

Abstract
Aims
Long QT syndrome (LQTS) is a heterogeneous collection of inherited cardiac ion channelopathies characterized by a prolonged electrocardiogram QT interval and increased risk of sudden cardiac death. β-Adrenergic blockers are the mainstay of treatment for LQTS. While their efficacy has been demonstrated in LQTS patients harbouring potassium channel mutations, studies of β-blockers in subtype 3 (LQT3), which is caused by sodium channel mutations, have produced ambiguous results. In this modelling study, we explore the effects of β-adrenergic drugs on the LQT3 phenotype.
Methods and results
In order to investigate the effects of β-adrenergic activity and to identify sources of ambiguity in earlier studies, we developed a computational model incorporating the effects of β-agonists and β-blockers into an LQT3 mutant guinea pig ventricular myocyte model. β-Activation suppressed two arrhythmogenic phenomena, transmural dispersion of repolarization and early after depolarizations, in a dose-dependent manner. However, the ability of β-activation to prevent cardiac conduction block was pacing-rate-dependent. Low-dose β-blockade by propranolol reversed the beneficial effects of β-activation, while high dose (which has off-target sodium channel effects) decreased arrhythmia susceptibility.
Conclusion
These results demonstrate that β-activation may be protective in LQT3 and help to reconcile seemingly conflicting results from different experimental models. They also highlight the need for well-controlled clinical investigations re-evaluating the use of β-blockers in LQT3 patients.
Keywords: Arrhythmia (mechanisms), Adrenergic (ant)agonists, Long QT syndrome, Computer modelling
1. Introduction
Inherited long QT syndrome (LQTS) is a cardiac genetic disease that leads to a prolonged QT interval and increased arrhythmia susceptibility. β-Adrenergic blocking drugs have been shown to suppress arrhythmias in patients with LQT subtypes 1 and 2,1–5 which are caused by mutations in cardiac potassium channels.6–8 However, trials of β-blockers in patients with subtype 3 of LQTS (LQT3) have not yielded the same beneficial results.
The pathophysiology of LQT3 differs from types 1 and 2, in that a mutation in the SCN5A-encoded sodium channel leads to sustained depolarizing sodium current that prolongs the action potential.9–11 One study comparing cardiac event rates before and after β-blocker treatment initiation in a small group of LQT3 patients found no significant difference in rates,4 and a second trial found a sustained high cardiac event rate during β-blocker treatment.5 However, these studies had very low numbers of LQT3 subjects; therefore, definitive conclusions about the benefit or harm of β-blocker treatment of LQT3 patients could not be established.
Investigations of the effects of the β-adrenergic agonist isoproterenol on isolated guinea pig myocytes12 and canine wedge preparations13 treated with toxins to simulate LQT3 have shown that β-adrenergic stimulation can induce a beneficial decrease in two factors thought to contribute to arrhythmogenesis: prolonged action potential duration and increased transmural dispersion of repolarization (TDR). The β-blocker propranolol was shown to reverse these benefits.13 Studies of isoproterenol-induced ventricular tachycardia (VT) in mouse models of the ΔKPQ LQT3 mutation have produced varying results. One study showed that isoproterenol can protect against the development of VT,14 whereas a second study showed no effect of isoproterenol on VT induction.15 Importantly, the pacing protocols used in these two studies differed substantially. Collectively, these studies suggest that in single cells and isolated tissue, β-adrenergic simulation may be beneficial in LQT3. However, whole mouse heart studies have demonstrated ambiguous results in regards to the effects of β-adrenergic activity on VT induction.14,15
In order to further investigate the efficacy of β-modulation in LQT3 and to identify sources of ambiguity in earlier studies, we have developed a computational model that incorporates a description of the human ΔKPQ LQT3 mutation and the effects of β-adrenergic stimulation and blockade into the Faber–Rudy16 model of the guinea pig cardiac ventricular myocyte. This model enables investigation of the pharmacology and pathophysiology of LQT3 mutant channels in the context of a human-like action potential.
Theoretical modelling approaches enable a comprehensive examination of a variety of factors such as pacing rates and ligand concentrations that may have contributed to the conflicting results obtained by different groups investigating VT in mouse models of LQT3. Also, by modelling a variety of drug doses, the dose-dependency of drug-induced phenomenon can be explored and the differential effects of on-target and off-target drug binding can be separated. Specifically, it has been shown that high-dose propranolol can block sodium channels17–19 in addition to its well-established β-blocking effects.
