

Abstract
Nodal, an embryonic morphogen belonging to the TGF-β superfamily, is an important regulator of embryonic stem cell fate. We have recently demonstrated that Nodal is expressed significantly in aggressive melanoma. Surprisingly, expression of the Nodal coreceptor, Cripto-1, was detected in only a small fraction of the melanoma tumor cell population, indicating a primary role for Cripto-1-independent signaling of Nodal in melanoma. In this review, we discuss how regulatory factors present in an embryonic environment, such as Lefty, can downregulate Nodal expression and inhibit tumorigenicity and plasticity of melanoma cells. Our translational studies show that antibodies against Nodal are capable of repressing melanoma vasculogenic mimicry and of inducing apoptosis in melanoma tumors in an in vivo lung-colonization assay. Our previous work and ongoing studies suggest that Nodal may represent a novel diagnostic marker and therapeutic target in melanoma.
Keywords: biomarker, Lefty, melanoma, Nodal, plasticity, therapy
The incidence of cutaneous melanoma is increasing, especially among Caucasians [1]. This aggressive skin malignancy can progress from a relatively localized and poorly invasive lesion characteristic of radial-growth phase (RGP) melanoma to the more aggressive and deeper lesion of vertical-growth phase (VGP) melanoma, which can lead to metastatic spread to regional lymph nodes and distant organs [2]. Although early diagnosed poorly aggressive lesions can be removed surgically, resulting in high cure rates, current treatment modalities have not significantly improved the 4–6-month survival range in patients with advanced-stage disease and distant metastases [3]. Numerous studies have begun to shed some light on the intricate molecular pathways involved in progression and metastatic spread of melanoma, resulting in the identification of novel biological markers. For instance, RAS–RAF–MAPK, RAS–PI3K–AKT and FAK signaling pathways have been found to be upregulated in melanoma, suggesting that the targeting of these molecules may lead to new treatment strategies [4-6]. Interestingly, low B-RAF and high KIT mutation rates have been detected in patients with cutaneous melanomas and chronic sun-induced damage, while patients with cutaneous melanomas without chronic sun damage have a higher frequency of B-RAF mutations and are less likely to have KIT mutations, suggesting that detection of B-RAF and KIT could help identify melanoma patients that would benefit most from specific targeting therapy against B-RAF or KIT [7]. Additional signaling pathways, such as BCL-2 and CDKN2A, have also been described as potential targets for melanoma therapy [8]. In this review, we will discuss the role of Nodal in human melanoma and how this may represent a potential biomarker for diagnosis and a new target for therapy.
Overview of Nodal function & signaling
Nodal & development
Nodal is a member of the TGF-β family and an important morphogen and regulator of cell fate in both embryological and adult systems [9,10]. TGF-β/activin/Nodal signaling has been demonstrated to play a role in maintaining pluripotency in human embryonic stem (ES) cells since disruption of this signaling pathway results in their differentiation [11,12]. In addition, Nodal has been demonstrated to inhibit differentiation of human ES cells along the neuroectodermal differentiation pathway, further supporting a role for Nodal in the maintenance of pluripotency in human ES cells [12]. These data appear to contrast with observations made in mouse ES cells, where TGF-β signaling is active but not required for stemness [11], suggesting that there are differences in the regulatory pathways involved during the maintenance of pluripotency between human and mouse ES cells. Nodal can also regulate mesoderm formation, establishment of the left–right (LR) axis and subsequent asymmetric development and positioning of visceral organs [10,13,14]. In the mouse, LR patterning depends on asymmetric expression of Nodal around the node, which leads to restriction of Nodal in the left lateral plate mesoderm (LPM). It has been suggested that this spatial restriction of Nodal expression may be determined by bone morphogenic protein (BMP) signaling activity on the right side of the LPM that inhibits Nodal, and higher levels of BMP antagonists, such as Noggin and Chordin, in the left LPM [15,16]. More recently, Man1, an inner nuclear membrane protein that regulates TGF-β signaling by interacting with receptor-associated Smads, has been demonstrated to control Nodal function by regulating its asymmetric expression in a node-independent manner [17]. Nodal expression also appears to be involved in mesoderm and/or endoderm development [18,19], although a recent study employing conditional mutagenesis experiments has demonstrated that abrogation of Nodal before the early somite stage does indeed result in LR developmental defects without adversely affecting mesodermal or endodermal development [20]. It is possible that Nodal may play an initial role during early specification of the different germ layers while other signaling pathways, such as BMP or Wnt/β-catenin[18,21], may be required for definite commitment of mesoderm and/or endoderm development. These studies highlight the complexity of the molecular mechanisms involved during early morphogenesis and the importance of identifying potential regulators that could play a role during proper development.
Regulation of Nodal expression
Nodal expression and signaling is finely tuned by transcriptional regulators as well as post-translational and extracellular modifications. In mice, Nodal expression is enhanced by at least three separate transcriptional regulatory regions: the node-specific enhancer (NDE), the left side-specific enhancer (LSE) and the asymmetric enhancer (ASE) [22-24]. Also in mice, the LSE and ASE are induced by Nodal via a feed-forward mechanism that ultimately activates transcription factors, such as FoxH1 (Figure 1A). Notch signaling through CBF1-binding elements in the promoter region of NDE has also been demonstrated to induce Nodal expression (Figure 1B) [25,26]. In humans, sequencing and alignment analysis indicates that the Nodal gene contains similar enhancer elements, which suggests that human Nodal expression may be regulated in a similar manner. In fact, our studies indicate that, similar to mouse Nodal, human Nodal is induced by Notch4 signaling in melanoma cells [27]. Moreover, a positive feed-forward activation, similar to that described for the LSE and ASE in mice, has been documented to sustain Nodal expression in human ES and melanoma cell types [28,29]. It is possible that Nodal expression may also be governed by gene methylation and miRNA-directed degradation. In this regard, we have detected a CpG island of more than 1300 base pairs in proximity to the transcription start site of the Nodal gene, which suggests that methylation/demethylation of this region may be involved during regulation of Nodal expression [27]. In addition, a novel miRNA (miR-430) was recently demonstrated to block the translation of the zebrafish Nodal homolog, Squint [30]. Comparable miRNA target sites are also present in the mammalian Nodal gene, suggesting that human Nodal expression may be similarly affected by miRNA-mediated degradation [10]. Post-translational modifications of Nodal are important for proper Nodal signaling. As in most TGF-β family members, Nodal is synthesized as a proprotein that is activated following proteolytic processing by subtilisin-like proprotein convertases, such as PACE-4 and Furin [31]. Interestingly, it was found that the Nodal precursor is also functionally active since it can bind and activate activin receptors participating in maintaining the expression of Furin and PACE-4 and, surprisingly, BMP4 in extraembryonic ectoderm distant from the Nodal source. In turn, BMP4 was found to induce Wnt3, thus amplifying Nodal expression in the epiblast and mediating the induction of mesoderm (Figure 1C) [18]. Removal of the prodomain is important since it reduces Nodal's stability and signaling range, which promotes local autocrine signaling, whereas glycosylation of mature Nodal stabilizes it and increases its capacity for paracrine signaling [32].
Figure 1. Nodal signaling pathways.
