | 12.1 | (11.7–12.4) |
| dN/dS ratio ω | 0.0274 | (0.0265–0.0284) |
| Rate variation dispersion α | 1.55 | (1.48–1.62) |
| Relaxed clock deviation σ | 0.28 | (0.16–0.74) |
We perform this codon-based analysis on a standard desktop PC sporting a 3.2 GHz Intel Core 2 Extreme (QX9770) CPU and 8 GB of 1.6 GHz DDR3 RAM. This CPU is the fastest available at the end of 2008 and, together with the high-end RAM, maximizes the speed at which the Java and C implementations run to provide a benchmark comparison. We equipped the desktop PC with three NVIDIA GTX280 cards each of which carries a single GPU with 240 processing cores running at 1.3 GHz and 1 GB of RAM. Exploiting all three GPUs, our analysis only requires 64 h to run.
Figure 3 compares runtime speeds using all three GPUs, a single GPU and Java and C implementations. For codon models, the C version is not available in the current BEAST release but our C implementation provides only a 1.6-fold speed increase over Java. To make these comparisons in reasonable time, we run short MCMC chains of 10 000 steps. Between the three GPUs and single GPU for the codon model, we observe a slightly <3-fold improvement. This factor compares well with the theoretical maximum, identifying that the replicated work in recomputing the finite-time transition probabilities is not a burden when fitting models with large state spaces. Between the three GPUs and the current BEAST implementation, we observe an over 140-fold improvement (a 90-fold improvement over the C implementation). One major difference between the default computations on the GPU and the CPU implementations is the precision, as modern CPUs provide double-precision at full performance. Fortunately, the GTX280 GPU also contains a very limited number (30) of double precision floating point units and it is possible to exploit double precision on the GPU, but at a performance cost. For the three GPU configuration, this cost is also ∼3-fold. To compute this data example in double precision requires more than the 1 GB of RAM available on a single card so we are unable to replicate this experiment using a single GPU. This is a limitation of our particular hardware; cards with 4 GB of RAM are now available. However, for this dataset, the performance hit comes with no scientific gain; posterior estimates at both double- and single precision remain unchanged.
Fig. 3.

Speed comparison of GPU, Java and C BEAST implementations. Speedup factors are on the log-scale.
One important scaling dimension of the parallelization is model state-space size S. We reanalyze the carnivore alignment at the nucleotide level using the GTR model for nucleotide substitution. For nucleotides, S=4 and we expect the speedup that the GPUs afford to become much more modest. However, we still observe ∼20-fold (three GPUs) and 12-fold (single GPU) performance improvements at single-precision, and single- and double-precision MCMC chains return the same estimates. These substantial increases definitely warrant using the GPUs even for small state spaces.
Two other scaling dimensions of practical concern are the numbers of alignment columns C and of taxa N. For very small C, the overhead in transferring data onto and off of the GPU may outweigh the computational benefit; performance should then rapidly improve to a point where the parallel resources on the GPU become fully occupied. After this saturation point, we suspect only moderate performance increase as C grows. The former cross-over point is critical in choosing whether or not to attempt a GPU analysis and the latter provides guidance in splitting datasets across multiple GPUs. On the other hand, changing N should have a much less pronounced effect with one additional taxon introducing one internal nodes and two branches to the phylogeny. Figure 4 reports how performance scales when analyzing 1–3600 unique codon alignment columns for all 62 carnivores and 200 columns for 3–62 taxa. Interestingly, even for a single codon column, the GPU implementation runs over 10-fold faster due to the performance gains in our parallel calculation of the finite-time transition probabilities. For this codon model, the saturation point occurs around 500 unique columns; therefore, optimal performance gains arise from distributing several hundred columns to each GPU in a multi-GPU system.
Fig. 4.

GPU performance scaling by number of unique alignment columns C and of taxa N. Speedup factors are on the log-scale.
4 DISCUSSION
The many-core algorithms we present in this article capably provide several orders of magnitude speedup in computing phylogenetic likelihoods. Given our algorithm design, performance is most impressive for larger state-space CTMCs, such as codon models. However, even nucleotide models demonstrate marked speed improvements, and our algorithms remain beneficial for small state spaces as well.
Codon models are particularly attractive as they can model the process of natural selection acting on different parts of the gene but are currently too prohibitively slow for use in inferring evolutionary relationships for non-trivial alignments. Through GPU computing, we are now able to infer the relationship among 62 carnivores using the M0 codon model applied to the entire protein-coding mtDNA genome. For this example, our algorithms compute nearly 150-fold faster than the currently available methods in the BEAST sampler. Using extrapolation to put this number in perspective, the current BEAST implementation would require 1.1 uninterrupted years to complete a 25 million MCMC sample run on our desktop PC, compared with the 64 h we endured.
For researchers looking to improve computational speed, one option is simply to purchase more computers of the same performance and link them into a cluster. At best this provides a linear speedup in speed proportional to the cost of each additional system. Even ignoring the costs of maintenance, space or air conditioning for such an increasingly large system, we suggest that upgrading the existing system with the currently available GPU boards could provide a similar performance at a fraction of the price. However, if budget is of limited concern, it is obviously possible to combine these approaches and equip cluster-computer nodes with GPU processing as well.
While our specific implementation of these algorithms is within a Bayesian framework, maximum likelihood inference requires the same calculations and a large number of software packages could benefit from the utilization of GPU computation. Indeed, we envisage the possibility and desirability of producing a general-purpose library and application programming interface (API) that abstracts the exact hardware implementation from the calling software package. In this way, improvements to these algorithms could be made or they could be implemented on other many-core hardware architecture, and all the supported packages would benefit. To this end, we have started an open source library, BEAGLE: Broad-platform Evolutionary Analysis General Likelihood Evaluator, that provides both an API and implementations in Java, C and for CUDA-based GPUs.
