

Abstract
The environmental pollutant 6-nitrochrysene (6-NC) is a potent mammary carcinogen in rats; it is more potent than numerous classical mammary carcinogens such as benzo[a]pyrene (BaP). The mechanisms that account for the remarkable carcinogenicity of 6-NC remain elusive. Similar to BaP, 6-NC is also known to induce DNA damage in rodents and in human breast tissues. As an initial investigation, we reasoned that DNA damage induced by 6-NC may alter the expression of p53 protein in a manner that differs from other DNA damaging carcinogens (e.g. BaP). Using human breast adenocarcinoma MCF-7 cells and immortalized human mammary epithelial MCF-10A cells, we determined the effects of 6-NC on the expression of p53 protein and its direct downstream target cyclin-dependent kinase inhibitor p21Cip1 as well as on the cell cycle progression. Western blot analysis demonstrated that treatments of MCF-7 and MCF-10A cells with 6-NC for 12, 24 or 48 h did not increase the level of total p53 protein; however, an increase of p21Cip1 protein and a commitment increase of G1 phase were observed in MCF-10A cells but not in MCF-7 cells. Further studies using 1, 2-dihydroxy-1, 2-dihydro-6-hydroxylaminochrysene (1, 2-DHD-6-NHOH-C), the putative ultimate genotoxic metabolite of 6-NC, was conducted and showed a significant induction of p53 (p < 0.05) in MCF-7 cells; however, this effect was not evident in MCF-10A cells, indicating the varied DNA damage responses between the two cell lines. By contrast to numerous DNA damaging agents such as BaP which is known to stimulate p53 expression, the lack of p53 response by 6-NC imply the lack of protective functions mediated by p53 (e.g. DNA repair machinery) after exposure to 6-NC and this may, in part, account for its remarkable carcinogenicity in the mammary tissue.
1. Introduction
Although breast cancer is second only to lung cancer as the leading cause of cancer-related deaths in American women [1], its etiology remains obscure. In addition to genetic disposition, most cancers including breast cancer in the U.S. are related to environmental factors and lifestyles [2, 3]. Consequently, the identification of carcinogens present in the human environment challenges both scientist and regulatory agencies. A number of ubiquitous environmental agents have been shown to induce mammary cancer in rodents; thus, they must be regarded as potential human risk factors and are candidates for a detailed evaluation [4]. Nitropolycyclic aromatic hydrocarbons (NO2-PAH) are widespread environmental contaminants which are normally produced from incomplete combustion of organic compounds such as diesel, gas, kerosene and liquid petroleum [5]. Exposure to environmental carcinogens, including NO2-PAH, has been implicated in the etiology of various types of cancer, including breast cancer [3, 6–8]. 6-NC is a representative member (Figure 1) of NO2-PAH. Previous studies demonstrate that under identical conditions, 6-NC is a more potent mammary carcinogen than benzo[a]pyrene (BaP) and the heterocyclic aromatic amine 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) [9–11]. Furthermore, the finding of 6-NC-hemoglobin adducts in humans [12] and the ability of human liver, lung and breast tissues to metabolize 6-NC into DNA reactive species [13, 14] have raised the concern about the effects of this compound on human health.
Fig. 1.
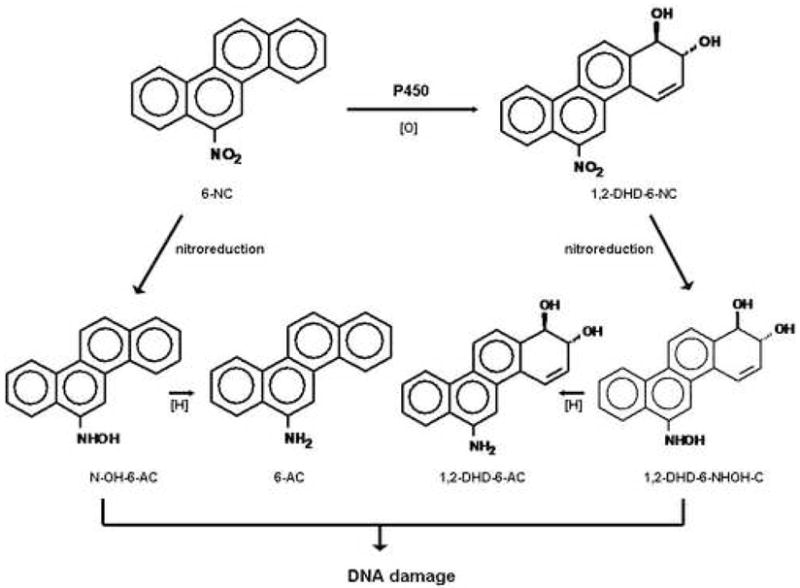
Metabolic activation of 6-NC to intermediate (N-OH-6-AC and 1, 2-DHD-6-NHOH) known to damage DNA.