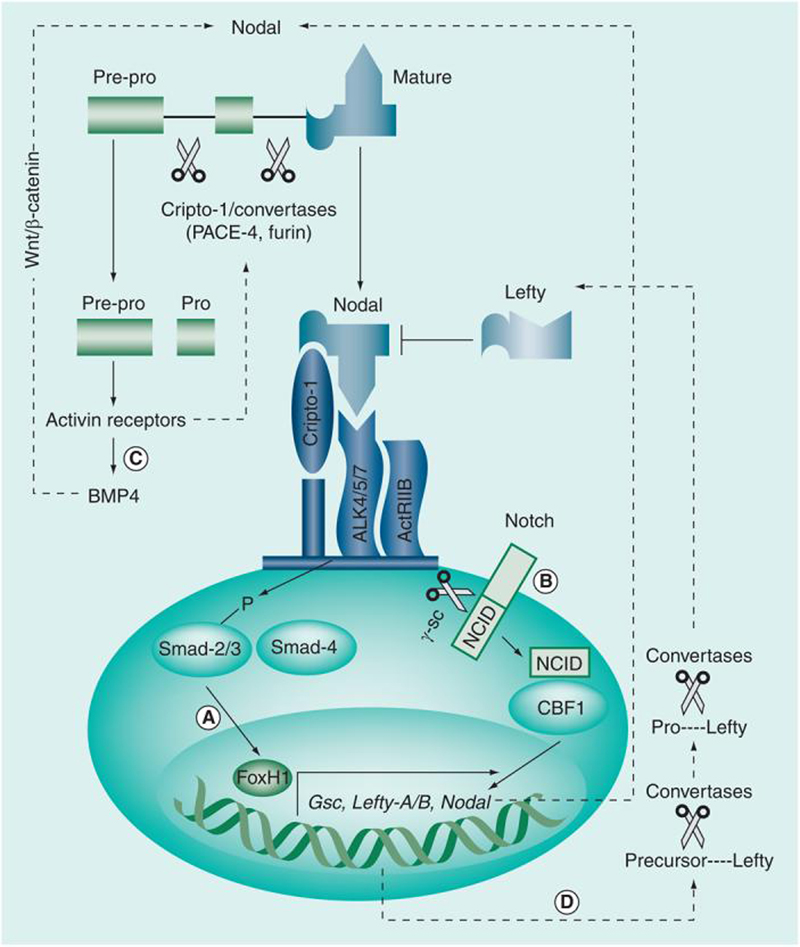
(A) Classic Nodal signaling involves binding with the coreceptor Cripto-1 and a heteromeric complex composed of Activin type I (ALK 4/5/7) and type II (ActRIIB) serine threonine kinase receptors. Type I and II receptor complexes induce phosphorylation of Smad-2 and -3, which then bind to Smad-4, forming a transcriptional complex that translocates to the nucleus, where it activates the transcription of target genes, such as FoxH1. (B) It is believed that Notch signaling can also induce Nodal expression through binding of the active NICD to CBF1, forming a transcriptional complex that enters the nucleus, inducing transcription of target genes including Nodal. (C) Nodal is synthesized by the cell as pre-pro-Nodal (∼55 kD), which is subject to cleavage by the subtilisin-like proprotein convertases PACE-4 and Furin, which generates the intermediate pro-Nodal (∼37 kD) and eventually active Nodal (∼18 kD). Recent data have suggested that Cripto-1 may play a role during the maturation process of Nodal by recruiting PACE-4/Furin and Nodal to the vicinity of the Nodal receptors on the cell membrane, thereby increasing both efficiency of Nodal processing and availability of the final active form of Nodal to its receptors. It is important to note that immature Nodal is capable of participating in BMP/Wnt/β-catenin signaling pathways, which can also influence Nodal function. Nodal also activates its own transcription via a feed-forward mechanism and the transcription of Lefty. (D) As Lefty exits the cell, it is processed to the active form by specific convertases and acquires the capability of downregulating the function and, ultimately, the expression of Nodal.
NCID: Notch intracellular domain.
Nodal & TGF-β signaling
While TGF-β activity has been demonstrated to act as a tumor suppressor in early stages of cancer, it has also been demonstrated to promote tumor cell proliferation at later stages of carcinogenesis [33]. This is largely due to downstream TGF-β signaling. For instance, TGF-β signaling through activation of Smad-2/3 is associated with increased cellular proliferation, while a decrease in cell proliferation can be observed with TGF-β-activated Smad-7 [34]. TGF-β can also signal independently of Smads in a ‘noncanonical’ fashion by inducing signaling molecule, such as B-RAF, RAS, MAPK and PI3K–AKT [34,35]. While TGF-β initiates signaling by binding and activating TGF-β type I and II receptors, Nodal signaling is initiated by binding to heterodimeric complexes composed of type I (ALK 4/7) and type II (ActRIIB) activin-like kinase receptors, which leads to the phosphorylation and activation of ALK 4/7 by ActRIIB and subsequent ALK 4/7-mediated phosphorylation of Smad-2 and Smad-3 [36]. Phosphorylated Smad-2/3 then associates with Smad-4, which translocates to the nucleus where it regulates gene expression through association with transcription factors such as FoxH1 and Mixer [10]. However, it cannot be excluded that Nodal can activate other important signaling molecules, such as MAPK and PI3K–AKT, as described earlier for noncanonical TGF-β signaling.
Nodal's coreceptor, Cripto-1
It is believed that the EGF colony-forming cell (CFC) members, primarily Cripto-1 in humans, are capable of potentiating this signaling pathway by acting as a coreceptor for Nodal [36] and even facilitating Nodal processing by recruiting Furin and/or PACE-4 in proximity to the other Nodal receptors where Nodal activity is required [37]. Interestingly, while Cripto-1 can be found both as a cell-associated and/or in an extracellular soluble form, it has been demonstrated that glycosylphosphatidylinositol (GPI) attachment of Cripto-1 is required for paracrine activity as a Nodal coreceptor [38]. However, it still remains unclear how essential Cripto-1 or cryptic are for Nodal signaling, since recent studies have demonstrated that the Nodal precursor can bind to ALK 4 in the extraembryonic ectoderm of the developing mouse embryo in a Cripto-1-independent manner and result in the expression of Nodal-responsive genes [18]. A recent study, in a murine knockout model, demonstrated that Nodal can signal and control axis specification in the absence of Cripto-1 so long as the Nodal antagonist Cerberus is also inhibited [39]. Additional findings support the role for Cripto-1 during anterior–posterior axis specification independently of Nodal signaling [40].
Inhibitors of Nodal
A potent morphogen such as Nodal requires tight control of its biological function, which is crucial for proper cueing of molecular signals during embryological development. Extracellular Nodal inhibitors control Nodal signaling by spatially and temporally restricting the Nodal-mediated activation of ALK 4/7. For example, Lefty-A and -B are highly divergent members of the TGF-β superfamily that specifically antagonize the Nodal signaling pathway by binding directly to and interacting with Nodal, or by binding to Cripto-1 and preventing Nodal from forming a more active signaling complex with the type I and II Activin receptors (Figure 1D) [41]. This restriction of Nodal signaling can occur in the extracellular microenvironment where Nodal and Cripto-1 are present, as well as at the cell surface. Of note, the Lefty proteins have not been found to bind ALK 4 or ActRIIB, which indicates that the Lefty proteins are not competitive inhibitors of the ALK receptor complex. Furthermore, in embryological systems, the Lefty genes are often downstream targets of Nodal signaling, which provides a powerful negative-feedback loop for this pathway [10,42]. Additionally, Tomoregulin (TMEFF1), a transmembrane protein containing two follistatin domains and an EGF motif, has been demonstrated to inhibit Nodal signaling in early Xenopus embryos by binding to the CFC domain of Cripto-1 and sequestering it from the ALK 4 receptor, thus preventing Cripto-1 from functioning as a coreceptor for Nodal [43]. Also, Cerberus, a member of the cysteine-knot superfamily [44], has been demonstrated to directly bind and block Nodal signaling [45], but was not expressed as abundantly as the Lefty proteins by human ES cells in our studies.
It is clear that regulation of development is a complex, highly regulated mechanism. Deregulation of the expression of key players involved during proper development, such as the morphogen Nodal, may result in embryonic lethality or even diseases in adults, such as cancer. Our research has identified Nodal in several human cancers and established that Nodal plays a key role during the growth and spread of cancer cells. Further studies will help elucidate whether some or all of the signaling pathways described earlier that have been demonstrated to be activated in the context of Nodal signaling (e.g., BMP, Wnt/β-catenin and Notch signaling) may also play a synergistic role with Nodal in human melanoma. As one example, inhibition of Notch signaling in C8161 aggressive human melanoma cells is associated with decreased Nodal expression in these cells, which suggests the possibility of crosstalk between Notch and Nodal signaling in melanoma [27]. Therefore, it is important to develop a better understanding of the regulatory processes involved in Nodal expression and function, which may help us to develop novel treatment strategies for targeting Nodal in human malignant disease.
