

Abstract
Terpene synthases (TPS) require divalent metal ion co-factors, typically magnesium, that are bound by a canonical DDXXD motif, as well as a putative second, seemingly less well conserved and understood (N/D)DXX(S/T)XXXE motif. Given the role of the Ser/Thr side chain hydroxyl group in ligating one of the three catalytically requisite divalent metal ions and the loss of catalytic activity upon substitution with Ala, it is surprising that Gly is frequently found in this ‘middle’ position of the putative second divalent metal binding motif in plant TPS. Here we report mutational investigation of this discrepancy in a model plant diterpene cyclase, abietadiene synthase from Abies grandis (AgAS). Substitution of the corresponding Thr in AgAS with Ser or Gly decreased catalytic activity much less than substitution with Ala. We speculate that the ability of Gly to partially restore activity relative to Ala substitution for Ser/Thr stems from the associated reduction in steric volume enabling a water molecule to substitute for the hydroxyl group from Ser/Thr, potentially in a divalent metal ion coordination sphere. In any case, our results are consistent with the observed conservation pattern for this putative second divalent metal ion binding motif in plant TPS.
Keywords: Terpene Synthase, Enzymatic Mechanism, Cyclization, Metal binding motifs, Labdane-related diterpenoids
1. Introduction
Terpene synthases (TPS)1 catalyze complex cyclization and/or rearrangement of isoprenyl pyrophosphate precursors (Christianson, 2006). This creates the diverse hydrocarbon skeletal/backbone structures that underlie the amazing variety of terpenoid natural products, and often comprises the committed step in particular biosynthetic pathways. TPS typically ionize the allylic pyrophosphate ester linkage of their substrate to initiate carbocationic reactions (i.e., class I TPS; EC 4.2.3.-). Catalysis is dependent on divalent metal ion co-factors, usually Mg2+, which were hypothesized to bind to and assist ionization of the pyrophosphate. Sequence analysis of initially cloned class I TPS led to the additional hypothesis that a DDXX(D/E) motif was involved in divalent metal binding based on previous findings in the mechanistically similar prenyltransferases (Facchini and Chappell, 1992). These hypotheses have been confirmed by mutational and structural analysis, and the DDXX(D/E) motif is now considered a characteristic feature of class I TPS (Christianson, 2006).
In addition to this DDXX(D/E) motif, based on the structure of trichodiene synthase from the fungus Fusarium sporotrichioides it has been hypothesized that class I TPS contain a second conserved metal binding motif, (N/D)DXX(S/T)XXXE (metal binding residues in boldface) (Rynkiewicz et al., 2001). These residues are generally conserved in class I TPS, and their importance for catalysis in plant class I TPS has been demonstrated by alanine scanning mutagenesis in the diterpene cyclase abietadiene synthase from Abies grandis (AgAS), wherein Ala substitution for any one of the identified metal binding residues reduces catalytic efficiency ~10,000–fold (Peters and Croteau, 2002). However, the ‘middle’ Ser/Thr position is not completely conserved, with Gly found in this position in a surprising number of plant class I TPS. For example, within the diterpene synthases that are a focus of our research, while AgAS contains Thr, the kaurene synthases from Cucurbita maxima (CmKS) and Arabidopsis thaliana (AtKS) contain Ser and Gly, respectively, at the corresponding position (Figure 1). Yet all three enzymes are catalytically active (Stofer Vogel et al., 1996; Yamaguchi et al., 1996; Yamaguchi et al., 1998). In the rice (Oryza sativa) family of eight functional kaurene synthase-like (OsKSL) class I diterpene cyclases, two have Gly in the otherwise conserved Thr/Ser position (Xu et al., 2007). Gly also is found in the Thr/Ser position in a number of plant mono- and sesqui-terpene class I TPS as well. Accordingly, investigators working with plant TPS often do not acknowledge this second motif. Nevertheless, the corresponding residues interact with a Mg2+ ion in all of the plant class I TPS structures known to date (Hyatt et al., 2007; Kampranis et al., 2007; Starks et al., 1997; Whittington et al., 2002). Although in each case, this second motif is conserved (i.e., contains a Ser or Thr at the appropriate position).
Figure 1.