6-NC requires metabolic activation to generate several electrophilic species before they can react with DNA and other macromolecules to initiate the carcinogenic process [4, 13, 14]. Our previous studies have shown that 1, 2-DHD-6-NHOH-C (Figure 1), a metabolite derived from ring oxidation and nitroreduction of 6-NC, is the ultimate genotoxic metabolite responsible for the formation of the major DNA adducts detected in the mammary gland of rats treated with 6-NC [4]. Earlier studies conducted in our laboratory demonstrated that human breast adenocarcinoma cell line MCF-7 [p53 wild type, estrogen receptor alpha (ERα)-positive] and immortalized human mammary epithelial cell line MCF-10A (p53 wild type, ERα-negative) [15] can metabolize 6-NC to intermediates that can damage DNA; furthermore, DNA adducts derived from 6-NC were detected in MCF-7 cells [14].
DNA damage induced by numerous carcinogens, if not repaired, can lead to mutagenesis and tumor initiation [16]. The p53 tumor suppressor protein is a transcription factor which regulates important cellular responses to DNA damage [17]. Following DNA damage, p53 can be activated leading to the repair of DNA damage or apoptosis. The major downstream target of p53 is the cyclin-dependent kinase inhibitor p21Cip1, which is responsible for exerting G1 arrest [18, 19]. Many PAH carcinogens and their reactive metabolites such as BaP or its ultimate carcinogen anti-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (BPDE), are known to induce the expression of p53 protein [20–24]; however, studies of the effects of 6-NC on molecular markers (e.g. p53) that are critical in the development of breast cancer are lacking. Thus, as an initial investigation we reasoned that using human breast cancer MCF-7 cells and immortalized MCF-10A cells, the effects of 6-NC on p53 and p21Cip1 protein expression as well as cell cycle progression may differ from those exerted by other chemical carcinogens such as BaP.
Our results demonstrate that by contrast to BaP, 6-NC failed to increase the levels of total p53 protein in both cell types. Since p53 is involved in the regulation of global nucleotide excision repair that is presumably responsive to DNA damage, these results may, in part, account for the remarkable carcinogenicity of 6-NC.
2. Materials and Methods
2.1. Chemicals and reagents
6-NC and 1, 2-DHD-6-NHOH-C were synthesized and purified as described previously [4]. Dimethyl sulfoxide (DMSO, cell culture grade), propidium iodide, cholera toxin, hydrocortisone, and insulin were purchased from Sigma Chemical Co. (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM)/F-12 was purchased from Clonetics. Penicillin/streptomycin, phosphate buffered saline (PBS), epidermal growth factor (EGF), trypsin-EDTA and horse serum were purchased from Gibco-Invitrogen. Fetal bovine serum (FBS) (heat inactivated) was purchased from Gemini Bio-Products (Woodland, CA).
Monoclonal antibody against p53 or p21Cip1 was purchased from Upstate (Lake Placid, NY). Mouse monoclonal antibody against β-actin was purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies against phospho-p53 at Ser15 and Ser20 were purchased from Cell Signaling Technology (Beverly, MA). Secondary anti-mouse or anti-rabbit IgG, conjugated with horse radish peroxidase (HRP), were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2. Cell culture and treatments
MCF-7 and MCF-10A cells were obtained from American Type Culture Collection (Manassas, VA) and grown in 75-cm2 cell culture flasks at 37°C in a humidified atmosphere with 5% CO2 in air. MCF-7 cells were maintained in DMEM/F12 (1:1 mixture) supplemented with 10% FBS and penicillin/streptomycin (50 μg/mL). MCF-10A cells were maintained in DMEM/F-12 supplemented with 5% horse serum, penicillin/streptomycin (50 μg/mL), insulin (10 μg/mL), hydrocortisone (500 ng/mL), EGF (20 ng/mL), and cholera toxin (100 ng/mL). Cells were seeded in 12 well plates at least 24 h before treatment. The medium was replaced with fresh medium (1 ml) before treating the cells with different concentrations of carcinogens or DMSO for the time indicated. 6-NC and 1, 2-DHD-6-NHOH-C were dissolved in DMSO, yielding a final DMSO concentration of 0.1% (v/v) in the medium. An initial growth inhibition experiment was conducted to determine the highest concentration of 6-NC that can be used in this study. In order to maximize the p53 response, we chose two concentrations of 6-NC (2 and 4 μM) at which more than 75% of the cells survived under the experimental conditions.