Nodal expression in human melanoma
Studies in human cancer have begun to address the importance of Nodal and/or its coreceptors during the growth and spread of malignant cells. Although Cripto-1 has been demonstrated to activate a common cancer-related signaling pathway, such as the c-Src–MAPK–AKT signaling pathway, independently of Nodal [46], the components of a Cripto-1-independent signaling pathway for Nodal remain to be clearly identified. However, recent data from our laboratory indicate that Nodal appears to play a predominant role in the growth of human cancers, such as melanoma [28]. Our studies have also demonstrated that, when detectable, only a minor subpopulation of melanoma cells actually express Cripto-1 [47,48]. While it is not clear what is regulating the expression of Cripto-1 in Nodal-expressing melanoma cells, one possible explanation may be that melanoma cells express high levels of BMPs, including BMP4 [49,50]. Specifically, since BMP4 has been demonstrated to downregulate Cripto-1 expression in human embryonal and colon cancer cell lines [51], BMP4 could also be negatively affecting Cripto-1 levels in melanoma cells.
As mentioned earlier, cell-associated Cripto-1 is required for proper paracrine activity for Nodal [38]. Our data indicate that less than 5% of aggressive human melanoma cell lines are Cripto-1 positive by FACS analysis (Figure 2A). Therefore, it seems unlikely that the growth and metastatic potential of melanoma would be entirely dependent on the capability of less than 5% of an entire melanoma population to bind Nodal and induce the protumorigenic activity. In addition, our experiments demonstrate that, by removing Cripto-1-expressing human melanoma cells from aggressive cell lines via FACS sorting, the Cripto-1-enriched population (<5%) (Figure 2A) appear to show characteristics reminiscent of cancer expressing a stem cell phenotype, such as slow growth rates, ability to form spherical colonies in vitro and express stem cell-related transcription factors such as Oct4 [48]. This ‘Cripto-1-high’ subpopulation also expressed higher levels of the multidrug resistant protein-1 (MDR-1), both at the protein [48] and mRNA level (Figure 2B). Interestingly, the Cripto-1-depleted melanoma cells were still capable after 2 weeks of inducing tumors (when injected orthotopically in nude mice) that were almost identical in size and histological morphology to the tumors formed by parental unsorted melanoma cells (Figure 2C). Collectively, these data support the concept of Cripto-1-independent Nodal signaling for melanoma as well as other ‘Nodal-high/Cripto-1-low’-expressing human cancers [39]. Studies are in progress to precisely determine the role of Cripto-1 and Nodal in human cancer, particularly melanoma.
Figure 2. Depletion of CR-expressing C8161 human melanoma cells and tumorigenicity.
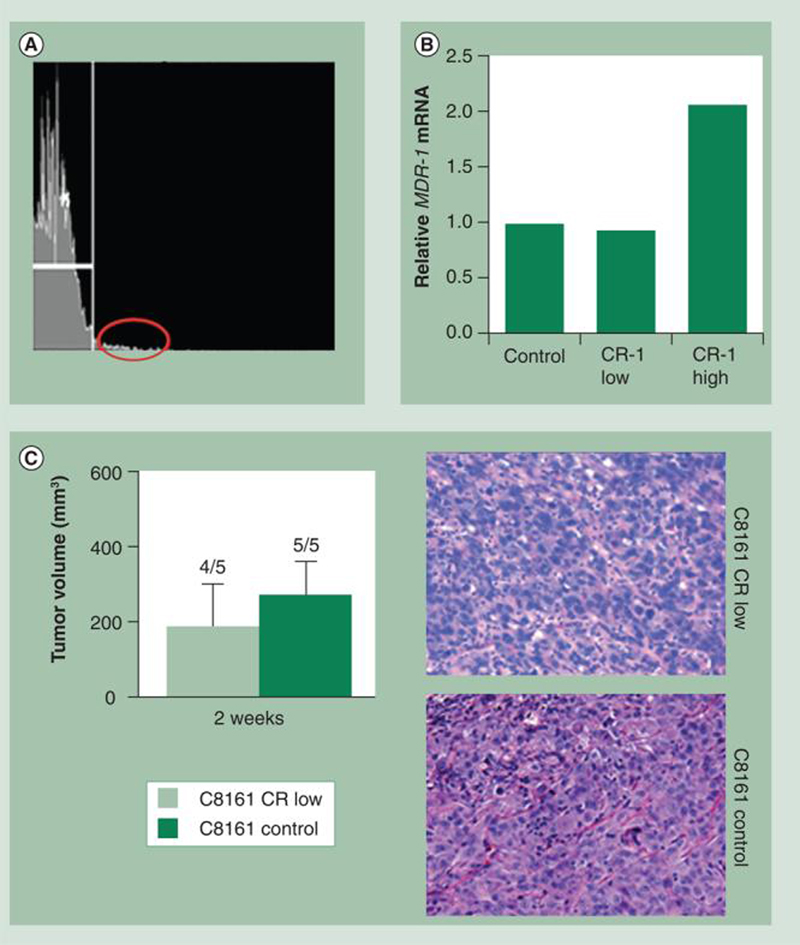
(A) C8161 human cutaneous metastatic melanoma cells were depleted of the CR-positive (<5%) subpopulation (red oval), which also expressed higher levels of MDR-1 mRNA, as detected by quantitative real-time PCR (B). The resulting C8161 CR-1-depleted as well as the starting C8161 parental population (control) were then injected subcutaneously into nude mice orthotopically to examine their ability to form tumors. (C) There were no significant differences (Mann–Whitney U test) in tumor volumes between tumors formed by parental C8161 cells and those formed by the CR-depleted C8161 cells, nor were there differences in histological morphology (original magnification 400×).
CR: Cripto-1.
Recent studies from our laboratory have characterized the expression pattern of the Nodal protein and transcript in a panel of human normal, neoplastic and stem cell types [47] and revealed that, similar to human ES cells, melanoma cells express Nodal. This contrasts with corresponding normal cells, such as melanocytes, in which Nodal was not detected [47]. We also determined that umbilical cord-derived mesenchymal stem cells (MSCs), amniotic fluid-derived stem cells and adult MSCs express negligible levels of Nodal. In addition, embryological studies in mice have demonstrated that Nodal expression is absent after the 12–14 somite stage [9,10], and online SAGE analyses using the Sage Anatomic Viewer of the Cancer Genome Anatomy Project [101] shows that Nodal expression is restricted to embryonic tissues, hESCs and cancer cells. Hence, Nodal expression is largely restricted to very early progenitor and reproductive cell types and re-emerges during tumorigenesis.
In addition to being expressed in tumor cells, our studies indicate that Nodal expression positively correlates with melanoma tumor progression toward a metastatic phenotype [28]. As indicated by Western blot analysis, metastatic melanoma cell lines (C8161, WM278 and 1205Lu) express high levels of Nodal, whereas Nodal is weakly expressed or absent in nonmetastatic melanoma cells (C81–61) [28]. Nodal expression also positively correlates with melanoma progression clinically (Figure 3A–C). Indeed, immuno histochemical analysis has demonstrated that Nodal protein is absent in normal skin and only rarely observed in poorly invasive RGP melanomas. This contrasts with invasive VGP melanomas and melanoma metastases, where Nodal expression is detectable in up to 60% of cases [28]. By comparison, staining for the Nodal coreceptor Cripto-1 in these sections is associated with only a very small subset of the tumor cell population, as demonstrated in VGP melanoma (Figure 3C). Collectively, these expression studies indicate that Nodal may prove to be a useful biomarker of melanoma progression – from a treatable RGP disease to a more aggressive VGP disease, to the presence of metastases.
Figure 3. Nodal as a biomarker for melanoma progression.