ACKNOWLEDGEMENTS
We thank Alexei Drummond for his impassioned support of the open source programming and critical insight on enabling GPU support in BEAST and Mark Silberstein for early discussions on implementing sum–product algorithms on GPUs. We thank John Welch for compiling the carnivore mitochondrial dataset.
Funding: Research gift from Microsoft Corporation, National Institutes of Health (R01 GM086887 to M.S.); The Royal Society (to A.R.)
Conflict of Interest: none declared.
REFERENCES
- Altekar G, et al. Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20:407–415. doi: 10.1093/bioinformatics/btg427. [DOI] [PubMed] [Google Scholar]
- Bininda-Emonds O, et al. Building large trees by combining phylogenetic information: a complete phylogeny of the extant Carnivora (Mammalia) Biol. Rev. 1999;74:143–175. doi: 10.1017/s0006323199005307. [DOI] [PubMed] [Google Scholar]
- Charalambous M, et al. Initial experiences porting a bioinformatics application to a graphics processor. In: Bozanis P, Houstis EN, editors. Proceedings of 10th Panhellenic Conference on Informatics (PCI2005). Vol. 3746. New York: Springer; 2005. pp. 415–425. Lecture Notes in Computer Science. [Google Scholar]
- Choi J, et al. PUMMA: parallel universal matrix multiplication algorithms on distributed memory concurrent computers. Concurrency Pract. Exp. 1994;6:543–570. [Google Scholar]
- Delisle I, Strobeck C. A phylogeny of the Caniformia (order Carnivora) based on 12 complete protein-coding mitochondrial genes. Mol. Phylogenet. Evol. 2005;37:192–201. doi: 10.1016/j.ympev.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Drummond A, et al. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Feng X, et al. Parallel algorithms for Bayesian phylogenetic inference. J. Parallel Distr. Comput. 2003;63:707–718. [Google Scholar]
- Feng X, et al. Parallel and Distributed Processing Symposium (IPDPS 2007). Washington, DC: 2007. Building the Tree of Life on terascale systems. [Google Scholar]
- Flynn J, et al. Whence the red panda? Mol. Phylogenet. Evol. 2000;17:190–199. doi: 10.1006/mpev.2000.0819. [DOI] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, et al. Dating the humanape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Keane T, et al. DPRml: distributed phylogeney reconstruction by maximum likelihood. Bioinformatics. 2005;21:969–974. doi: 10.1093/bioinformatics/bti100. [DOI] [PubMed] [Google Scholar]
- Lanave C, et al. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 1984;20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- Lange K. Mathematical and Statistical Methods for Genetic Analysis. New York: Springer; 1997. [Google Scholar]
- Lee H-J, et al. ICS '97: Proceedings of the 11th International Conference on Supercomputing. New York, NY: ACM; 1997. Generalized cannon's algorithm for parallel matrix multiplication; pp. 44–51. [Google Scholar]
- Manavski SA, Valle G. CUDA compatible GPU cards as efficient hardware accelerators for Smith-Waterman sequence alignment. BMC Bioinformatics. 2008;9:S10. doi: 10.1186/1471-2105-9-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B, et al. pIQPNNI: parallel reconstruction of large maximum likelihood phylogenies. Bioinformatics. 2005;21:3794–3796. doi: 10.1093/bioinformatics/bti594. [DOI] [PubMed] [Google Scholar]
- Moore G. Cramming more components onto integrated circuits. Proc. IEEE. 1998;86:82–85. [Google Scholar]
- Moret B, et al. High-performance algorithm engineering for computational phylogenetics. J. Supercomput. 2002;22:99–111. [Google Scholar]
- Muse S, Gaut B. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with applications to the chloroplast genome. Mol. Biol. Evol. 1994;11:715–725. doi: 10.1093/oxfordjournals.molbev.a040152. [DOI] [PubMed] [Google Scholar]
- NVIDIA. NVIDIA CUDA Compute Unified Device Architecture: Programming Guide Version 2.0. 2008. Available at http://developer.download.nvidia.com/compute/cuda/ 2_0/docs/NVIDIA_CUDA_Programming_Guide_2.0.pdf (last accessed date March 16, 2009) [Google Scholar]
- Ren F, et al. An empirical examination of the utility of codon-substitution models in phylogeny reconstruction. Syst. Biol. 2005;54:808–818. doi: 10.1080/10635150500354688. [DOI] [PubMed] [Google Scholar]
- Schadt E, et al. Computational advances in maximum likelihood methods for molecular phylogeny. Genome Res. 1998;8:222–233. doi: 10.1101/gr.8.3.222. [DOI] [PubMed] [Google Scholar]
- Schmidt H, et al. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Shapiro B, et al. Choosing appropriate substitution models for the phylogenetic anaylsis of protein-coding sequences. Mol. Biol. Evol. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- Silberstein M, et al. Efficient computation of sum-products on GPUs through software-managed cache. In: Zhou P, editor. Proceedings of the 22nd Annual International Conference on Supercomputing. New York: ACM; 2008. pp. 309–318. [Google Scholar]
- Stamatakis A, et al. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Yang Z. Estimating the pattern of nucleotide substitution. J. Mol. Evol. 1994;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum likelihood estimation on large phylogeneis and analysis of adaptive evolution in human influenza virus A. J. Mol. Evol. 2000;51:423–432. doi: 10.1007/s002390010105. [DOI] [PubMed] [Google Scholar]
- Yang Z, et al. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, et al. Phylogenetic relationships within mammalian order Carnivore indicated by sequences of two nuclear DNA genes. Mol. Phylogenet. Evol. 2004;33:694–705. doi: 10.1016/j.ympev.2004.08.001. [DOI] [PubMed] [Google Scholar]