2.3. Cell lysis and Western blotting
Total cellular protein was homogenized in lysis buffer containing 20 mM Tris buffer, pH 7.4, 150 mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM Na3VO4, 1 mM NaF, 10 μg/ml leupeptin, 1 mM PMSF, 10 μg/ml aprotinin and 10 μg/ml pepstatin A. Cell debris and particulate fractions were removed by centrifugation at 13,000 × g for 10 min at 4 °C. Protein concentration was measured using Bio-Rad DC protein assay kit. Equal amounts of protein (approximately 25 μg) were diluted with 3 × sample buffer containing β-mercaptoethanol and bromophenol blue and then heated at 100 °C for 5 min. Proteins were separated by 10% SDS-PAGE, transferred onto PVDF membranes (Bio-Rad, Hercules, CA), and subjected to immunoblot analysis using the respective antibody against: phospho-p53, p53, p21Cip1, and β-actin. Immunoreactive proteins were subsequently detected with appropriate secondary antibodies conjugated with HRP and enhanced chemilluminescence kits and exposed in the SynGene Gene Gnome. Band density was determined using SynGene GeneTools software. The relative densities of phospho-p53, p53, and p21Cip1 to β-actin were calculated. Statistical analyses were performed using the Student’s t test and p < 0.05 was considered statistically significant. All values represent mean ± S.D. (n ≥ 3).
2.4. Flow Cytometry
Cells were grown to exponential phase and treated with the indicated concentration of compounds that had been dissolved in DMSO or DMSO only for 24 h. Cells were harvested, fixed in ice-cold 70% ethanol, and stored at −20°C. The fixed cells were washed with phosphate-buffered saline, treated with RNase A (3 units/mL) at 37°C for 30 min, and stained with propidium iodide (50 μg/mL) for 5 min. DNA content for 250,000 cells per analysis was monitored with a Becton-Dickinson FACScan flow cytometer and Modfit software (LT version 2.0) was used for analysis of cell cycle status.
2.5. siRNA Transfection
Since 6-NC had no measurable effect on p53 expression in both cell lines, this experiment was conducted to determine the linkage between p21Cip1 expression and G1 accumulation in MCF-10A cells treated with 6-NC. The p21Cip1 siRNAs kit was purchased from Cell Signaling Technology, Inc (SignalSilence p21waf1/cip1 siRNA kit). According to the manufacturer, the targeting sequence of human p21Cip1 gene is 5′-AACUUCGACUUUGUCACCGAG-3′ that corresponds to the 108 nucleotides from the start codon [25]. MCF-10A cells were plated at a density of 5 × 105 cells per well of 6-well plates. Cells were transfected with 100 nM of siRNA targeted against p21Cip1 according to the manufacturer’s suggested procedures. Briefly, 5 μL of TransIT-TKO (Mirus) were diluted in 250 μL of serum-free media and incubated for 5 min followed by addition of siRNA. The resulting mixture was then incubated for an additional 5 min at room temperature prior to the addition to MCF-10A cells. At 24 h post-transfection, cells were treated with 4 μM of 6-NC for an additional 24 h before harvest. The dose of 6-NC and the incubation time were selected based on the results of cell cycle analysis. These cells were then analyzed by flow cytometry or Western blot as described above.
3. Results
3.1. Effects of 6-NC on the expression of phosphorylated p53, p53 and p21Cip1 proteins in MCF-7 and MCF-10A cells
Lysates from cells treated with 2 or 4 μM 6-NC for 12, 24 or 48 h were subjected to Western blot analysis to determine the levels of phosphorylated p53, p53 and p21Cip1 proteins. The results of BaP are included for comparison. Our results showed that by contrast to BaP, treatments of MCF-7 and MCF-10A cells with 6-NC did not result in an increase in the level of total p53 protein despite weak induction of phosphorylated p53 at Serine 15(Ser15) (Figure 2). Phosphorylation of p53 protein at Ser20 was not detected in either cell line (data not shown). By contrast to MCF-7 cells, treatments of MCF-10A cells with 6-NC led to an increase in the level of p21Cip1 protein which is independent of p53 expression (Figures 2 and 3). BaP has already shown to induce p53 and p21Cip1 expression in MCF-7 cells [24] but similar studies using MCF-10A cells have not been reported. Our results show that BaP also increases the levels of p53 and p21Cip1 in MCF-10A cells but to a less extent than in MCF-7 cells (Figure 2).