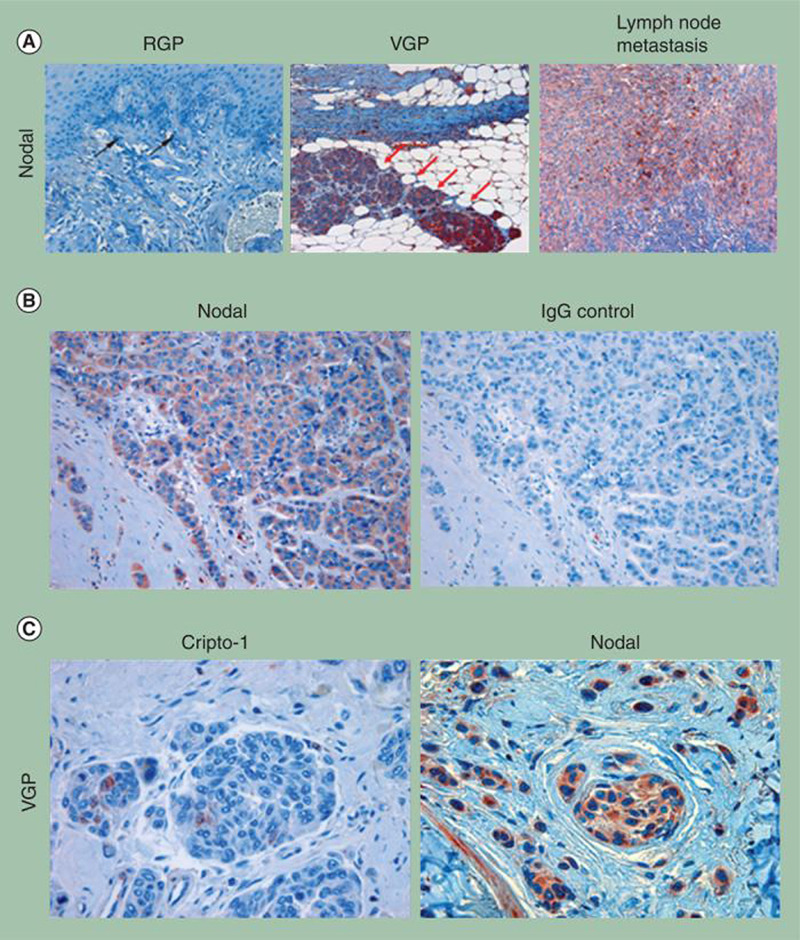
(A) Immunohistochemical localization of Nodal (red) in human melanoma. Sections represent RGP melanoma, VGP melanoma and a melanoma metastasis in a lymph node biopsy. Note that RGP melanoma cells express barely detectable Nodal (black arrows) (original magnification 200×). By contrast, strong expression of Nodal is observed in VGP melanoma cells (original magnification 100×) that have invaded subcutaneous connective or adipose tissues (red arrows). Nodal staining is also strong in melanoma cells that have metastasized to the lymph node (original magnification 100×). (B) Detection of Nodal by immunohistochemistry is demonstrated in an example of aggressive human melanoma invading subcutaneous tissue (original magnification 200×). (C) Melanoma cells in advanced VGP melanoma show relatively weak staining for Cripto-1 compared with staining for Nodal from a similar histological area (original magnification 400×).
RGP: Radial growth phase; VGP: Vertical growth phase.
Downregulation of Nodal abrogates melanoma plasticity & aggressive behavior
In addition to serving as a useful biomarker for melanoma progression, Nodal has also been demonstrated to be important for maintaining melanoma cell plasticity, invasiveness and tumorigenicity [28]. Using comparative global gene analyses, work from our laboratory and others have demonstrated that aggressive melanoma cells manifest a functional plasticity characterized by the expression of genes from a number of different cell types, as well as a reduction in the expression of melanocyte-associated genes. For example, aggressive melanoma cells aberrantly express genes (and proteins) such as VE-Cadherin, which are normally associated with endothelial cells and keratins, which are intermediate filaments characteristically associated with epithelial cells [52]. Furthermore, Melan-A (a marker for melanocytes) is reduced by more than fivefold and tyrosinase, which catalyses the conversion of tyrosine to the pigment melanin, is reduced by more than 35-fold in aggressive melanomas relative to their poorly aggressive counterparts [52]. Collectively, this gene-expression pattern confers a functional plasticity upon aggressive melanoma cells that enables them to escape normal physiological control and regulation. For example, VE-Cadherin expression by melanoma cells is essential for the formation of tumor-derived vascular networks, thought to provide rapidly growing tumors with a paravascular perfusion pathway, while the expression of keratins is associated with enhanced invasion and metastasis [53,54]. Our studies have demonstrated that Nodal may be an important mediator of this plasticity based on the following experimental evidence. Treatment of metastatic C8161 melanoma cells with the small-molecule inhibitor of ALK 4, 5 and 7 (SB431542), which is a general inhibitor of TGF-β/Activin/Nodal signaling, resulted in a reduction in tumor cell invasion through a defined extracellular matrix (Figure 4A) [28]. Using Morpholino (MONodal) to specifically knockdown Nodal expression in C8161 melanoma cells resulted in decreased tumorigenesis in nude mice (Figure 4B), re-expression of tyrosinase and downregulation of VE-Cadherin and keratin 8/18 (Figure 4C) [28]. The C8161 cells treated with MONodal were also less migratory in vitro as well as in vivo in the neural crest microenvironment of a developing chick [55]. Interestingly, our results indicated that downregulation of Nodal expression using MONodal lasted for approximately 14 days – during which time there was no significant tumor formation (Figure 4B). By 17 days, Nodal was re-expressed in the melanoma cells and tumorigenicity resumed [28]. In order to establish a mechanism for the reduction in tumorigenicity, we have subsequently examined the effects of this treatment on in vivo tumor cell proliferation and apoptosis [47]. Using immunohistochemical staining for Ki67 as a measure of proliferation and terminal deoxynucleotidyl transferase biotin–dUTP nick-end labeling (TUNEL) as a measure of apoptosis, we determined that inhibition of Nodal expression with MONodal decreases proliferation and increases apoptosis in orthotopic melanoma tumors [47]. These in vivo data support a role for Nodal in the maintenance of melanoma tumorigenicity and potential involvement in suppressing apoptosis.
Figure 4. Nodal inhibition abrogates aberrant gene expression, invasiveness and tumorigenicity in metastatic melanoma cells.
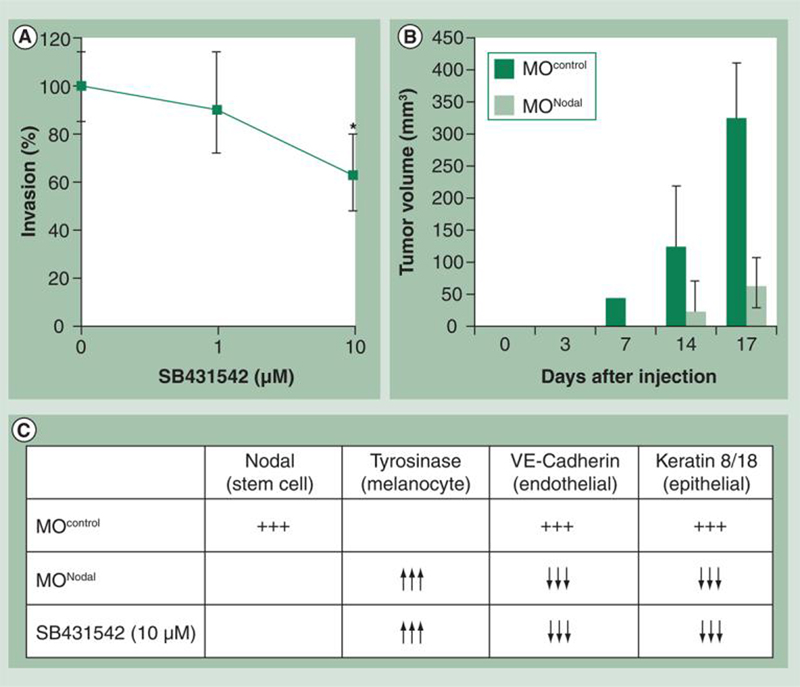
(A) In vitro invasion assay of C8161 cells cultured in the absence or presence of SB431542 (1 μM or 10 μM), a small molecule inhibitor of Nodal signaling. Invasion was calculated as a percentage of cells able to invade through a defined matrix (collagen IV, laminin and gelatin)-coated membrane during a 24-h period using an in vitro invasion assay. Bars represent the mean normalized invasion indices ± standard deviations. The values indicated by an asterisk are significantly different from the invasion index of control cells (n = 11 wells; p < 0.05, Mann–Whitney U test). Nodal inhibition significantly decreases the invasiveness of C8161 cells. (B) In vivo tumor formation in a mouse injected with C8161 cells treated with either MOControl or MONodal. Values represent the median tumor volume (mm3) ± interquartile range, and the MOControl and MONodal tumor volumes were significantly different at the time points indicated by an asterisk (n = 5; p < 0.05, Mann–Whitney U test). (C) Summary of results from Western blot and reverse transcriptase (RT)-PCR analyses of Nodal, tyrosinase, VE-Cadherin and keratin 8/18 expression in C8161 cells cultured on 3D type I collagen matrices for 6 days in the presence of vehicle (dimethyl sulfoxide), an inhibitor of the Nodal signaling pathway (SB431542, 10 μM) or a Morpholino designed to inhibit Nodal expression (MONodal). Inhibition of Nodal expression resulted in the re-expression of the melanocyte-specific marker tyrosinase concomitant with a reduction in the expression of VE-Cadherin, an endothelial lineage marker, and keratin 8/18, an epithelial lineage marker.