The simulations presented in this study demonstrate that β-adrenergic activation can decrease the susceptibility of model of LQT3 cells and transmural fibres to two major hypothesized arrhythmogenic triggers:20 early after depolarizations (EADs) and increased TDR. However, the ability of isoproterenol to prevent conduction block was dependent on the pacing method used to evoke block. Finally, the β-blocker propranolol was only efficacious in the treatment of LQT3 when given at a dose that was high enough to produce off-target blockade of sodium channels.
2. Methods
2.1. Single cell model
The Faber–Rudy16 version of the Luo–Rudy21 guinea pig model incorporating the Dumaine22 model of the transient outward current, Ito, was used as the basic model. A guinea pig model was chosen so that our simulated action potentials could be validated by comparison to the guinea pig experimental results reported in ref. 12. Also, studies have demonstrated that the model is capable of realistically producing LQT3 mutation-induced EADs,23 unlike some other ventricular myocyte models.24,25 Epicardial, mid-myocardial, and endocardial cells were created by varying the maximal conductance of the slow outward potassium current, IKs, by a ratio of 0.433:0.125:0.289 (Endo:Mid:Epi).26 Ito was varied by a ratio of 0:0.2125:0.25 (Endo:Mid:Epi).22,27 A previously published Markov description of the ΔKPQ mutant sodium channel (or a wild-type channel)28 was incorporated into the model. (For a review of mutation modelling, please see ref. 29.)
2.2. Isoproterenol model
A comprehensive model of β-adrenergic signalling in cardiac myocytes30 was used to evaluate the effects of isoproterenol on different sarcolemmal currents. The model computed the fraction of phosphorylated:non-phosphorylated L-type calcium channels (FracICa,L-p) and phosphorylated:non-phosphorylated outward potassium channels (FracIK-p) in response to a given concentration of isoproterenol. (The fractions of rapid and slow outward potassium, IKr and IKs, phosphorylation were set to be equivalent.31) Baseline FracICa,L-p was assumed to be 0.23, whereas baseline FracIK-p was assumed to be 0.13. These fractions were then used to compute scaling factors that modify the corresponding Faber–Rudy currents to account for influences of β-adrenergic activation.23,32
The scaling factor A was calculated using:23
| 1 |
The ratio of the maximum channel conductance in the phosphorylated to non-phosphorylated state for potassium channels (gK-p:gK-np) was 1.6523. Scaling factor A was used to modify the standard IKr and IKs equations as follows:
| 2 |
| 3 |
where IKr* and IKs* are the currents that have been modified to incorporate the effects of isoproterenol.
Scaling factor B was calculated using:
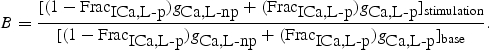 |
4 |
The ratio of maximum channel conductance in the phosphorylated to non-phosphorylated state for calcium channels (gCa-p:gCa-np) was 1.62. Scaling factor B was used to modify the standard ICa,L equation as follows:
| 5 |
where ICa,L* is the L-type calcium current that has been modified to incorporate the effects of isoproterenol.
2.3. Propranolol model
In order to investigate the effects of propranolol on the cardiac ventricular myocyte, two concentrations of propranolol were modelled. At a low dose (33 nM), 50% of the maximal cAMP generation through the β-adrenergic receptor is suppressed.33 This dosage was simulated by calculating the half-maximal cAMP value in response to 1 µM isoproterenol, and then computing factors A and B that correspond to this cAMP value. High-dose (3 µM) and very high-dose (33 µM) propranolol models were also created. At high and very high doses, complete β-adrenergic receptor blockade was assumed.
The sodium channel blocking effects of propranolol were also incorporated into the Markov model of the ΔKPQ sodium current. It has been reported in an abstract18 that propranolol acts as both an inactivated-state (tonic) sodium channel blocker (Kd = 37.6 µM) and an open-state (use-dependent) sodium channel blocker (Kd = 2.9 µM). Using an approach that has previously been used to model sodium channel block by other drugs,34,35 both tonic and use-dependent blocks were incorporated into the model. New Markov states resulting from the block of inactivated channels and open channels were added to model inactivated-state and open-state blocks, respectively. The forward transition rates for each type of block are the same and limited by diffusion of the drug (1 × 108 M−1 s−1) and are given by: [Propranolol] * 1 × 105 ms−1. The reverse transition rates differ between the types of block and are given by: Kd * 1 × 105 ms−1.
