

Abstract
Many chemicals in the environment, in particular those with estrogenic activity, can disrupt the programming of endocrine signaling pathways that are established during development and result in adverse consequences that may not be apparent until much later in life. Most recently, obesity and diabetes join the growing list of adverse consequences that have been associated with developmental exposure to environmental estrogens during critical stages of differentiation. These diseases are quickly becoming significant public health issues and are fast reaching epidemic proportions worldwide. In this review, we summarize the literature from experimental animal studies documenting an association of environmental estrogens and the development of obesity, and further describe an animal model of exposure to diethylstilbestrol (DES) that has proven useful in studying mechanisms involved in abnormal programming of various differentiating estrogen- target tissues. Other examples of environmental estrogens including the phytoestrogen genistein and the environmental contaminant Bisphenol A are also discussed. Together, these data suggest new targets (i.e., adipocyte differentiation and molecular mechanisms involved in weight homeostasis) for abnormal programming by estrogenic chemicals, and provide evidence that support the scientific hypothesis termed “the developmental origins of adult disease”. The proposal of an association of environmental estrogens with obesity and diabetes expands the focus on the diseases from intervention/treatment to include prevention/avoidance of chemical modifiers especially during critical windows of development.
Keywords: metabolic disease, diabetes, xenoestrogens, developmental exposure, endocrine disruptor, Bisphenol A
Introduction
Obesity and overweight are quickly becoming a significant human health problem worldwide (Ogden et al. 2007; Oken E and Gillman MW 2003). The prevalence of obesity has risen dramatically in wealthy industrialized countries over the last 2 to 3 decades, but it is also on the rise in poorer underdeveloped nations. In the United States, the Center for Disease Control (CDC) reported in 2008 that obesity has reached epidemic proportions with more than 60% of adults being either obese or overweight (CDC 2008). Not only is obesity a problem for adults, but it is of particular concern in children since most obese and overweight children grow up to be obese adults. The number of children and adolescents who are considered overweight or at risk for being overweight has increased similarly to adults (Ogden et al. 2002). Obesity has proven to be a challenge to treat effectively once it is established. Further, obesity and overweight are known to have adverse health effects, and to impact the risk and prognosis for a number of serious medical conditions such as Type 2 diabetes, hyperinsulinemia, insulin resistance, coronary heart disease, high blood pressure, stroke, gout, liver disease, asthma and pulmonary problems, gall bladder disease, kidney disease, reproductive problems, osteoarthritis, and some forms of cancer (Collins 2005; Mokdad et al. 2003; Mokdad et al. 1999). Unfortunately, these illnesses are starting to be more frequently reported in obese and overweight children whereas in the past, these were diseases of older adults. Health professionals warn that the current generation of children may be the first in history to experience a shorter life expectancy than their parents due to the impact of obesity-related diseases.
Obesity is caused by a complex interaction between genetic, behavioral, and environmental factors. The most common causes are thought to be overeating high caloric fatty diets combined with a sedentary lifestyle which is imposed on a background of genetic predisposition for the disease. Although much interest has focused on these factors including the need to incorporate healthy foods in our diets and more exercise into our lifestyle, these factors can not solely explain the alarming rise in obesity. Further, it remains puzzling why some people are more successful in dieting and loosing weight than others. Obesity is most definitely a multi-factorial disease. Although many of the causative factors remain unknown, health experts agree that since obesity is so difficult to treat, prevention becomes of the upmost importance.
Until the 1990s, fat cells or “adipocytes” were considered to be just storage depots for excess metabolic fuel. However, following the discovery of an adipocyte-derived hormone termed “leptin” that communicates energy reserve information from adipocytes to other organs of the body including the central nervous system, a new appreciation emerged that these “fat storage cells” actually function as an endocrine organ (Collins 2005). Today it is well accepted that adipocytes have an endocrine function in addition to fat storage. Since the discovery of leptin, evidence has shown that adipocytes secrete many other cytokines and growth factors that play important roles in growth and differentiation of the organism, as well as, in the feedback of information to other endocrine organs. Considering the newly identified endocrine function of adipocytes and recognizing that their endocrine signaling pathways are established during perinatal development, it is of interest to us to investigate whether exposure to endocrine disrupting chemicals (EDCs), in particular those with estrogenic activity, during critical period of development, is related to obesity or any of its associated diseases including diabetes.