hESC-derived Lefty inhibits Nodal-positive melanoma tumors in vivo
Based on our previous work demonstrating that hESC-derived Lefty inhibits the expression of Nodal in melanoma and breast carcinoma cells as well as their clonogenicity in vitro [47], we examined the effects of injecting hESC-derived Lefty into palpable tumors formed 2 weeks after injecting 250,000 C8161 cells orthotopically into nude mice. These tumors were injected with hESC-derived Lefty (estimated <50 ng), recombinant Lefty (rLefty; 30 ng) or a control (carrier minus Lefty) once every other day over the following 2-week period. The tumors were then harvested and examined immunohistochemically for tumor cell apoptosis by TUNEL labeling (red) (Figure 5A–C), and for proliferation by staining for the proliferation marker Ki67 (brown) (Figure 5D–F). As seen in Figure 5, melanoma cells in the tumor treated with hESC-derived Lefty appear apoptotic (Figure 5A) c ompa r ed w it h no detectable apoptosis in tumor cells in either the control (Figure 5B) or rLefty-treated (Figure 5C) tumors. As we have discussed previously, the difference between rLefty and hESC-derived Lefty in relation to the effects observed on melanoma cells appears to be related to differences in post-translational modification, including the glycosylation state of hESC-derived Lefty [47]. By contrast, tumor cells in the hESC-derived Lefty-treated tumors did not stain for Ki67 (Figure 5D), while tumor cells in the control group (Figure 5E) or those treated with rLefty (Figure 5F) show elevated levels of Ki67 staining. Taken together, these results demonstrate that tumors formed orthotopically in nude mice by a Nodal-expressing human metastatic melanoma cell line (C8161) and injected with hESC-derived Lefty, but not rLefty, demonstrate decreased cell proliferation and increased apoptosis.
Figure 5. hESC-derived Lefty downregulates Nodal expression and induces apoptosis and decreases proliferation in metastatic melanoma cells in vivo.

C8161 human cutaneous metastatic melanoma cells were injected (250,000 cells per animal) orthotopically into a nude mouse model and allowed to form a palpable primary tumor mass for 2 weeks. Subsequently, hESC-derived Lefty (estimated <50 ng), rLefty (30 ng) or carrier minus Lefty (control) was delivered intratumorally over a 2-week period. (A–C) Tumor cell apoptosis was determined by immunohistochemical staining for terminal deoxynucleotidyl transferase biotin–dUTP nick-end labeling (TUNEL), which appears red in the color photomicrographs. (D–F) Immunohistochemistry of the Ki67 proliferation marker appears as a brown staining product. (A) Apoptosis is observed in C8161 melanoma tumor cells injected with Lefty derived from hESCs. (B) No apoptosis is evident in the control tumor receiving a sham injection regime of control or rLefty (C). (D) The proliferation marker Ki67 is not observed in the C8161 melanoma tumors treated with hESC-derived Lefty. (E) Ki67 is abundant in the control tumor receiving a sham injection regime of control or (F) rLefty (original magnification 400×).
hESC: Human embryonic stem cell; rLefty: Recombinant Lefty.
In light of our observations concerning the role Nodal plays in cancer progression and the aggressive phenotype of tumor cells [27,28] as well as Lefty's role as an inhibitor of Nodal, these in vivo results clearly suggest that targeting Nodal as a therapeutic treatment for aggressive tumors by one of its naturally derived inhibitors, Lefty, show great promise in a preclinical model.
Anti-Nodal antibody therapy
We have demonstrated that aggressive VGP melanoma and melanoma metastases express Nodal, whereas it was not detected in normal skin or in normal melanocytes, and was absent in non invasive RGP melanomas [28]. This expression not only correlated with melanoma progression but was also found to be important for maintaining tumor cell plasticity. As discussed previously, it is possible to reprogram the melanoma tumorigenic phenotype by blocking Nodal expression or function with Lefty or synthetic inhibitors of the Nodal receptor complex or with an anti-Nodal-specific morpholino. These results suggest the feasibility of treating melanoma or other Nodal-expressing human cancers with agents that can inhibit and block its activity, such as function-blocking antibodies. As a proof-of-principle, data obtained from our laboratory have demonstrated that human melanoma cells treated with function-blocking antibodies against Nodal demonstrate a significant reduction in their capacity to engage in vasculogenic mimicry in vitro (Figure 6A) [52,56]. Most noteworthy, function-blocking anti-Nodal antibody was demonstrated to reduce the ability of metastatic melanoma cells (C8161) to colonize lungs in mice in an in vivo tumor colonization assay as follows: C8161 metastatic melanoma cells were injected retroorbitally in nude mice and characteristically colonize first to the lung. After 2 days, the mice were injected intra peritoneally with either a function-blocking anti-Nodal antibody or an isotype control antibody (control IgG). The mice were subject to a total of five injections over a 10-day period. We found that the tumor cell colonies on the lung surface were macroscopically more evident in the control IgG-treated mice compared with anti-Nodal antibody-treated mice (Figure 6B). Also, melanoma cells in the lungs of mice treated with anti-Nodal antibody were more likely to show signs of cellular distress, such as cytoplasmic swelling and vacuolization, apoptosis (Figure 6C) and decreased expression of Nodal, as determined by immunohistochemistry, when compared with mice treated with control IgG (Figure 7). These findings implicate Nodal not only as a diagnostic or prognostic marker but also as a potential new therapeutic target.
Figure 6. Function-blocking anti-Nodal antibody inhibits vasculogenic mimicry in vitro and lung colonization in nude mice by metastatic melanoma cells.