Action potentials were evoked with suprathreshold stimuli of 0.5 ms duration. For numerical integration of the model equations, we used a forward Euler scheme with a time step of 0.01 ms.
2.4. Transmural cable simulations
One-dimensional transmural fibre simulations were performed. The 1.65 cm cable was constructed of the model cells described above. One hundred and sixty-five cells were linked together by gap junctions. Cells 1–60 were endocardial, 61–105 were mid-myocardial, and 105–165 were epicardial. Gap junction conduction was 1.73 µS throughout the cable except at the M-cell to epicardial cell transition zone where there was a five-fold decrease in gap junction conductance in cells 104–107.27 Simulations were solved as in ref. 36. TDR in the one-dimensional cables was measured by taking the difference between the action potential durations (APDs) of cell 83 (in the middle of the M-cell region) and cell 154 (in the epicardial region). All cables were paced from the endocardium for at least 100 beats prior to the start of each experiment.
3. Results
3.1. Effects of isoproterenol on APD and currents
To demonstrate the effects of both the ΔKPQ mutation and isoproterenol, we simulated wild-type, ΔKPQ, and ΔKPQ/isoproterenol-treated model endocardial cells at a basic cycle length (BCL) of 500 ms and measured action potential duration and major currents (Figure 1). The ΔKPQ mutation prolonged the APD to 90% repolarization (APD90) from the wild-type 158.3 to 179.1 ms. One micromolar of isoproterenol shortened the mutant APD90 to 165.9 ms (Figure 1A). Performing our simulations in a guinea pig ventricular model allowed us to confirm that these findings are consistent with earlier experimental data from isolated toxin-treated guinea pig myocytes.12 In our model, the ΔKPQ mutation produced a persistent sodium current that was absent in wild-type cells (Figure 1B). This sodium current prolongs the action potential. Peak ICa,L, IKr, and IKs are all increased by treatment with 1 µM isoproterenol (Figure 1C–E). A recent investigation of a cellular model of LQT3 demonstrated that augmentation of IKr could be anti-arrhythmic.37
Figure 1.

Isoproterenol can reverse ΔKPQ mutation-induced action potential durations (APD) prolongation and early after depolarization (EAD) formation. (A–E) Wild-type (grey), ΔKPQ (black), and 1 µM isoproterenol-treated ΔKPQ (dashed) endocardial model myocytes were paced at a basic cycle length (BCL) of 500 ms for 200 beats. APD was prolonged in the mutant cell; this prolongation was partially reversed through treatment with isoproterenol (A). The ΔKPQ mutation causes a late sodium current (B). ICa,L (C), IKr (D), and IKs (E) are all affected by isoproterenol treatment. (F–H) Mid-myocardial (black), endocardial (red), and epicardial (blue) cells were paced with a dynamic restitution protocol in the presence (dashed) and absence (solid) of 1 µM isoproterenol. Wild-type (F), heterozygous ΔKPQ mutant (G), and homozygous ΔKPQ mutant (H) cells were examined. Pacing began at a BCL of 1500 ms and was decreased by 100 ms after 50 beats at each BCL until a final BCL of 400 ms was reached. Only BCLs where no EADs occurred are plotted. Conditions where an EAD occurred were further examined by performing a second pacing protocol that began at the last tested BCL where EADs occurred. A BCL range of 100 ms was then probed in 10 ms increments with 50 beats at each step.
3.2. Isoproterenol decreases APD90 and suppresses the development of early after depolarizations
Prolongation of the action potential in LQT3 cells was more pronounced at slower heart rates. In order to investigate the effects of isoproterenol on rate-dependent APD accommodation, a dynamic restitution protocol was performed. Cells were paced at a starting BCL of 1500 ms, and every 50 beats, the BCL was decreased by 100 ms until a BCL of 400 ms was reached. Then, if an EAD was present in the initial experiment, a second dynamic restitution protocol was performed with 10 ms BCL steps that spanned the 100 ms window at the transition from the presence to absence of EADs in order to accurately locate the first EAD-free BCL.