Fig. 2.

Representative Western blot analysis of phospho-p53(ser15), p53 and p21Cip1 proteins in MCF-7 cells (A) and MCF-10A (B) cells after exposure to 6-NC (2 μM and 4 μM) or BaP (2 μM) for 12, 24 and 48 h. Cells treated with DMSO alone were used as negative control. β-Actin was used as a loading control.
Fig. 3.

Changes in phospho-p53, p53 and p21Cip1 proteins following 12, 24 and 48 h treatment with 6-NC (2 and 4 μM) in MCF-7 (A) and MCF-10A (B) cells. The data represent the means of at least three determinations ± S.D. and are expressed as fold of induction relative to DMSO control.
3.2. Effects of 1, 2-DHD-6-NHOH-C on phosphorylated p53, p53 and p21Cip1 protein expression in MCF-7 and MCF-10A cells
To bypass the metabolic activation of 6-NC, we examined the effects of its putative ultimate genotoxic metabolite 1, 2-DHD-6-NHOH-C on p53 and p21Cip1 protein expression in MCF-7 and MCF-10A cells. The results of BPDE are included for comparison. Our results demonstrate that treatment of MCF-7 cells with 1, 2-DHD-6-NHOH-C (0.5, 1, 2 and 4 μM) resulted in a significant increase of phospho-p53(Ser15) (p < 0.05) or p53 protein (p < 0.05) at 24 h (Figure 4A); however, the effects were not dose-dependent. A non-significant increase of p21Cip1 protein was detected in MCF-7 cells. The p53 response observed in MCF-10A cells was much weaker than that found in MCF-7 cells (Figure 4B); however, induction of p21Cip1 was detected and comparable to that observed in MCF-7 cells.
Fig. 4.
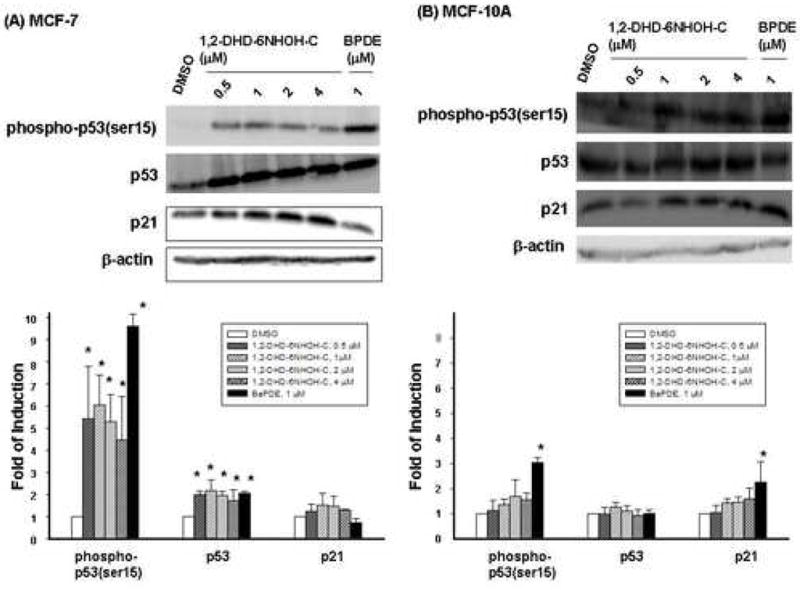
Western blot analysis of phospho-p53(ser15), p53 and p21Cip1 proteins in MCF-7 cells (A) and MCF-10A cells (B) after exposure to 1,2-DHD-6-NHOH-C [0.5 ( ), 1 (
), 1 ( ),2 (
),2 ( ) and 4 (
) and 4 ( ) μM] or BPDE ( 2 μM
) μM] or BPDE ( 2 μM  ) for 24 h. Cells treated with DMSO (
) for 24 h. Cells treated with DMSO ( ) alone were used as control. β-Actin was used as a loading control. Top panels: representative Western blots; Bottom panels: quantitative analysis of protein changes (mean ± S.D, n=3). * indicates p < 0.05.