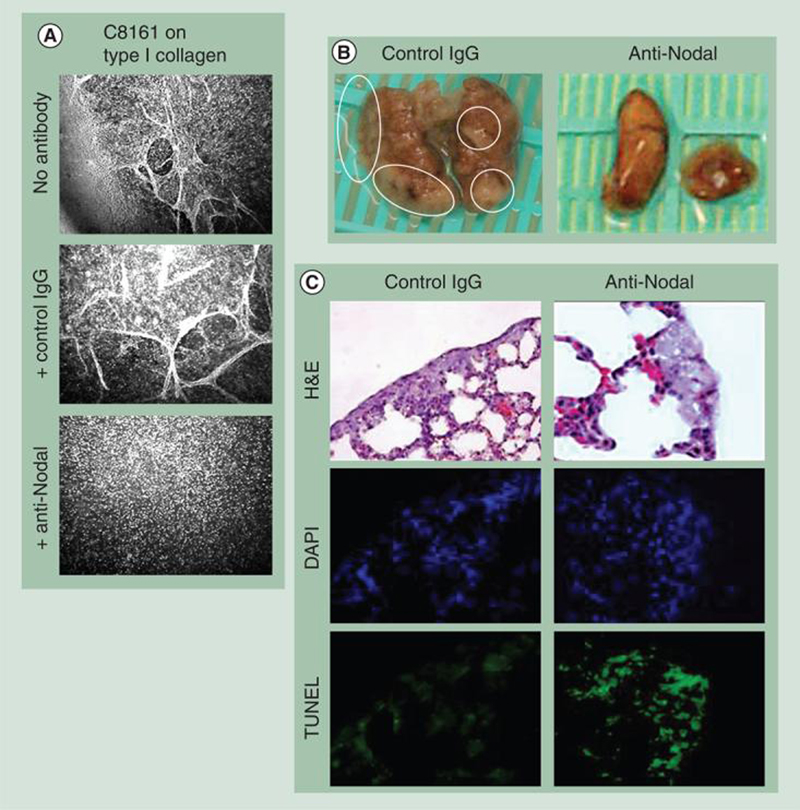
(A) C8161 metastatic melanoma cells treated with a function-blocking anti-Nodal antibody were inhibited from engaging in vasculogenic mimicry in vitro compared with no antibody or a nonfunction-blocking IgG control antibody (original magnification 200×). (B & C) C8161 cells were injected retro-orbitally in nude mice and characteristically colonize first to the lung. The mice were then injected intraperitoneally with either a function-blocking anti-Nodal antibody or control IgG over the next 10 days. At the end of this period, the mice were sacrificed and their lungs examined macroscopically, histologically and immunohistochemically. (B) Macroscopic examination of the lungs revealed a significant reduction in the number and size of the tumor cell colonies in the lungs of mice treated with the anti-Nodal antibody compared with the mice treated with the control IgG antibody. (C) Histological examination of H&E-stained lung tissue revealed that the metastatic C8161 cells forming the colonies in the anti-Nodal antibody-treated mice showed evidence of cellular distress characterized by cytoplasmic swelling and vacuolization not observed in the control antibody-treated mice; staining by TUNEL (green stain) showed an increase in the level of apoptosis in the melanoma cells in response to the anti-Nodal antibody compared with the mice treated with the control antibody (original magnification 200×). DAPI staining for cell nuclei was used as a control to locate each cell in the field of view.
DAPI: 4′,6-diamidino-2-phenylindole; H&E: Hemotoxylin and eosin; TUNEL: TdT-mediated biotin 16–dUTP nick-end labeling.
Figure 7. Expression of Nodal in lung colonies formed by C8161 human melanoma cells treated with anti-Nodal antibody.
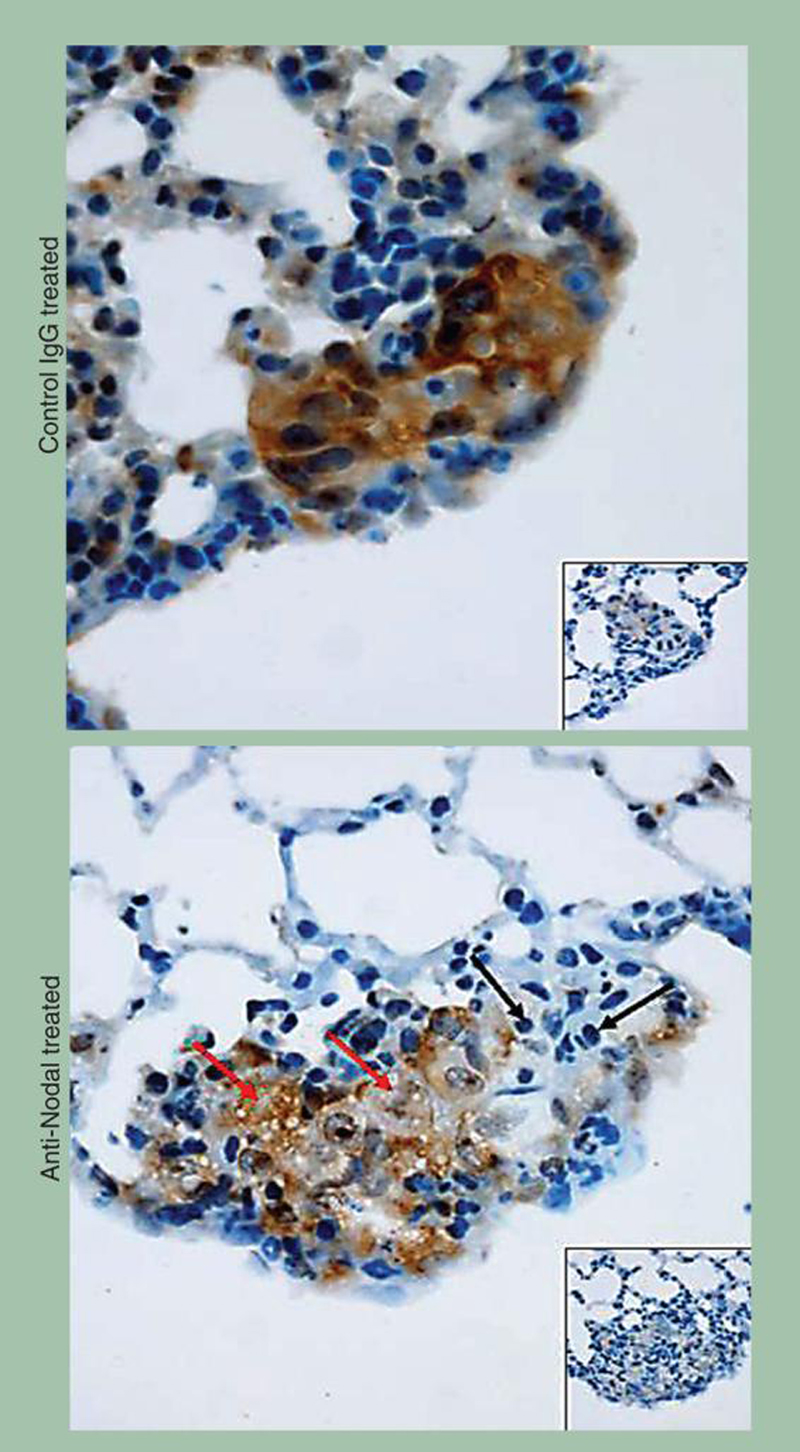
Immunohistochemistry shows strong staining for Nodal in the C8161 lung colonies of control IgG-treated mice. Staining for Nodal is less intense in the C8161 lung colonies of mice treated with the anti-Nodal antibody. Of note is the diffuse foam-like appearance of cytoplasmic vacuolization (red arrows) and areas of apoptosis containing evident apoptotic bodies (black arrows) in the anti-Nodal antibody-treated C8161 lung colonies. Insets show tissues stained with irrelevant isotype IgG used as negative control (original magnification 400×).
Expert commentary
Human melanoma is the most rapidly increasing malignant skin disease in Caucasians and frequent exposure to UV radiation from the sun due to increased outdoor activity seems to be a major contributing factor. Early diagnosis and surgical excision of the primary lesion often lead to high cure rates. However, the progression and metastatic spread of malignant melanoma to regional lymph nodes and distant organs can drastically reduce survival to months. Staging of melanoma is important since this will often dictate therapeutic options and prognosis. For instance, high-dose IFN-α-2b is often chosen as an adjunct to surgical removal of nonmetastatic melanomas and has been demonstrated to improve relapse-free survival. Unfortunately, treatment with specific anticancer agents alone or in combination has not demonstrated any significant survival advantage for patients with advanced-stage or metastatic melanoma. Studies aimed at dissecting the molecular pathways involved in promoting growth, metastasis and drug resistance in malignant melanoma are needed to help identify more accurate biomarkers for early diagnosis and disease progression and to properly design novel therapies that will specifically target melanoma cells, thus increasing therapeutic efficacy and limiting unwanted bystander effects that can compromise surrounding normal cells and tissues.