In the wild-type case, all three cardiac cell types (epicardial, endocardial, and mid-myocardial) demonstrated reduced APD90 in response to 1 µM isoproterenol at all BCLs examined (Figure 1F). Heterozygous (ΔKPQ +/−) cells showed a more pronounced reduction in APD90 in response to isoproterenol (Figure 1G). Untreated heterozygous mid-myocardial cells (M-cells) developed EADs at BCLs >730 ms (reflected in Figure 1G by the absence of APD90 values for BCL >730 ms), while isoproterenol-treated M-cells only developed EADs at BCLs longer than 1050 ms. This large isoproterenol-induced increase in the EAD-free BCL window could substantially decrease the arrhythmogenicity of LQT3 cells. In homozygous (ΔKPQ −/−) cells, isoproterenol shortened the APD90 and enlarged the EAD-free BCL window in all three cell types (Figure 1H).
Isoproterenol increased the EAD-free BCL window in mutant cells in a dose-dependent manner (Figure 2A–C). The range of isoproterenol doses used includes those used in earlier experimental studies.12,13 Even low (1 nM) and intermediate (0.01 µM) doses were successful in prolonging the BCL where an EAD first occurred. In endocardial cells, isoproterenol doses of 0.01 µM and higher successfully prevented the development of EADs at every BCL examined.
Figure 2.

Isoproterenol increases the early after depolarization (EAD)-free BCL window in a dose-dependent manner. Endocardial (A), mid-myocardial (B), and epicardial (C) homozygous ΔKPQ mutant cells were paced with a dynamic restitution protocol as described for Figure 1 in the presence of varying concentrations of isoproterenol. The range of EAD-free BCLs is indicated by the black bars.
3.3. In transmural cables, isoproterenol decreases transmural dispersion of repolarization and prevents early after depolarization formation
In order to investigate the effects of isoproterenol on tissues, transmural cables, which contained endocardial, mid-myocardial, and epicardial cells, were paced at a BCL of 1000 ms. Untreated ΔKPQ cables (Figure 3B) showed APD prolongation when compared with wild-type cables (Figure 3A) in all cell types, with the most pronounced prolongation occurring in the M-cell region. (APD90 of cell 83 in the M-cell domain was 183.0 ms in wild-type and 269.4 ms in untreated ΔKPQ.) Treatment of the ΔKPQ cable with 1 µM isoproterenol reduced the APDs of all cell types (Figure 3C). (APD90 in the M-cell domain of treated cables was reduced to 209.8 ms). At a BCL of 1500 ms (Figure 3D–F), in addition to reducing APDs, isoproterenol was able to suppress the development of EADs in the endocardial region of mutant cables.
Figure 3.

Isoproterenol suppresses early after depolarization (EAD) formation and decreases transmural dispersion of repolarization in a mutant model transmural cable. Transmural cardiac cables were paced from the endocardium (top of each panel, position 0 on the cable), and action potentials propagated down through the mid-myocardial and epicardial regions [as labelled in (A)]. A wild-type cable paced at 1000 ms (A) demonstrated shorter action potentials than an untreated ΔKPQ cable (B). One micromolar of isoproterenol reduced ΔKPQ APDs (C). Similar mutation-induced prolongation occurred at a BCL of 1500 ms (E as compared to D), with a non-propagating EAD occurring in the endocardial region. Isoproterenol was able to suppress mutation-induced EADs and APD prolongation (F).
Mutant, untreated cells demonstrated a large increase in TDR (Figure 4A). This increase was rate-dependent, with larger increases happening at slower pacing rates. Isoproterenol was able to reduce TDR at BCLs of both 1000 and 1500 ms. These decreases in TDR are consistent with earlier experimental findings in canine wedge preparations of drug-induced LQT3 syndrome.13
Figure 4.

Isoproterenol decreases transmural dispersion of repolarization (TDR) and the vulnerable window for failure to capture by an S2 stimulus. The ΔKPQ mutation increased TDR in a transmural cable. One micromolar of isoproterenol partially reversed this increase (A). The vulnerable window for failure to capture by an S2 stimulus was measured in mutant cables in the presence and absence of isoproterenol at BCLs of 1000 ms (grey) and 1500 ms (black) (B). Failure to capture was defined as a stimulus that failed to evoke an action potential.