It is well established that many environmental chemicals can interfere with complex endocrine signaling pathways and cause adverse consequences in the developing organism (Bern 1992; Colborn et al. 1996). Although concern initially focused on reproductive and carcinogenic effects, we now know that multiple organ systems are affected by EDCs including the cardiovascular and neuro-endocrine systems. Most recently, an association of environmental chemicals with the development of obesity has been proposed (Baillie-Hamilton 2002; Heindel 2003; Heindel and Levin 2005; Newbold et al. 2008; Newbold et al. 2005; Newbold et al. 2007; Newbold et al. 2007) Mechanistic studies have further described the disruptive effects of environmental chemicals on normal adipocyte development, and homeostatic control over adipogenesis and early energy balance (Grun and Blumberg 2006; Grun et al. 2006). Although uncertainties remain about the full extent of health consequences that follow exposure to environmental chemicals, especially low dose exposures that are relevant to the general population, we are just beginning to understand that the complexities and interactions of endocrine signaling mechanisms include adipocytes and weight controlling mechanisms.
The Developmental Origins of Adult Disease
The developing fetus and neonate are uniquely sensitive and can be easily disturbed by exposure to chemicals with hormone-like activity (Bern 1992). The protective mechanisms that are available to the adult such as DNA repair mechanisms, a fully competent immune system, detoxifying enzymes, liver metabolism, and the blood/brain barrier are not fully functional in the fetus or neonate. Further, the developing fetus and neonate have an increased metabolic rate as compared to an adult which in some cases may make them more sensitive to chemical toxicity. Numerous examples document that developmental exposure to certain chemicals during critical periods of differentiation can cause adverse effects; some of these effects may not be apparent until much later in life.
The idea that adult health and disease can have an etiology that arises in fetal or early neonatal life is not exclusive to the field of endocrine disruption and chemical exposures. In the field of maternal nutrition, low birth weight resulting from suboptimal fetal nutrition (the thrifty phenotype hypothesis) and subsequent early adipogenic catchup growth is associated with increased risk of non-communicable diseases, coronary heart disease, type 2 diabetes, osteoporosis, and metabolic dysfunction later in adult life (Barker et al. 2002). Chronic stress has also been associated with similar responses; for example, experimental studies using Macque monkeys demonstrate that early life stress results in enhanced visceral fat deposition and increased incidences of metabolic diseases later in life (Kaufman et al. 2007). Maternal smoking is another fetal stressor which has been linked to the subsequent development of obesity and disease later in life. Many of these topics are discussed in further detail by other authors in this series. Taken together, these findings have lead to the “developmental origins of health and disease (DOHaD)” paradigm in which a substantial research effort has focused on perinatal influences and subsequent chronic disease (Gluckman and Hanson 2004).
Thus, both the fields of endocrine disruptor toxicology and maternal nutrition have independently provided numerous examples documenting perinatal factors can alter the developing organism and cause long-term effects in the adult. Exposure to the estrogenic chemical diethylstilbestrol (DES), a well-known perinatal carcinogen, is one example of DOHaD effects [for review, see (Bern 1992; NIH 1999)]. Although DES is often referred to as a potent estrogenic chemical due to its high binding affinity to estrogen receptor (ER) alpha as compared to estradiol, very low doses of DES (in the range of .001 micrograms/kg) can be used to study the effects of weaker binding environmental chemicals with estrogenic activity. Further, DES binds with similar affinity to ER beta as some phytoestrogens and it binds to a newly described membrane bound receptor similar to Bisphenol A (BPA) (Welshons et al. 2006). Thus, DES has numerous properties that recommend it as a good model compound to study the effects of environmental estrogenic chemicals.
The Developmental Exposed DES Animal Model to Study Obesity
DES, a potent synthetic estrogen, was widely prescribed to pregnant women from the 1940s through the 1970s with the mistaken belief that it could prevent threatened miscarriages. It was estimated that a range of 2 to 8 million pregnancies worldwide were exposed to DES. Today, it is well known that prenatal DES treatment resulted in a low but significant increase in neoplastic lesions, and a high incidence of benign lesions in both the male and female offspring exposed during fetal life. To study the mechanisms involved in DES toxicity, we developed experimental mouse models of perinatal (prenatal or neonatal) DES exposure over 30 years ago (Newbold 1995). Outbred CD-1 mice were treated with DES by subcutaneous injections on days 9–16 of gestation (the period of major organogenesis in the mouse) (McLachlan et al. 1980) or days 1–5 of neonatal life (Newbold 2004; Newbold et al. 1990) (a period of cellular differentiation of the reproductive tract, and a critical period of immune, behavioral, and adipocyte differentiation). These perinatal DES animal models have successfully duplicated, and in some cases, predicted, many of the alterations (structural, function, cellular and molecular) observed in similarly DES- exposed humans.