Alignment of the TPS divalent metal binding motifs for selected diterpene synthases (‡ above the alignment indicates the divalent metal binding residues; * below, the ‘middle’ Ser/Thr position of the second binding motif investigated here).
AgAS is a bifunctional enzyme that catalyzes two sequential cyclization reactions in distinct active sites (Peters et al., 2001). This includes a canonical class I TPS domain with both divalent metal binding motifs, whose importance for catalysis has been previously demonstrated by alanine scanning mutagenesis (Peters and Croteau, 2002). To investigate the ability of Gly to functionally substitute for Ser/Thr in the putative second divalent metal binding motif, we replaced the Thr at this position in AgAS with Ser, Ala, or Gly. Kinetic assays specifically examining class I TPS activity revealed that substitution by Ser or Gly decreases the activity of AgAS much less than substitution with Ala. This observation is consistent with the observed conservation pattern in plant class I TPS. We further speculate that the ability of Gly to partially restore activity relative to Ala substitution for Ser/Thr stems from the associated reduction in steric volume enabling a water molecule to substitute for the hydroxyl group from Ser/Thr, potentially in a divalent metal ion coordination sphere.
2. Results
2.1 Development of a coupled assay to examine AgAS class I activity
AgAS catalyzes protonation-initiated cyclization (i.e., a class II TPS reaction) of the universal diterpene precursor GGPP to CPP prior to catalyzing class I cyclization of CPP to abietadienes in a separate active site (Figure 2). To selectively assay class I activity, CPP was produced from GGPP with a mutant AgAS wherein the first Asp of the DDXXD motif has been substituted by Ala (AgAS:D621A), which essentially completely eliminates class I activity (~106 reduction) (Peters et al., 2001). The resulting CPP was then utilized to selectively assay AgAS class I activity. These assays were carried out using recombinant AgAS encoded with an N-terminal 6xHis tag for ease of purification. Because it has been shown that a KR motif at the N-terminus of AgAS forms part of the class I active site (Peters et al., 2003), the 6xHis tag was separated from the beginning of the AgAS protein sequence by a 25 amino acid linker peptide. This change in protein structure did lead to a reduction in catalytic efficiency, with an increase in KM from 0.4 to 1.2 μM and decrease in kcat from ~2 to 0.4 s−1, resulting in an overall ~17-fold decrease in kcat/KM from 5 to 0.3 (×106) M−1 s−1 relative to untagged AgAS. Nevertheless, given the ease of purification and relative nature of mutant analysis, we chose to carry out our studies in the context of this 6xHis tag.
Figure 2.
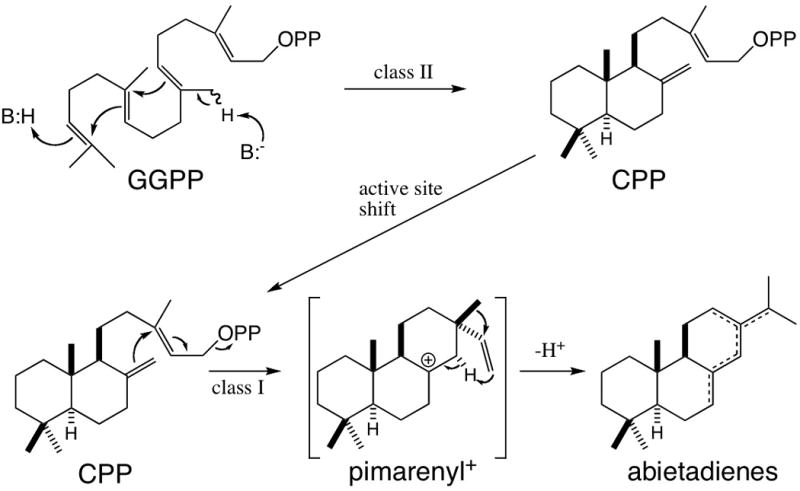
Sequential cyclization reactions catalyzed by AgAS.