3.4. Isoproterenol modulates the effects of premature stimuli on mutant cables
In order to understand how isoproterenol may decrease arrhythmogenesis in a mutant cable, a number of different pacing protocols were examined. First, the vulnerable window for ‘failure to capture’ by an S2 stimulus from the endocardium was quantified (Figure 4B). Failure to capture resulted when a local depolarization due to a stimulus occurred in the first few cells of the cable but did not trigger an action potential. In a mutant cable paced at 1000 ms, a premature stimulus given <274 ms after the original stimulus will fail to capture. However, in the presence of isoproterenol, an S2 stimulus given at least 234 ms after the S1 stimulus will capture and propagate the entire length of the cable. Isoproterenol reduces the vulnerable window for failure to capture even more substantially at a BCL of 1500 ms. Clinically, failure to capture may set up a cardiac substrate capable of sustaining re-entrant arrhythmias.
Next, the development of conduction block in untreated mutant cables was investigated. Conduction block occurs when an action potential is triggered at the stimulus site (unlike in failure to capture) but fails to propagate down the entire length of the cable. Conduction block can increase susceptibility to lethal arrhythmias such as ventricular fibrillation by increasing gradients of repolarization in the heart.38
We first tried to induce conduction block through application of one premature stimulus. An S2 stimulus given at the endocardium 273 ms after the original stimulus results in failure to capture (see Supplementary material online, Figure S1A). If the S2 stimulus is given just 1 ms later (see Supplementary material online, Figure S1B), an action potential fires and is able to propagate the entire length of the cable. Rather than blocking as one may expect in the M-cell domain, which has a prolonged APD, the conduction velocity of the wave slowed considerably as it passed into and through the M-cell region and then increased again upon exiting the region. This ‘slow and go’ behaviour has been seen in other mutant one-dimensional model simulations.39 In fact, when a single S2 endocardial stimulus was given, block never occurred.
Because we could not evoke conduction block with one S2, we stimulated the cable with a train of 10 premature S2 stimuli from the endocardium. As multiple stimuli are applied, repolarization gradients form across the cable, and eventually conduction block occurs (Figure 5). In order to accurately compare block in the treated and untreated states, rather than measuring the timing of our S2 stimulus based on an interval since the S1 stimulus, we timed our S2 stimulus to be given a set interval after repolarization of the previous action potential.
Figure 5.

The ability of isoproterenol to protect against conduction block depends on the S2 pacing interval. ΔKPQ mutant cables paced at a BCL of 1500 ms were given 10 S2 stimuli at intervals varying from 25 to 40 ms after repolarization of the previous action potential. At an interval of 40 ms, block occurs at the 6th beat in an untreated cable (A) and 1 µM isoproterenol protected against block at this interval (B). The results of experiments at several pacing intervals in the presence and absence of isoproterenol are summarized in (C). Black circles indicate beats where cable-wide propagation occurred; open triangles indicate beats where failure to capture occurred, and grey squares indicate beats where conduction block occurred.
In the untreated, mutant case, if the S2 stimulus is given 40 ms after repolarization of the previous beat, the sixth beat blocks (Figure 5A). However, in the presence of isoproterenol (Figure 5B), block never occurs with an S2 interval of 40 ms.
The results of experiments at several different S2 intervals are summarized in Figure 5C. At short intervals (25 ms) in both the treated and untreated case, the first two action potentials propagate the entire length of the cable (black circles). However, beat 3 in the treated case and beat 4 in the untreated case fail to capture (i.e. the local depolarization is unable to fire an action potential; white triangles). Isoproterenol-treated cells have shorter action potentials, and therefore, the gates of the sarcolemmal channels have less time to recover from inactivation before the S2 stimulus; therefore, at very short S2 intervals, failure to capture occurs more easily in treated cables.
At slightly longer intervals (30 ms), block (grey squares) occurs in both the treated and untreated cables at the same beat numbers (4 and 8). At a 35 ms S2 interval, block occurs at both beats 4 and 8 in untreated cables, but only at beat 6 in treated cables. Finally, at a 40 ms interval, isoproterenol completely suppresses block in the mutant cable. This phenomenon of rate-dependent variation in the ability of isoproterenol to prevent mutation-induced block may account for some of the conflicting results seen by groups using different pacing protocols to evoke arrhythmias in experimental models of LQT3.