Although our major focus was initially on reproductive tract abnormalities and subfertility/infertility, we also examined the relationship of perinatal DES treatment with the development of obesity later in life. We sought to determine if DES was an obesogen as well as a reproductive toxicant, and if so, what were its molecular targets and the mechanisms through which it might act. For our obesity experiments, mice were treated with DES on days 1–5 of neonatal life using a low dose of 0.001 mg/day (1 μg/kg/day); this dose did not affect body weight during treatment but was associated with a significant increase in body weight as adults. Figure 1A is a representative photomicrograph of control and neonatal DES treated female mice at 4–6 months of age; male mice treated as neonates did not demonstrate this increase in body weight (Newbold et al. 2008). Unlike the lower dose of DES (0.001 mg/day = 1 μg/kg/day), a higher dose of DES (1000 μg/kg/day =1 mg/kg/day) caused a significant decrease in body weight during treatment which was followed by a “catch up” period around puberty and then finally resulted in an increase in body weight of the DES treated mice compared to controls after ~2 months of age. This is reminiscent of the thrifty phenotype described earlier for humans. Figure 2 shows the weight patterns of DES treated (1000 μg/kg/day =1 mg/kg/day) mice and their corresponding controls. Additional studies indicated that the increase in body weight in these DES-exposed mice was associated with an increase in the percent of body fat as determined by mouse densitometry (Lunar PIXIMUS, GE Healthcare, Waukesha, Wi) (Figure 1B and Table 1).
Figure 1. Representative photograph of Control and DES-treated Mice.

A. Photograph shows the difference in body size of the two groups at ~ 6months of age. B. Images of Control and DES treated mice as generated by Piximus densitometry. Note that the DES mouse is much larger than the control at 6 months of age. Image was published in (Newbold et al. 2005)
Figure 2. Body Weight of Mice Following DES Exposure.
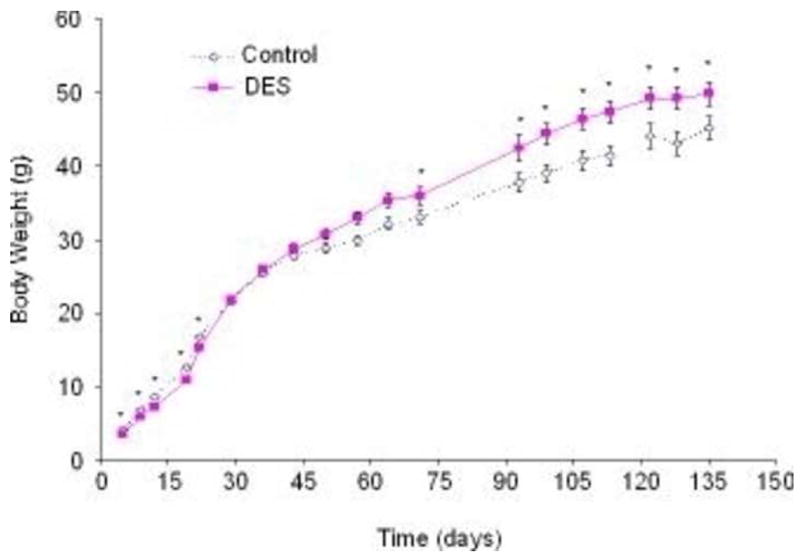
Body weights in grams plotted on the y axis were measured at various ages indicated on the x axis. Mice treated with DES (1000 μg/kg/day =1 mg/kg/day) had significantly lower body weights during treatment as compared to controls. By1 month, DES mice caught up to controls and by 2 months, they had surpassed the controls and gained a significantly higher body weight (n+16 mice per group); numbers are the mean ± S.E.M.; * denotes significance using ANOVA followed by Dunnett’s test (p <0.05). Data was published in (Newbold et al. 2007)
Table 1.