Five-year view
Strategies for targeting melanoma, especially metastatic melanoma, are rapidly evolving from simply killing tumor cells (with adverse side effects involving normal cells) to selectively targeting tumor cells only expressing unique antigens. The discovery of a novel embryonic signaling pathway, Nodal, underlying aggressive melanoma cell plasticity and tumorigenicity has allowed us to postulate a new therapeutic target and possible biomarker for disease status. Based on the promising results of the preclinical studies summarized in this review, we now have a better understanding regarding the translational significance of our work and future therapeutic opportunities. In particular, knowing that less than 5% of Nodal-positive aggressive melanoma cells express Cripto-1, a coreceptor for Nodal, suggests that this subpopulation may represent a stem cell-like phenotype with drug-resistance capabilities – and merits further investigation. Equally noteworthy are the findings demonstrating a direct relationship between the downregulation of Nodal in melanoma cells and their inability to form tumors in vivo, together with the induction of apoptosis in melanoma cells resulting from the intratumoral administration of Lefty (Nodal's inhibitor). From a clinical perspective, the most promising results to date emanate from the Nodal antibody experiments, which clearly demonstrate a complete abrogation of vasculogenic mimicry and a selective in vivo targeting of human melanoma lesions in mouse lungs, accompanied by tumor cell apoptosis. Collectively, these studies support the significant potential of targeting Nodal-positive melanoma cells with humanized Nodal-specific antibodies and with specialized forms of Lefty to selectively neutralize the expression of this aberrantly expressed embryonic pathway by aggressive melanoma tumor cells.
Key issues.
Nodal is an embryonic morphogen belonging to the TGF-β superfamily.
Nodal signaling is important for the maintenance of pluripotent cells and for proper embryonic development.
Nodal is poorly detected in normal postnatal tissues.
Nodal has been demonstrated to be highly expressed in advanced-stage melanoma compared with early-stage melanoma and breast cancer.
Nodal expression underlies tumorigenicity and plasticity of melanoma cells.
Human embryonic stem cell-derived factors, such as Lefty, can downregulate Nodal expression and function, causing reduced tumorigenicity and apoptosis of melanoma cells.
Anti-Nodal antibodies can downregulate Nodal-induced plasticity in vitro and induce apoptosis in melanoma lung colonization assays in mice.
Nodal may have potential as a novel biomarker and therapeutic target for human melanoma.
Acknowledgments
This work was supported by US NIH grants CA59702 and CA121205 and by the Eisenberg Scholar Research Award.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Luigi Strizzi, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6327 Fax: +1 773 755 6534 lstrizzi@childrensmemorial.org.
Lynne-Marie Postovit, Department of Anatomy & Cell Biology, Schulich School of Medicine, University of Western Ontario, London, Ontario, Canada Tel.: +1 519 661 2111 lynne.postovit@schulich.uwo.ca.
Naira V Margaryan, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6340 Fax: +1 773 755 6534 nmargaryan@childrensmemorial.org.
Alina Lipavsky, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6570 Fax: +1 773 755 6534 alipavsky@childrensmemorial.org.
Jules Gadiot, The Netherlands Cancer Institute, Antoni van Leeuwenhoek Ziekenhuis (NKI-AVL) Department of Medical Oncology/Immunology, Amsterdam, The Netherlands Tel.: +31 20 512 2085 j.gadiot@nki.nl.
Christian Blank, The Netherlands Cancer Institute, Antoni van Leeuwenhoek Ziekenhuis (NKI-AVL) Department of Medical Oncology/Immunology, Amsterdam, The Netherlands Tel.: +31 20 512 2570 c.blank@nki.nl.
Richard EB Seftor, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6377 Fax: +1 773 755 6534 rseftor@childrensmemorial.org.
Elisabeth A Seftor, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6366 Fax: +1 773 755 6534 eseftor@childrensmemorial.org.
Mary JC Hendrix, Children's Memorial Research Center, 2300 Children's Plaza, Box 222, Chicago, IL 60614, USA Tel.: +1 773 755 6328 Fax: +1 773 755 6534 mjchendrix@childrensmemorial.org.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N. Engl. J. Med. 2004;351(10):998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 2.Clark WH, Jr, Elder DE, Guerry D, 4th, et al. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984;15(12):1147–1165. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- 3.Moan J, Porojnicu AC, Dahlback A. Ultraviolet radiation and malignant melanoma. Adv. Exp. Med. Biol. 2008;624:104–116. doi: 10.1007/978-0-387-77574-6_9. [DOI] [PubMed] [Google Scholar]
- 4.Collisson EA, De A, Suzuki H, Gambhir SS, Kolodney MS. Treatment of metastatic melanoma with an orally available inhibitor of the Ras–Raf–MAPK cascade. Cancer Res. 2003;63(18):5669–5673. [PubMed] [Google Scholar]
- 5.Haluska F, Pemberton T, Ibrahim N, Kalinsky K. The RTK/RAS/BRAF/PI3K pathways in melanoma: biology, small molecule inhibitors, and potential applications. Semin. Oncol. 2007;34(6):546–554. doi: 10.1053/j.seminoncol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Hess AR, Postovit LM, Margaryan NV, et al. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 2005;65(21):9851–9860. doi: 10.1158/0008-5472.CAN-05-2172. [DOI] [PubMed] [Google Scholar]
- 7.Kalinsky K, Haluska FG. Novel inhibitors in the treatment of metastatic melanoma. Expert Rev. Anticancer Ther. 2007;7(5):715–724. doi: 10.1586/14737140.7.5.715. [DOI] [PubMed] [Google Scholar]
- 8.Hocker TL, Singh MK, Tsao H. Melanoma genetics and therapeutic approaches in the 21st Century: moving from the benchside to the bedside. J. Invest. Dermatol. 2008;128(11):2575–2595. doi: 10.1038/jid.2008.226. [DOI] [PubMed] [Google Scholar]
- 9.Schier AF, Shen MM. Nodal signalling in vertebrate development. Nature. 2000;403(6768):385–389. doi: 10.1038/35000126. • Excellent review on the role of Nodal during development. [DOI] [PubMed] [Google Scholar]
- 10.Schier AF. Nodal signaling in vertebrate development. Annu. Rev. Cell. Dev. Biol. 2003;19:589–621. doi: 10.1146/annurev.cellbio.19.041603.094522. [DOI] [PubMed] [Google Scholar]
- 11.James D, Levine AJ, Besser D, Hemmati-Brivanlou A. TGFβ/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132(6):1273–1282. doi: 10.1242/dev.01706. •• Shows that the role for TGF-β in human embryonic stem (ES) cells is different from that in mouse ES cells. [DOI] [PubMed] [Google Scholar]
- 12.Vallier L, Reynolds D, Pedersen RA. Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev. Biol. 2004;275(2):403–421. doi: 10.1016/j.ydbio.2004.08.031. •• Describes the role of Nodal for maintaining human ES cells in an undifferentiated state. [DOI] [PubMed] [Google Scholar]
- 13.Smith WC, McKendry R, Ribisi S, Jr, Harland RM. A nodal-related gene defines a physical and functional domain within the Spemann organizer. Cell. 1995;82(1):37–46. doi: 10.1016/0092-8674(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 14.Tian T, Burrage K. Stochastic models for regulatory networks of the genetic toggle switch. Proc. Natl Acad. Sci. USA. 2006;103(22):8372–8377. doi: 10.1073/pnas.0507818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Zhang W, Chen G, et al. Combinatorial signals of activin/nodal and bone morphogenic protein regulate the early lineage segregation of human embryonic stem cells. J. Biol. Chem. 2008;283(36):24991–25002. doi: 10.1074/jbc.M803893200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mine N, Anderson RM, Klingensmith J. BMP antagonism is required in both the node and lateral plate mesoderm for mammalian left-right axis establishment. Development. 2008;135(14):2425–2434. doi: 10.1242/dev.018986. [DOI] [PubMed] [Google Scholar]