3.5. Low-dose propranolol worsens the LQT3 phenotype, whereas high-dose propranolol is beneficial
β-Blockers, such as propranolol, are the first-line treatment for LQT patients; therefore, we simulated the effects of propranolol on APD and TDR. As seen in Figures 1–4, isoproterenol can ameliorate mutation-induced increases in APD, TDR, and EAD formation. When low-dose propranolol (33 nM) is given in addition to isoproterenol, it has the detrimental effects of increasing both APD and TDR (Figure 6A–D). However, when a higher dose (3 or 33 µM) of propranolol was given, both APDs and TDR decreased, due to blockade of the late sodium current that causes the ΔKPQ phenotype (data not shown).
Figure 6.

Low-dose propranolol worsens the LQT3 phenotype, but higher doses can decrease both APD and transmural dispersion of repolarization (TDR). Isoproterenol-treated mutant transmural cable models were paced at a BCL of 1000 ms, and treated with 0 µM, 33 nM, 3 µM, or 33 µM propranolol. APD90 in the endocardium (cell 10) (A), mid-myocardium (cell 83) (B), and epicardium (cell 154) (C) were measured. TDR (D) was measured as in Figure 4.
4. Discussion
Efficacy of β-blockers in LQT3 patients has not been demonstrated.4,5 Early experimental studies indicated that β-adrenergic activation may actually be protective in LQT3.12,13 The proposed mechanism of arrhythmogenesis in LQT3 is that mutation-induced late sodium current leads to APD prolongation that can trigger the development of EADs, which in the context of increased TDR can lead to the development of re-entrant arrhythmias.20
To date, experiments investigating LQT3 have either used toxin-treated guinea pigs or dogs12,13,40 or genetically engineered mouse models of specific LQT3 mutations.14,15 While these systems have provided valuable insights, they both have serious limitations. Namely, toxins will not perfectly recreate the behaviour of specific LQT3 mutations, while mouse and human cardiac electrodynamics differ significantly.41
Experimental studies examining the effects of isoproterenol in mouse models of LQT3 have yielded conflicting results. Our results suggest that pacing protocol differences contributed to these different outcomes. We demonstrated that when multiple premature stimuli were applied to a mutant model cable at some pacing intervals (i.e. 30 ms), there was no difference in the pattern of conduction block between isoproterenol-treated and untreated cables. However, at slightly longer S2 intervals, isoproterenol protected against the development of conduction block (Figure 5). This result suggests that the anti-arrhythmic effects of isoproterenol in LQT3 syndrome are pacing-pattern-dependent.
Our simulations also demonstrate that differences in β-blocker concentrations can complicate interpretations of experimental studies of adrenergic activity in LQT3 syndrome. While the study of ref. 42 suggests that degree of sodium channel block by propranolol can affect arrhythmogenesis, it does not explicitly compare the β-adrenergic and sodium-channel blocking effects of propranolol on LQT3 myocytes. In order to investigate these distinct effects, we created both low (β-adrenergic blocking) and high (β-adrenergic and sodium channel blocking) dose propranolol models. In mutant transmural cable models, low-dose propranolol increased arrhythmogenicity by increasing APDs and TDR; however, higher doses of propranolol were able to decrease both APD and TDR (Figure 6). This finding is consistent with data that shows that 40% of the patients with propranolol-responsive ventricular arrhythmias (due to a variety of causes) require doses that are significantly higher than those known to block β-receptors.43
The level of adrenergic stimulation in LQT3 patients during cardiac events is unknown. Presumably because many events happen during times of rest, sympathetic activation levels are lower than during events in LQT1 and LQT2 patients, which often occur during periods of excitation. By modelling a wide range of isoproterenol concentrations, we have tried to capture the variety of endogenous β-adrenergic stimulation levels. Also, even during sleep, there can be substantial levels of adrenergic stimulation in humans.44 Our results indicate that even at low levels of β-adrenergic activation, isoproterenol could protect against the development of arrhythmogenic triggers (i.e. EADs and increased TDR) in LQT3 cells (Figures 1 and 2).
As in all modelling investigations, a limitation of this study is that our model may be overly simplistic and may not fully recreate all aspects of cardiac electrodynamics. For example, our model only includes ventricular myocardial cells; however, it has been suggested that LQT arrhythmias may actually be triggered in the Purkinje fibres.45 Future modelling studies of LQT3 could incorporate both ventricular and Purkinje model cells.