Increased body fat in mice treated neonatally with DES as determined by PIXImus™ Mouse Densitometry.
|
2 months | ||||
|---|---|---|---|---|
| Treatmenta | Estimated Body Weight (g)b | Estimated Fat Weight (g) | % Fat | % Lean |
| Control | 26.8±0.7 | 5.7±0.3 | 21.2±0.8 | 78.8±0.8 |
| DES | 29.3±0.9* | 6.3±0.3 | 21.4±0.7 | 78.6±0.7 |
|
6 months | ||||
| Treatmenta | Estimated Body Weight (g)b | Estimated Fat Weight (g) | % Fat | % Lean |
|
| ||||
| Control | 37.9±2.5 | 11.5±1.3 | 29.9±1.8 | 70.1±0.8 |
| DES | 45.0±2.5 * | 18.4±1.8 * | 40.8±1.4 * | 59.2±1.8* |
Mice were treated on days 1–5 with DES 1 mg/kg; n= 4–8 mice per group.
Using the PIXImus™ mouse densitometer, an estimate of the body weight was obtained at 2 and 6 months of age; this measurement did not include the head region since the mice were so large, the entire animal could not be screened at one time.
p<0.05 using ANOVA followed by Dunnett’s test.
Data was previously published in Newbold et al, 2007.
Increased body weight in all DES treated mice, both low and high doses, was maintained throughout adulthood; however, by 18 months of age, statistical differences in body weight between DES treated and controls were difficult to show because individual animal variability within groups increased so much as they aged (data not included). Since various doses of DES resulted in obesity whether or not pups were underweight during treatment, most likely multiple pathways are involved in programming for obesity by environmental estrogens.
Densitometry images suggested DES treated mice had excessive abdominal fat (Fig. 1B) which had been previously reported to be associated with cardiovascular disease and diabetes (Gillum 1987), therefore, we determined the weights of various fat pads to determine if specific fat pads were affected by DES treatment or whether it was a generalized change throughout the mouse. Fat pad weights were compared in DES treated mice (1000 μg/kg/day =1 mg/kg/day) and controls at 6–8 months of age; inguinal, parametrial, gonadal, and retroperitoneal fat pads were all increased in DES treated mice as compared to controls, but, brown fat weights were not significantly different at this age (Table 2)(Newbold et al. 2005).
Table 2.
Fat Pad Weights of Mice Treated Neonatally with DES
| Treatment a | Inguinal Fat (g) | Parametrial Fat (g) | Gonadal Fat (g) | Retroperitoneal Fat (g) |
|---|---|---|---|---|
| Control | 0.108 ± 0.015 | 1.744 ± 0.211 | 0.608 ± 0.129 | 0.291 ± 0.029 |
| DES | 0.207 ± 0.041 * | 2.084 ± 0.254 | 0.667 ± 0.071 | 0.555 ± 0.080 * |
Mice were treated on days 1–5 with DES 1 mg/kg and sacrificed at 6–8 months of age. n=8 per group
p<0.05 using ANOVA followed Dunnett’s test.
Data was previously published in Newbold et al. 2007.
Although DES- treated mice were not statistically different in weight to controls at 2 months of age, DES (1000 μg/kg/day =1 mg/kg/day) mice exhibited elevated serum levels of leptin, adiponectin, IL-6, and triglycerides prior to becoming overweight and obese. This suggests that these endpoints may be important early markers of subsequent adult disease. The elevated levels of leptin are not surprising considering the increase number and size of the adipocytes in the DES treated mice but the increase in adiponectin was not expected since low levels usually correlate with diabetes. However, this may indicate insensitivity to these hormones and/or a loss of the negative feedback mechanisms that regulate adipogenesis. At 6 months of age, insulin and all of the serum markers except triglycerides were found to be significantly elevated as compared to controls (Table 3) (Newbold et al. 2007).
Table 3.
Serum Profiles of Mice Treated Neonatally with DES
| Treatment a |
|||
|---|---|---|---|
| 2 months | Control | DES | |
| Leptin (ng/ml) | 4.8 ± 0.5 | 25.0 ± 1.4 * | |
| Adiponectin (μg/ml) | 6.6 ± 0.6 | 38.1 ± 3.6 * | |
| IL-6 (pg/ml) | 6.3 ± 0.9 | 60.4 ± 5.0 * | |
| Insulin (μU/ml) | 7.4 ± 0.7 | 1.3 ± 0.3 * | |
| Triglycerides (mg/ml) | 97.6 ± 3.2 | 122.9 ± 3.5 * | |
|
|
|||
| 6 months | Control | DES | |
|
|
|||
| Leptin (ng/ml) | 8.1 ± 0.4 | 60.7 ± 2.3 * | |
| Adiponectin (μg/ml) | 9.3 ± 0.6 | 39.2 ± 1.6 * | |
| IL-6 (pg/ml) | 10.1 ± 0.4 | 93.8 ± 1.9 * | |
| Insulin (μU/ml) | 8.6 ± 0.3 | 10.8 ± 0.3 * | |
| Triglycerides (mg/ml) | 116.9 ± 1.7 | 106.8 ± 1.5 | |
|
|
|||
Mice were treated on days 1–5 with DES 1 mg/kg; n= 8 per group at 2 months and 16 per group at 6 months of age.
p<0.05 using ANOVA followed by Dunnett’s test.