2.2 Mutational analysis of the Ser/Thr position in the second TPS divalent metal binding motif
Given the observed conservation pattern and previous mutational analysis of the Ser/Thr position in the (N/D)DXX(S/T)XXXE second divalent metal binding motif, Ser, Ala, or Gly were substituted for the corresponding Thr769 in recombinantly 6xHis tagged AgAS, creating AgAS:T769S, AgAS:T769A, and AgAS:T769G, respectively. Much as previously reported (Peters and Croteau, 2002), AgAS:T769A exhibits a large reduction in catalytic activity, with kcat reduced to <10−4 s−1. By contrast, AgAS:T769S and AgAS:T769G retain more significant amounts of catalytic activity (Table 1). Ser substitution reduces kcat ~13-fold, from 0.4 to 0.03 s−1, with little effect on the pseudo-substrate binding constant KM, for only a ~17-fold reduction in catalytic efficiency. Even Gly substitution has relatively little effect, reducing kcat ~130-fold, to 0.003 s−1, again with little effect on KM, for an overall ~167-fold reduction in catalytic efficiency. Again as previously reported (Peters and Croteau, 2002), there is a slight change in product profile with the AgAS:T769A mutant, which exhibits some redistribution in the ratio of various abietadiene double bond isomers. However, substitution with Ser or Gly did not lead to significant changes in product profile (data not shown). Finally, comparison of the circular dichroism spectra for wild type and mutant AgAS indicate that these substitutions do not significantly alter protein structure (Figure 3).
Table 1.
Kinetic parameters
| Abietadiene synthase | kcat (s−1) | KM (μM) | kcat/KM (×105 M−1 s−1) |
|---|---|---|---|
| Wild type | 0.4 ± 0.1 | 1.2 ± 0.3 | 3 |
| T769S | 0.03 ± 0.01 | 1.5 ± 0.4 | 0.2 |
| T769A | < 10−4 | NDa | < 0.0001a |
| T769G | 0.003 ± 0.001 | 1.5 ± 0.4 | 0.02 |
Previous analysis indicates T769A increases KM 5-fold (ND, not determined).
Figure 3.
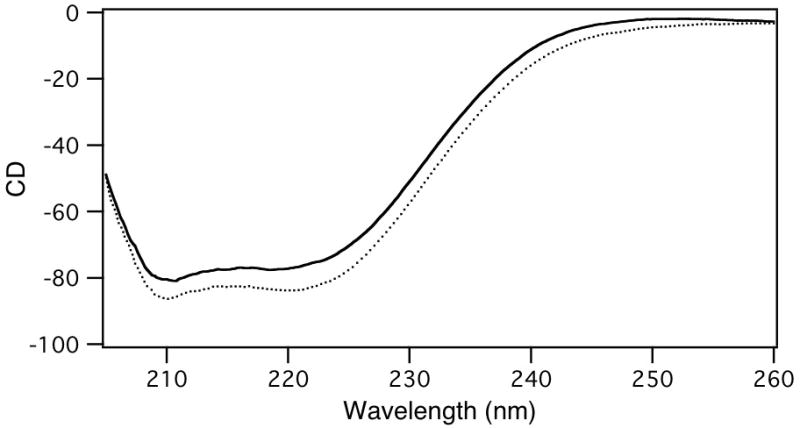
Circular dichroism spectra for wild-type AgAS (solid line) and AgAS:T769G (dotted line).
4. Discussion
Previous mutational analysis of the divalent metal binding motifs in AgAS (Peters and Croteau, 2002), as well as an aristolochene (sesquiterpene) synthase from the fungus Penicillium roqueforti (Felicetti and Cane, 2004), only examined the effect of Ala substitution for the Ser/Thr position in the (N/D)DXX(S/T)XXXE putative second TPS divalent metal binding motif. More recently, a detailed structure-function investigation of this motif in the trichodiene (sesquiterpene) synthase from F. sporotrichioides reported that Thr substitution for the Ser found at the Ser/Thr position leads to a ~700-fold decrease in catalytic efficiency. This largely stemmed from a relatively large 77-fold increase in KM, which crystallographic structure analysis suggested arises from steric incompatibility of the introduced γ-methyl of the Thr side chain with other elements of the active site (note that Ser is typically found at this position in fungal class I TPS such as this) (Vedula et al., 2008). However, in contrast to these mutational analyses, Gly is occasionally found in the Ser/Thr position in functional class I TPS from plants (e.g., Figure 1). Here we tried to reconcile these disparate findings by specifically examining the functional plasticity of the Ser/Thr position in the putative second TPS divalent metal binding motif of AgAS, a model plant diterpene synthase.