In summary, we investigated the effects of β-adrenergic agonists and antagonists in LQT3 syndrome using a mathematical model of a human-like ventricular myocyte (e.g. a guinea pig myocyte model) that incorporates the ΔKPQ mutant sodium current. We found that isoproterenol can decrease the arrhythmogenicity of LQT3 mutant cells and transmural cables by decreasing APD and TDR and suppressing EAD development. However, the ability of isoproterenol to prevent conduction block depended on the pacing protocol used. This result implicates pacing differences as one major cause of ambiguity in previous experimental studies of isoproterenol in LQT3 syndrome. Low-dose propranolol was shown to worsen the LQT3 phenotype, whereas high dose that also blocked sodium channels was beneficial.
The modelling results presented in the study could inform future clinical investigations. Specifically, a larger randomized controlled trial of β-blockers in the treatment of LQT3 patients is needed. These findings suggest that in addition to a control group, such a study should include a low-dose propranolol group, a high-dose propranolol group, and a group receiving a β-blocker that does not have any known off-target sodium channel effects. In addition to anti-arrhythmic efficacy, it would be important to monitor confounding variables such as serum drug levels, heart rate, and level of sympathetic activity. Finally, findings about the effects of β-adrenergic modulation in LQT3 may provide insights into other more common conditions where late sodium current has been implicated to contribute to disease pathogenesis, such as ischemia–reperfusion injury.46
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by National Institutes of Health pre-doctoral training awards [GM07739 for R.C.A.-N.; 1F30HL094073-01 for R.C.A.-N.] and the Kenny Gordon Foundation.
Supplementary Material
Acknowledgements
We would like to thank Jun Xu and Zheng Zhu for model code and assistance.
Conflict of interest: none declared.
References
- 1.Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation. 1991;84:503–511. doi: 10.1161/01.cir.84.2.503. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 3.Shimizu W, Noda T, Takaki H, Kurita T, Nagaya N, Satomi K, et al. Epinephrine unmasks latent mutation carriers with LQT1 form of congenital long-QT syndrome. J Am Coll Cardiol. 2003;41:633–642. doi: 10.1016/s0735-1097(02)02850-4. [DOI] [PubMed] [Google Scholar]
- 4.Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 6.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 8.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–1607. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 11.Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 12.Priori SG, Napolitano C, Cantu F, Brown AM, Schwartz PJ. Differential response to Na+ channel blockade, beta-adrenergic stimulation, and rapid pacing in a cellular model mimicking the SCN5A and HERG defects present in the long-QT syndrome. Circ Res. 1996;78:1009–1015. doi: 10.1161/01.res.78.6.1009. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 14.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 15.Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, et al. Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J Cardiovasc Electrophysiol. 2005;16:1329–1340. doi: 10.1111/j.1540-8167.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- 16.Faber GM, Rudy Y. Action potential and contractility changes in Na+ overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tarr M, Luckstead EF, Jurewicz PA, Haas HG. Effect of propranolol on the fast inward sodium current in frog atrial muscle. J Pharmacol Exp Ther. 1973;184:599–610. [PubMed] [Google Scholar]
- 18.Wang DW, George AL., Jr Abstract 505: propranolol block of human cardiac sodium channels is enhanced by SCN5A mutations in long-QT syndrome type 3. Circulation. 2007;116:II_88. [Google Scholar]
- 19.Woosley RL, Kornhauser D, Smith R, Reele S, Higgins SB, Nies AS, et al. Suppression of chronic ventricular arrhythmias with propranolol. Circulation. 1979;60:819–827. doi: 10.1161/01.cir.60.4.819. [DOI] [PubMed] [Google Scholar]
- 20.Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476. [DOI] [PubMed] [Google Scholar]
- 21.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74:1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 22.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura H, Kurokawa J, Bai CX, Asada K, Xu J, Oren RV, et al. Progesterone regulates cardiac repolarization through a nongenomic pathway: an in vitro patch-clamp and computational modeling study. Circulation. 2007;116:2913–2922. doi: 10.1161/CIRCULATIONAHA.107.702407. [DOI] [PubMed] [Google Scholar]