Data was previously published in Newbold et al, 2007.
Glucose levels were also measured in DES (1000 μg/kg/day =1 mg/kg/day) and control mice prior to the development of obesity at approximately 2 months of age (Newbold et al. 2007). Interestingly, 25% of the DES-treated mice had significantly higher glucose levels than controls; these mice also showed a slower clearance rate of glucose from the blood since higher levels were seen throughout the experiment (Newbold et al. 2007). It is important to note that altered glucose levels were observed in these mice before they developed excessive weight. Additional glucose measurements in older mice may help determine if a higher percentage of mice are affected with age, and if higher and sustained levels of glucose can be demonstrated. To date, however, our data suggest that overweight and obesity observed in perinatal DES- treated mice will be associated with the development of diabetes, similar to the association of obesity with diabetes in humans. Earlier studies from our laboratory support a role for altered glucose metabolism since we have shown a high prevalence of islet cell hyperplasia in the pancreas of mice exposed to DES or other environmental estrogens including BPA and genistein treated mice (unpublished).
Since, the balance of activity levels and food intake are known contributors to obesity, activity was measured in DES (1000 μg/kg/day =1 mg/kg/day) and control mice at 2 months of age before a difference in body weight could be detected. Individual mice were placed in an Opto-Max motor activity chamber (Columbus Instruments, Columbus, OH) and their ambulatory activity measured. Overall, there was no statistical difference in this parameter between the two groups although the DES group showed less movement as compared to controls as the experiment progressed. This difference, however, was not sufficient to explain the enhanced weight gain in DES mice as they age (Newbold et al. 2007). Additional measures of activity including the running wheel measured during the dark photoperiod are being determined and are necessary before the role of activity in the development of obesity can be fully accessed.
Feed consumption was also measured in control and DES-treated mice (1000 μg/kg/day =1 mg/kg/day). DES-treated mice consumed more than controls over the course of the experiment (~3 grams more), but the amounts were not statistically different between the groups (Newbold et al. 2007). Taken into account, both the marginal decrease in activity and the increase in food intake in DES treated mice as compared to controls, it is unlikely these two measurements can solely explain the development of obesity in DES treated mice.
A recent study describes a role for developmental genes in the origins of obesity and body fat distribution in mice and humans (Gesta et al. 2006). Therefore, exposure to environmental chemicals with hormonal activity may be altering gene expression involved in programming adipocytes during development. Several genes have been implicated in altering adipocyte differentiation and function such as Hoxa5, Gpc4 and Tbx15 and fat cell distribution such as Thbd, Nr2f1 and Sfrp2. We investigated changes in gene expression by microarray analysis in uterine samples from DES treated mice (1000 μg/kg/day =1 mg/kg/day) compared to controls at 19 days of age. In these samples, genes involved in adipocyte differentiation were not altered in the uterus following neonatal DES exposure, however, genes involved in fat distribution were. Thbd and Nr2f1 were significantly down regulated and Sfrp2 was significantly up regulated in DES treated uteri compared to controls (data taken from study published by (Newbold et al. 2007). These findings support the idea that environmental estrogens may play a role in regulating the expression of obesity-related genes in development.
Although the data summarized in this review describes only neonatal exposure to a high dose of DES, lower doses and exposure during prenatal life have also been shown to be associated with obesity later in life. Interestingly, high prenatal DES doses caused lower birth weight compared to controls, followed by a “catch-up period”, and finally resulted in obesity; low prenatal DES doses had no effect on birth weight but it still resulted in obesity later in life (Newbold et al. 2005). Thus, it appears that the effects of DES on adipocytes may depend on the time of exposure and the dose, and that multiple mechanisms maybe altered resulting in the same obesity phenotype.