Our results demonstrate that, at least in the context of the AgAS active site, substitution of the Thr divalent metal ligand with Ser has relatively little effect on catalysis, either efficiency or reaction outcome. Selective removal of the directly ligating hydroxyl group by Ala substitution drastically reduces catalytic efficiency. However, complete side chain removal by Gly substitution partially restores catalytic activity. From these results, we speculate that replacement of Thr with Gly, but not Ala, creates sufficient space within the active site for a water molecule to bind and substitute for the Ser/Thr hydroxyl group, potentially as a divalent metal ion ligand (Figure 4), albeit with significantly reduced efficiency. In particular, the methyl group of the Ala side chain would prevent a water molecule from occupying a position approximating that of a Ser/Thr hydroxyl group (i.e., the space occupied by the oxygen of such a hydroxyl group containing side chain are occluded by the methyl protons of Ala). We also note that the postulated water molecule would be shielded from the reactive carbocation intermediates by the divalent metal and ionized pyrophosphate complex, consistent with the observed lack of hydroxylated products.
Figure 4.
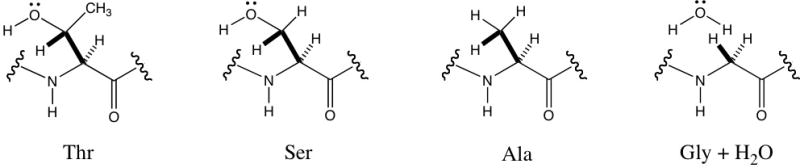
Hypothesized interaction of various side chains in the ‘middle’ position of the second TPS divalent metal binding site.
It has been noted that there are subtle, but functionally important differences between the active sites of microbial and plant derived class I TPS (Christianson, 2006). Thus, given the absence of detailed structural information for AgAS, it is difficult to resolve the structure-function relationships underlying the differences between the results reported here and those reported for similar mutational analysis of the fungal trichodiene synthase (Vedula et al., 2008). While other deviations from this putative second TPS divalent metal binding motif can be found in plant TPS [e.g., see (Martin et al., 2004), wherein several functional class I TPS contain a Gly in place of the ‘first’ N/D position], previous results (Peters and Croteau, 2002), along with those reported here, are consistent with an important role for this motif in plant, as well as fungal, TPS, despite a more divergent composition. Given the significant reduction in activity observed upon Gly substitution (Table 1), secondary changes are presumably required to restore full enzymatic activity. Nevertheless, our results provide biochemical observations consistent with the observed conservation pattern of this putative second TPS divalent metal binding motif in plants.
4. Experimental
4.1 General procedures
Unlabeled [E,E,E]-geranylgeranyl pyrophosphate (GGPP) was purchased from Isoprenoids, LC (Tampa, FL). [1-3H]GGPP was purchased from American Radiolabeled Chemicals (St. Louis, MO), Ni-NTA His-bind matrix was purchased from Novagen (Madison, WI), and molecular biology reagents were purchased from Invitrogen (Carlsbad, CA). Unless otherwise noted, all other chemicals were purchased from Fisher Scientific (Loughborough, Leicestershire, UK).
4.2 Recombinant Constructs
The recombinant pseudo-mature AgAS gene has been previously described (Peters et al., 2000), and transferred into the Gateway (Invitrogen) vector system via directional topoisomerization into pENTR/SD/D-TOPO (Cyr et al., 2007). Site-directed mutagenesis was carried out via PCR amplification of the pENTR/AgAS construct with overlapping mutagenic primers, and the mutant genes verified by complete sequencing. The resulting wild type and mutant genes were then transferred via directional recombination to the T7-promoter and N-terminal 6xHis fusion expression vector pDEST17. Use of the pDEST17 vector results in a 25 amino acid residue linker between the 6xHis tag and the cloned protein (here pseudo-mature AgAS). The linker peptide sequence is LESTSLYKKAGSAAALFNFKKEPFT.