- 24.Verkerk AO, Wilders R, de Geringel W, Tan HL. Cellular basis of sex disparities in human cardiac electrophysiology. Acta Physiol (Oxf) 2006;187:459–477. doi: 10.1111/j.1748-1716.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 25.Berecki G, Zegers JG, Bhuiyan ZA, Verkerk AO, Wilders R, van Ginneken AC. Long-QT syndrome-related sodium channel mutations probed by the dynamic action potential clamp technique. J Physiol (Lond) 2006;570:237–250. doi: 10.1113/jphysiol.2005.096578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 27.Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90:889–896. doi: 10.1161/01.res.0000016960.61087.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 29.Rudy Y. From genetics to cellular function using computational biology. Ann N Y Acad Sci. 2004;1015:261–270. doi: 10.1196/annals.1302.022. [DOI] [PubMed] [Google Scholar]
- 30.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003;278:47997–48003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 31.Choe CU, Schulze-Bahr E, Neu A, Xu J, Zhu ZI, Sauter K, et al. C-terminal HERG (LQT2) mutations disrupt IKr channel regulation through 14-3-3epsilon. Hum Mol Genet. 2006;15:2888–2902. doi: 10.1093/hmg/ddl230. [DOI] [PubMed] [Google Scholar]
- 32.Terrenoire C, Clancy CE, Cormier JW, Sampson KJ, Kass RS. Autonomic control of cardiac action potentials: role of potassium channel kinetics in response to sympathetic stimulation. Circ Res. 2005;96:e25–e34. doi: 10.1161/01.RES.0000160555.58046.9a. [DOI] [PubMed] [Google Scholar]
- 33.Galandrin S, Bouvier M. Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol. 2006;70:1575–1584. doi: 10.1124/mol.106.026716. [DOI] [PubMed] [Google Scholar]
- 34.Wang DW, Yazawa K, Makita N, George AL, Jr, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–1720. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clancy CE, Zhu ZI, Rudy Y. Pharmacogenetics and anti-arrhythmic drug therapy: a theoretical investigation. Am J Physiol Heart Circ Physiol. 2007;292:H66–H75. doi: 10.1152/ajpheart.00312.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 37.Diness JG, Hansen RS, Nissen JD, Jespersen T, Grunnet M. Antiarrhythmic effect of IKr activation in a cellular model of LQT3. Heart Rhythm. 2009;6:100–106. doi: 10.1016/j.hrthm.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 38.Gelzer AR, Koller ML, Otani NF, Fox JJ, Enyeart MW, Hooker GJ, et al. Dynamic mechanism for initiation of ventricular fibrillation in vivo. Circulation. 2008;118:1123–1129. doi: 10.1161/CIRCULATIONAHA.107.738013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henry H, Rappel WJ. The role of M cells and the long QT syndrome in cardiac arrhythmias: simulation studies of reentrant excitations using a detailed electrophysiological model. Chaos. 2004;14:172–182. doi: 10.1063/1.1636272. [DOI] [PubMed] [Google Scholar]
- 40.Chinushi M, Izumi D, Iijima K, Ahara S, Komura S, Furushima H, et al. Antiarrhythmic vs. pro-arrhythmic effects depending on the intensity of adrenergic stimulation in a canine anthopleurin-A model of type-3 long QT syndrome. Europace. 2008;10:249–255. doi: 10.1093/europace/eun002. [DOI] [PubMed] [Google Scholar]
- 41.Salama G, London B. Mouse models of long QT syndrome. J Physiol (Lond) 2007;578:43–53. doi: 10.1113/jphysiol.2006.118745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas G, Killeen MJ, Grace AA, Huang CL. Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in DeltaKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol (Oxf) 2008;192:505–517. doi: 10.1111/j.1748-1716.2007.01770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duff HJ, Roden DM, Brorson L, Wood AJ, Dawson AK, Primm RK, et al. Electrophysiologic actions of high plasma concentrations of propranolol in human subjects. J Am Coll Cardiol. 1983;2:1134–1140. doi: 10.1016/s0735-1097(83)80340-4. [DOI] [PubMed] [Google Scholar]
- 44.Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med. 1993;328:303–307. doi: 10.1056/NEJM199302043280502. [DOI] [PubMed] [Google Scholar]
- 45.Ben Caref E, Boutjdir M, Himel HD, El-Sherif N. Role of subendocardial Purkinje network in triggering torsade de pointes arrhythmia in experimental long QT syndrome. Europace. 2008;10:1218–1223. doi: 10.1093/europace/eun248. [DOI] [PubMed] [Google Scholar]
- 46.Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92(Suppl. 4):iv1–iv5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.