Other Environmental Estrogens and EDCs Role in Obesity
In 2002, Baillie-Hamilton postulated a role for chemical toxins in the etiology of obesity by showing that the obesity epidemic coincided with the marked increase of industrial chemicals in the environment over the past 40 years (Baillie-Hamilton 2002). She further speculated that the current obesity epidemic could not be explained solely by alterations in food intake and/or decrease in physical activity. She cited numerous studies where chemicals including pesticides, organophosphates, polychlorinated biphenyls, polybrominated biphenyls, phthalates, Bisphenol A, heavy metals, and some solvents caused weight gain, and proposed that these chemicals were interfering with weight homeostasis by altering weight–controlling hormones, altering sensitivity to neurotransmitters, or altering activity of the sympathetic nervous system (Baillie-Hamilton 2002). It is interesting that in a few of the studies she cited, the chemicals were actually designed to have growth-promoting properties such as with DES, which was widely used by the livestock industry specifically for this role (Raun and Preston 2002).
Since the Baille-Hamilton review (Baillie-Hamilton 2002), an increasing number of studies has been specifically designed to address the effects of environmental chemical exposure on weight gain and loss. Numerous studies have shown that exposure to numerous EDCs during critical periods of differentiation, at low environmentally-relevant doses, can alter developmental programming resulting in obesity. Figure 3 shows the body weight increase associated with developmental exposure to some estrogenic chemicals that we have studied in our lab.
Figure 3. Neonatal Exposure to Various Environmental Estrogens Causes Obesity Later in Life.

Neonatal treatment was on days 1–5; Panel A: 6 month old animals: 2OH estradiol (20mg/kg/day) and 4OH estradiol (0.1 mg/kg/day) compared to their age matched controls; Panel B: 4 month old animals: DES (0.001mg/kg/day) and genistein (50 mg/kg/day); doses were chosen to be approx. equal in estrogenic activity as determined by the immature mouse uterotropic assay. Data is summarized from (Newbold et al. 2005).
Phytoestrogens, contained in various food and food supplements, in particular soy products, are another class of chemicals that are receiving attention. Genistein and daidzein are two of the most abundant phytoestrogens in the human diet and, genistein, because of its estrogenic activity, has been proposed to have a role in the maintenance of health by regulating lipid and carbohydrate homeostasis. However, a recent study showed that genistein at pharmacologically high doses did indeed inhibit adipose deposition but, at low doses similar to that found in Western and Eastern diets, in soy milk, or in food supplements containing soy, it induced adipose tissue deposition especially in males. Further, this increase in adipose tissue deposition by genistein was correlated with mild peripheral insulin resistance. Interestingly, like our findings with DES, genistein did not significantly affect food consumption (Penza et al. 2006) suggesting an abnormal programming of factors involved in weight homeostasis. Phytoestrogens are further discussed in detail in another chapter.
Another novel and interesting class of chemicals is called organotins which includes persistent organic pollutants with endocrine-disrupting properties. Tributyl tin chloride and triphenyl tin chloride have been identified as nanomolar agonist ligands for retinoid X receptor (RXR) and peroxisome proliferator–activated receptor γ (PPARγ), nuclear receptors that play important roles in lipid homeostasis and adipogenesis; tributyl tin (TBT) was shown to disrupt normal development and homeostatic controls over adipogenesis and energy balance, resulting in obesity (Grun et al. 2006);. Interestingly, TBT was shown to cause permanent physiological changes in both male and female mice exposed during prenatal life resulting in a predisposition to weight gain. The mechanisms describing how these chemicals actually program for the development of obesity was eloquently described (Tabb and Blumberg 2006) and is also covered in detail in another chapter in this series.
In vitro studies using 3T3-L1 cells (mouse fibroblasts that can differentiate into adipocytes) also show a link between environmental chemicals including TBT(Inadera and Shimomura 2005), Bisphenol A (Masuno et al. 2005; Masuno et al. 2002; Sakurai et al. 2004), nonylphenol (Masuno et al. 2003), and genistein (Dang et al. 2003) in the development of overweight and obesity. Studies of pancreatic cells in both primary culture and in vivo also suggest that environmentally- relevant doses of Bisphenol A and DES affect the normal physiology of the endocrine pancreas by altering the regulation of glucose and lipid metabolism (Alonso-Magdalena et al. 2005; Alonso-Magdalena et al. 2006).