4.3 Enzyme Expression and Purification
Transformed bacterial cells including pDEST17/AgAS (wild type or one of the various mutants) were grown in 1 L of NZY medium at 37°C with shaking to an A600 of 0.6–0.8, transferred to 16°C for 1 h, then induced with 0.5 mM IPTG and incubated with shaking for an additional 16–20 h. Bacterial cells were harvested by centrifugation and resuspended in 20 mL lysis buffer (50 mM Bis-Tris, pH 6.8, 150 mM KCl, 10 mM MgCl2, 10% Glycerol). After sonication (Branson Sonifier 450: 3 × 5 s continuous output at a setting of 5), the lysate was clarified by centrifugation at 15000g for 25 min at 4°C. AgAS was purified using Ni-NTA His-bind resin in batch mode. In brief, clarified lysate was added to 1 mL of Ni-NTA that had been pre-equilibrated in wash buffer (50 mM Bis-Tris, pH 6.8, 1mM DTT), 6xHis tagged AgAS bound by gently shaking at 4°C for 1 h, then washed with 50 mL wash buffer with 20 mM imidazole, and AgAS eluted with 2 × 1 ml elution buffer (50 mM Bis-Tris, pH 6.8, 250 mM imidazole, 1 mM DTT). The imidazole was removed by concentration through a 50 kDa molecular weight cutoff centricon (Millipore, Billerica, MA) to ~0.2 mL, followed by dilution in wash buffer to 2 mL. AgAS protein concentration was determined by A280 using the calculated extinction coefficient 138,350 M−1 cm−1. Structural integrity was measured by circular dichroism. For this purpose, AgAS was purified and analyzed at 0.5 mg/mL in 10 mM PBS, 10 mM MgCl2, 1 mM DTT, 10% glycerol.
4.4 Enzymatic Analysis
Kinetic analysis was carried out using the D621A mutant of AgAS, which eliminates class I activity (Peters et al., 2001), to convert [1-3H]GGPP to [1-3H]copalyl pyrophosphate (CPP). In brief, 1 ml reactions in assay buffer (50 mM Hepes, pH 7.2, 0.1 mM MgCl2, 5% glycerol, 5 mM DTT, 100 mM KCl and 0.1 mg/ml α-casein) containing 50 μM [1-3H]GGPP and 1 μM AgAS:D621A were run for 2 h at 30°C, which is sufficient to completely convert this amount of unlabeled GGPP to CPP. Kinetic assays were then performed similar to the method described previously (Peters et al., 2000). In brief, duplicate 1 ml reactions in assay buffer containing 3 nM wild type AgAS were initiated by the addition of [1-3H]CPP, allowed to react for 1 min at 30°C, then stopped by the addition of KOH to 0.2 M and EDTA to 15 mM. For analysis of the mutants, the enzymatic concentration was increased to 10 nM, 50 nM, or 100 nM, and incubation times increased to 5 min., 2 h, or 20 h for T769S, T769G, or T769A, respectively. The produced diterpenes were then extracted by hexane, the pooled extract passed over a short silica gel column, and product formation assessed by scintillation counting. The resulting data was analyzed using Kaleidagraph (Synergy, Reading, PA).
Diterpene products were analyzed by co-expressing AgAS (wild type or one of the various mutants) with GGPP synthase in E. coli, as previously described (Cyr et al., 2007). The resulting organic solvent extract of these recombinant cells were analyzed by gas chromatography with mass spectral detection (GC-MS), which was performed with a VF-1 column on a Varian (Palo Alto, CA) 3900 GC with Saturn 2100 ion trap MS system in electron ionization (70 eV) mode. Samples (1 μL) were injected in splitless mode at 50°C and, after holding for 3 min. at 50°C, the oven temperature was raised at a rate of 14°C/min. to 300°C, where it was held for an additional 3 min. MS data from 90 to 600 m/z were collected starting 12 min. after injection until the end of the run.
Acknowledgments
We thank Meimei Xu for assistance with molecular biology and Francis Mann for assistance with the GC-MS analyses. This work was supported by NIH grant GM076324 to R.J.P.