- 17.Ishimura A, Chida S, Osada SI. Man1, an inner nuclear membrane protein, regulates left–right axis formation by controlling nodal signaling in a node-independent manner. Dev. Dyn. 2008;237(12):3565–3576. doi: 10.1002/dvdy.21663. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Haim N, Lu C, Guzman-Ayala M, et al. The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Dev. Cell. 2006;11(3):313–323. doi: 10.1016/j.devcel.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Mesnard D, Guzman-Ayala M, Constam DB. Nodal specifies embryonic visceral endoderm and sustains pluripotent cells in the epiblast before overt axial patterning. Development. 2006;133(13):2497–2505. doi: 10.1242/dev.02413. [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, Lualdi M, Lewandoski M, Kuehn MR. Broad mesodermal and endodermal deletion of Nodal at postgastrulation stages results solely in left/right axial defects. Dev. Dyn. 2008;237(12):3591–3601. doi: 10.1002/dvdy.21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sumi T, Tsuneyoshi N, Nakatsuji N, Suemori H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/β-catenin, Activin/Nodal and BMP signaling. Development. 2008;135(17):2969–2979. doi: 10.1242/dev.021121. [DOI] [PubMed] [Google Scholar]
- 22.Norris DP, Robertson EJ. Asymmetric and node-specific nodal expression patterns are controlled by two distinct cis-acting regulatory elements. Genes Dev. 1999;13(12):1575–1588. doi: 10.1101/gad.13.12.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saijoh Y, Oki S, Tanaka C, et al. Two nodal-responsive enhancers control left–right asymmetric expression of Nodal. Dev. Dyn. 2005;232(4):1031–1036. doi: 10.1002/dvdy.20192. [DOI] [PubMed] [Google Scholar]
- 24.Vincent SD, Norris DP, Le Good JA, Constam DB, Robertson EJ. Asymmetric Nodal expression in the mouse is governed by the combinatorial activities of two distinct regulatory elements. Mech. Dev. 2004;121(11):1403–1415. doi: 10.1016/j.mod.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Raya A, Kawakami Y, Rodriguez-Esteban C, et al. Notch activity induces Nodal expression and mediates the establishment of left–right asymmetry in vertebrate embryos. Genes Dev. 2003;17(10):1213–1218. doi: 10.1101/gad.1084403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krebs LT, Iwai N, Nonaka S, et al. Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev. 2003;17(10):1207–1212. doi: 10.1101/gad.1084703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. Targeting Nodal in malignant melanoma cells. Expert Opin. Ther. Targets. 2007;11(4):497–505. doi: 10.1517/14728222.11.4.497. [DOI] [PubMed] [Google Scholar]
- 28.Topczewska JM, Postovit LM, Margaryan NV, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat. Med. 2006;12(8):925–932. doi: 10.1038/nm1448. [DOI] [PubMed] [Google Scholar]
- 29.Hendrix MJ, Seftor EA, Seftor RE, et al. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer. 2007;7(4):246–255. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 30.Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318(5848):271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- 31.Beck S, Le Good JA, Guzman M, et al. Extraembryonic proteases regulate Nodal signalling during gastrulation. Nat. Cell Biol. 2002;4(12):981–985. doi: 10.1038/ncb890. [DOI] [PubMed] [Google Scholar]
- 32.Le Good JA, Joubin K, Giraldez AJ, et al. Nodal stability determines signaling range. Curr. Biol. 2005;15(1):31–36. doi: 10.1016/j.cub.2004.12.062. • Describes processing of Nodal and highlights the signaling properties of the different processed forms of Nodal. [DOI] [PubMed] [Google Scholar]
- 33.Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr. Opin. Genet. Dev. 2002;12(1):22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 34.Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-β in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21(2):123–132. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem. Photobiol. 2008;84(2):289–306. doi: 10.1111/j.1751-1097.2007.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo C, Whitman M. Nodal signals to Smads through Cripto-dependent and Cripto-independent mechanisms. Mol. Cell. 2001;7(5):949–957. doi: 10.1016/s1097-2765(01)00249-0. [DOI] [PubMed] [Google Scholar]
- 37.Blanchet MH, Le Good JA, Mesnard D, et al. Cripto recruits Furin and PACE4 and controls Nodal trafficking during proteolytic maturation. EMBO J. 2008;27(19):2580–2591. doi: 10.1038/emboj.2008.174. • Describes a new role for Cripto-1 during Nodal processing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe K, Hamada S, Bianco C, et al. Requirement of glycosylphosphatidylinositol anchor of Cripto-1 for trans activity as a Nodal co-receptor. J. Biol. Chem. 2007;282(49):35772–35786. doi: 10.1074/jbc.M707351200. [DOI] [PubMed] [Google Scholar]
- 39.Liguori GL, Borges AC, D'Andrea D, et al. Cripto-independent Nodal signaling promotes positioning of the A–P axis in the early mouse embryo. Dev. Biol. 2008;315(2):280–289. doi: 10.1016/j.ydbio.2007.12.027. • Recent study that describes Cripto-1-independent signaling for Nodal during development. [DOI] [PubMed] [Google Scholar]
- 40.D'Andrea D, Liguori GL, Le Good JA, et al. Cripto promotes A–P axis specification independently of its stimulatory effect on Nodal autoinduction. J. Cell Biol. 2008;180(3):597–605. doi: 10.1083/jcb.200709090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Shen MM. Two modes by which Lefty proteins inhibit nodal signaling. Curr. Biol. 2004;14(7):618–624. doi: 10.1016/j.cub.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 42.Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007;134(6):1023–1034. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- 43.Harms PW, Chang C. Tomoregulin-1 (TMEFF1) inhibits nodal signaling through direct binding to the nodal coreceptor Cripto. Genes Dev. 2003;17(21):2624–2629. doi: 10.1101/gad.1127703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu DR, Economides AN, Wang X, Eimon PM, Harland RM. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell. 1998;1(5):673–683. doi: 10.1016/s1097-2765(00)80067-2. [DOI] [PubMed] [Google Scholar]
- 45.Piccolo S, Agius E, Leyns L, et al. The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature. 1999;397(6721):707–710. doi: 10.1038/17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bianco C, Strizzi L, Rehman A, et al. A Nodal- and ALK4-independent signaling pathway activated by Cripto-1 through Glypican-1 and c-src. Cancer Res. 2003;63(6):1192–1197. [PubMed] [Google Scholar]
- 47.Postovit LM, Margaryan NV, Seftor EA, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl Acad. Sci. USA. 2008;105(11):4329–4334. doi: 10.1073/pnas.0800467105. •• Reports for the first time the effect of human ES cell-derived Lefty on cancer cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strizzi L, Abbott DE, Salomon DS, Hendrix MJ. Potential for cripto-1 in defining stem cell-like characteristics in human malignant melanoma. Cell Cycle. 2008;7(13):1931–1935. doi: 10.4161/cc.7.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothhammer T, Poser I, Soncin F, et al. Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 2005;65(2):448–456. [PubMed] [Google Scholar]
- 50.Rothhammer T, Bataille F, Spruss T, Eissner G, Bosserhoff AK. Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene. 2007;26(28):4158–4170. doi: 10.1038/sj.onc.1210182. [DOI] [PubMed] [Google Scholar]
- 51.Mancino M, Strizzi L, Wechselberger C, et al. Regulation of human Cripto-1 gene expression by TGF-β1 and BMP-4 in embryonal and colon cancer cells. J. Cell Physiol. 2008;215(1):192–203. doi: 10.1002/jcp.21301. [DOI] [PubMed] [Google Scholar]
- 52.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat. Rev. Cancer. 2003;3(6):411–421. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 53.Hendrix MJ, Seftor EA, Meltzer PS, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc. Natl Acad. Sci. USA. 2001;98(14):8018–8023. doi: 10.1073/pnas.131209798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendrix MJ, Seftor EA, Chu YW, et al. Coexpression of vimentin and keratins by human melanoma tumor cells: correlation with invasive and metastatic potential. J. Natl Cancer Inst. 1992;84(3):165–174. doi: 10.1093/jnci/84.3.165. [DOI] [PubMed] [Google Scholar]
- 55.Kasemeier-Kulesa JC, Teddy JM, Postovit LM, et al. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev. Dyn. 2008;237(10):2657–2666. doi: 10.1002/dvdy.21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am. J. Pathol. 1999;155(3):739–752. doi: 10.1016/S0002-9440(10)65173-5. •• First report that describes vasculogenic mimicry in human melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.The Cancer Genome Anatomy Project: SAGE Anatomic Viewer (SAV) http://cgap.nci.nih.gov/SAGE/AnatomicViewer.