Summary and Conclusions
The data included in this review supports the idea that brief exposure, early in development to environmental chemicals with estrogenic activity, increases body weight gain with age and alters markers predictive of obesity in experimental animals. Epidemiology studies support the findings in experimental animals and show a link between exposure to environmental chemicals (such as PCBs, DDE, and persistent organic pollutants) and the development of obesity (Smink et al. 2008). Furthermore, the use of soy-based infant formula containing the estrogenic component genistein has been positively associated with obesity later in life (Stettler et al. 2005). Using the DES animal model as an important research tool to study “obesogens”, the mechanisms involved in altered weight homeostasis (direct and /or endocrine feedback loops, i.e., ghrelin, leptin, etc.) by environmental estrogens can be elucidated. In addition, hopefully this animal model may shed light on areas of prevention. Public health risks can no longer be based on the assumption than overweight and obesity are just personal choices involving the quantity and kind of foods we eat combined with inactivity, but rather that complex events including exposure to environmental chemicals during development may be contributing to the obesity epidemic.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. The authors gratefully acknowledge Dr. Terry Phillips, Division of Bioengineering and Physical Sciences, Office of Research Services, Office of the Director, NIH, DHHS, Bethesda, MD for the serum measurements. The authors would also like to thank Ms. Sherry Grissom for the microarray analysis of gene expression changes in the uterus following neonatal DES treatment and Mr. Ryan Snyder for his technical expertise in carrying out this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, Soria B, et al. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect. 2005;113(8):969–977. doi: 10.1289/ehp.8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ Health Perspect. 2006;114(1):106–112. doi: 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8(2):185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31(6):1235–1239. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- Bern B. The Fragil Fetus. Princeton, NJ: Princeton Scientific Publishing Co; 1992. [Google Scholar]
- CDC. Report on overweight and obesity. Centers for Disease Control and Prevention; 2008. http://www.cdc.gov/nccdphp/dnpa/obesity. [Google Scholar]
- Colborn T, Dumanoski D, Myers JP. Our Stolen Future. Penguin Books USA, Inc; 1996. [Google Scholar]
- Collins S. Overview of clinical perspectives and mechanisms of obesity. Birth Defects Res A Clin Mol Teratol. 2005;73(7):470–471. doi: 10.1002/bdra.20140. [DOI] [PubMed] [Google Scholar]
- Dang ZC, Audinot V, Papapoulos SE, Boutin JA, Lowik CW. Peroxisome proliferator-activated receptor gamma (PPARgamma ) as a molecular target for the soy phytoestrogen genistein. J Biol Chem. 2003;278(2):962–967. doi: 10.1074/jbc.M209483200. [DOI] [PubMed] [Google Scholar]
- Gesta S, Bluher M, Yamamoto Y, Norris AW, Berndt J, Kralisch S, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A. 2006;103(17):6676–6681. doi: 10.1073/pnas.0601752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillum RF. The association of the ratio of waist to hip girth with blood pressure, serum cholesterol and serum uric acid in children and youths aged 6–17 years. J Chronic Dis. 1987;40(5):413–420. doi: 10.1016/0021-9681(87)90174-3. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004;15(4):183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147(6 Suppl):S50–55. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20(9):2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- Heindel JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci. 2003;76(2):247–249. doi: 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- Heindel JJ, Levin E. Developmental origins and environmental influences--Introduction. NIEHS symposium. Birth Defects Res A Clin Mol Teratol. 2005;73(7):469. doi: 10.1002/bdra.20141. [DOI] [PubMed] [Google Scholar]
- Inadera H, Shimomura A. Environmental chemical tributyltin augments adipocyte differentiation. Toxicol Lett. 2005;159(3):226–234. doi: 10.1016/j.toxlet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Kaufman D, Banerji MA, Shorman I, Smith EL, Coplan JD, Rosenblum LA, et al. Early-life stress and the development of obesity and insulin resistance in juvenile bonnet macaques. Diabetes. 2007;56(5):1382–1386. doi: 10.2337/db06-1409. [DOI] [PubMed] [Google Scholar]
- Masuno H, Iwanami J, Kidani T, Sakayama K, Honda K. Bisphenol a accelerates terminal differentiation of 3T3-L1 cells into adipocytes through the phosphatidylinositol 3-kinase pathway. Toxicol Sci. 2005;84(2):319–327. doi: 10.1093/toxsci/kfi088. [DOI] [PubMed] [Google Scholar]