Footnotes
Abbreviations: AgAS, abietadiene synthase from Abies grandis; CPP, copalyl diphosphate; FID, flame ionization detection; GC, gas chromatography; GGPP, [E,E,E]-geranylgeranyl diphosphate; MS, mass spectrometry; TPS, terpene synthase.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- Cyr A, Wilderman PR, Determan M, Peters RJ. A Modular Approach for Facile Biosynthesis of Labdane-Related Diterpenes. J Am Chem Soc. 2007;129:6684–6685. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchini PJ, Chappell J. Gene family for an elicitor-induced sesquiterpene cyclase in tobacco. Proc Natl Acad Sci U S A. 1992;89:11088–11092. doi: 10.1073/pnas.89.22.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felicetti B, Cane DE. Aristolochene Synthase: Mechanistic Analysis of Active Site Residues by Site-Directed Mutagenesis. J Am Chem Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- Hyatt DC, Youn B, Zhao Y, Santhamma B, Coates RM, Croteau RB, Kang C. Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc Natl Acad Sci USA. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampranis SC, Ioannidis D, Purvis A, Mahrez W, Ninga E, Katerelos NA, Anssour S, Dunwell JM, Degenhardt J, Makris AM, Goodenough PW, Johnson CB. Rational Conversion of Substrate and Product Specificity in a Salvia Monoterpene Synthase: Structural Insights into the Evolution of Terpene Synthase Function. Plant Cell. 2007;19:1994–2005. doi: 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DM, Faldt J, Bohlmann J. Functional characterization of nine norway spruce TPS genes and evolution of gymnosperm terpene synthases of the TPS-d subfamily. Plant Physiol. 2004;135:1908–1927. doi: 10.1104/pp.104.042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters RJ, Carter OA, Zhang Y, Matthews BW, Croteau RB. Bifunctional abietadiene synthase: Mutual structural dependence of the active sites for protonation-initiated and ionization-initiated cyclizations. Biochemistry. 2003;42:2700–2707. doi: 10.1021/bi020492n. [DOI] [PubMed] [Google Scholar]
- Peters RJ, Croteau RB. Abietadiene synthase catalysis: Mutational analysis of a prenyl diphosphate ionization-initiated cyclization and rearrangement. Proc Natl Acad Sci USA. 2002;99:580–584. doi: 10.1073/pnas.022627099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters RJ, Flory JE, Jetter R, Ravn MM, Lee HJ, Coates RM, Croteau RB. Abietadiene synthase from grand fir (Abies grandis): Characterization and mechanism of action of the “pseudomature” recombinant enzyme. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- Peters RJ, Ravn MM, Coates RM, Croteau RB. Bifunctional abietadiene synthase: Free diffusive transfer of the (+)-copalyl diphosphate intermediate between two distinct active sites. J Am Chem Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- Rynkiewicz MJ, Cane DE, Christianson DW. Structure of trichodiene synthase from Fusarium sporotrichioides provides mechanistic inferences on the terpene cyclization cascade. Proc Natl Acad Sci U S A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- Stofer Vogel B, Wildung MR, Vogel G, Croteau R. Abietadiene synthase from grand fir (Abies grandis) J Biol Chem. 1996;271:23262–23268. doi: 10.1074/jbc.271.38.23262. [DOI] [PubMed] [Google Scholar]
- Vedula LS, Jiang J, Zakharian T, Cane DE, Christianson DW. Structural and mechanistic analysis of trichodiene synthase using site-directed mutagenesis: Probing the catalytic function of tyrosine-295 and the asparagine-225/serine-229/glutamate-233–Mg2+B motif. Arch Biochem Biophys. 2008;469:184–194. doi: 10.1016/j.abb.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Bornyl diphosphate synthase: structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci U S A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Wilderman PR, Morrone D, Xu J, Roy A, Margis-Pinheiro M, Upadhyaya N, Coates RM, Peters RJ. Functional characterization of the rice kaurene synthase-like gene family. Phytochemistry. 2007;68:312–326. doi: 10.1016/j.phytochem.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Saito T, Abe H, Yamane H, Murofushi N, Kamiya Y. Molecular cloning and characterization of a cDNA encoding the gibberellin biosynthetic enzyme ent-kaurene synthase B from pumpkin (Cucurbita maxima L.) Plant J. 1996;10:101–111. doi: 10.1046/j.1365-313x.1996.10020203.x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Sun T, Kawaide H, Kamiya Y. The GA2 locus of Arabidopsis thaliana encodes ent-kaurene synthase of gibberellin biosynthesis. Plant Physiol. 1998;116:1271–1278. doi: 10.1104/pp.116.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]