- Masuno H, Kidani T, Sekiya K, Sakayama K, Shiosaka T, Yamamoto H, et al. Bisphenol A in combination with insulin can accelerate the conversion of 3T3-L1 fibroblasts to adipocytes. J Lipid Res. 2002;43(5):676–684. [PubMed] [Google Scholar]
- Masuno H, Okamoto S, Iwanami J, Honda K, Shiosaka T, Kidani T, et al. Effect of 4-nonylphenol on cell proliferation and adipocyte formation in cultures of fully differentiated 3T3-L1 cells. Toxicol Sci. 2003;75(2):314–320. doi: 10.1093/toxsci/kfg203. [DOI] [PubMed] [Google Scholar]
- McLachlan JA, Newbold RR, Bullock BC. Long-term effects on the female mouse genital tract associated with prenatal exposure to diethylstilbestrol. Cancer Research. 1980;40:3988–3999. [PubMed] [Google Scholar]
- Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003;289(1):76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the United States, 1991–1998. Jama. 1999;282(16):1519–1522. doi: 10.1001/jama.282.16.1519. [DOI] [PubMed] [Google Scholar]
- Newbold R. Lessons Learned from Perinatal Exposure to Diethylstilbestrol (DES) Toxicology and Applied Pharmacology. 2004;199:142–150. doi: 10.1016/j.taap.2003.11.033. [DOI] [PubMed] [Google Scholar]
- Newbold RR. Cellular and molecular effects of developmental exposure to diethylstilbestrol: implications for other environmnetal estrogens. Environmental Health Perspectives. 1995;103(7):83–87. doi: 10.1289/ehp.95103s783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold RR, Bullock BC, McLachlan JA. Uterine adenocarcinoma in mice following developmental treatment with estrogens: a model for hormonal carcinogenesis. Cancer Research. 1990;50(23):7677–7681. [PubMed] [Google Scholar]
- Newbold RR, Jefferson WN, Grissom SF, Padilla-Banks E, Snyder RJ, Lobenhofer EK. Developmental exposure to diethylstilbestrol alters uterine gene expression that may be associated with uterine neoplasia later in life. Mol Carcinog. 2007;46(9):783–796. doi: 10.1002/mc.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Jefferson WN, Heindel JJ. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31(2):201–208. doi: 10.1111/j.1365-2605.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73(7):478–480. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Perinatal exposure to environmental estrogens and the development of obesity. Mol Nutr Food Res. 2007;51(7):912–917. doi: 10.1002/mnfr.200600259. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Padilla-Banks E, Snyder RJ, Phillips TM, Jefferson WN. Developmental exposure to endocrine disruptors and the obesity epidemic. Reprod Toxicol. 2007;23(3):290–296. doi: 10.1016/j.reprotox.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. DES Research Update. Bethesda, MD: 1999. NIH Publication No. 00-4722. [Google Scholar]
- Ogden CL, Flegal KM, Carroll MD, Johnson CL. Prevalence and trends in overweight among US children and adolescents, 1999–2000. Jama. 2002;288(14):1728–1732. doi: 10.1001/jama.288.14.1728. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132(6):2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Oken E, Gillman MW. Fetal origins of obesity. Obes Res. 2003;11:496–506. doi: 10.1038/oby.2003.69. [DOI] [PubMed] [Google Scholar]
- Penza M, Montani C, Romani A, Vignolini P, Pampaloni B, Tanini A, et al. Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology. 2006;147(12):5740–5751. doi: 10.1210/en.2006-0365. [DOI] [PubMed] [Google Scholar]
- Raun A, Preston R. History of diethylstilbestrol use in livestock. 2002 http://wwwasasorg/oldsite/Bios/Raunhistpdf.
- Sakurai K, Kawazuma M, Adachi T, Harigaya T, Saito Y, Hashimoto N, et al. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br J Pharmacol. 2004;141(2):209–214. doi: 10.1038/sj.bjp.0705520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smink A, Ribas-Fito N, Garcia R, Torrent M, Mendez MA, Grimalt JO, et al. Exposure to hexachlorobenzene during pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr. 2008;97(10):1465–1469. doi: 10.1111/j.1651-2227.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- Stettler N, Stallings VA, Troxel AB, Zhao J, Schinnar R, Nelson SE, et al. Weight gain in the first week of life and overweight in adulthood: a cohort study of European American subjects fed infant formula. Circulation. 2005;111(15):1897–1903. doi: 10.1161/01.CIR.0000161797.67671.A7. [DOI] [PubMed] [Google Scholar]
- Tabb MM, Blumberg B. New modes of action for endocrine-disrupting chemicals. Mol Endocrinol. 2006;20(3):475–482. doi: 10.1210/me.2004-0513. [DOI] [PubMed] [Google Scholar]
- Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147(6 Suppl):S56–69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]