) alone were used as control. β-Actin was used as a loading control. Top panels: representative Western blots; Bottom panels: quantitative analysis of protein changes (mean ± S.D, n=3). * indicates p < 0.05.
3.3 Effect of 6-NC on cell cycle regulation in MCF-7 and MCF-10A cells
Cell cycle distribution was determined by flow cytometric analysis of propidium iodide stained cells. MCF-7 and MCF-10A cells were treated with 6-NC under experimental conditions identical to those employed in the above mentioned studies that were aimed at determining the effect of this carcinogen on the expression of p53 and p21Cip1 protein. Figure 5 shows the percentage of MCF-7 (A) and MCF-10A cells (B) in different phases of cell cycle after treatment with (1) DMSO; (2) 2 μM 6-NC; (3) 4 μM 6-NC for 24 h. Figures 6 summarizes the changes in cell cycle for MCF-7 (A) and MCF-10A (B) cells, respectively. 6-NC induced a G1 increase, which was more pronounced in MCF-10A cells than in MCF-7 cells; however, a G2 increase was only noticed in MCF-7 cells. Reduction in S phase was observed in both cell types.
Fig. 5.
Representative flow cytometry histogram in (A) MCF-7 cells and (B) MCF-10A cells treated with 6-NC (2 and 4 μM) for 24 h. Cells treated with DMSO were used as control.
Fig. 6.
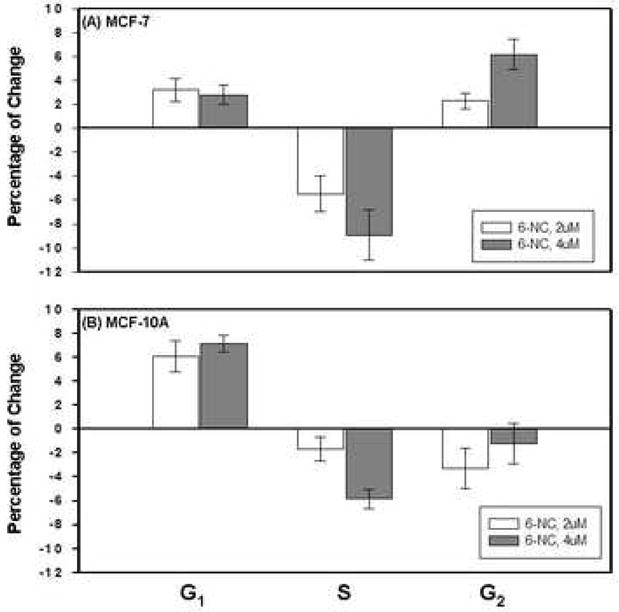
Percentage of changes of (A) MCF-7 (B) MCF-10A cells (mean ± S.D) in G1 phase, S phase and G2 phase after exposure to 6-NC for 24 h.
3.3 Effect of p21Cip1siRNA on cell cycle regulation in MCF-10A cells treated with 6-NC
To test whether the observed increase in G1 phase in MCF-10A cells treated with 6-NC is mediated by the increased expression of p21Cip1 protein, cells were treated with p21Cip1 siRNA as described in Materials and Methods section. Our results, although preliminary, showed that transfection with siRNA in MCF-10A cells reduced the effects of 6-NC (4 μM, 24 h) on p21Cip1 protein expression by ~50%; treatment with siRNA also reduced the effect if 6-NC on G1 accumulation by ~11%.
4. Discussion
The p53 protein is a transcription factor which regulates important cellular responses to DNA damages [17]. By contrast to other DNA damaging agents such as BaP, we showed that 6-NC did not increase the level of total p53 protein in both MCF-7 and MCF-10A cells. Although the mechanisms that can account for the remarkable mammary carcinogenicity of 6-NC in rodents remain unclear, the lack of p53 response by 6-NC in these cells imply the lack of protective functions mediated by p53 (e.g. DNA repair machinery) and this may, in part, contribute to the potent mammary carcinogenicity of 6-NC.
Further studies using 1, 2-DHD-6-NHOH-C, the putative ultimate genotoxic metabolite of 6-NC, clearly demonstrate the ability of this metabolite to increase the levels of phosphorylated as well as total p53 proteins in MCF-7 cells (Figure 4A). These results indicate that the weak induction of p53 by 6-NC may be at least partially due to the low efficiency of MCF-7 cells in metabolizing 6-NC into 1, 2-DHD-6-NHOH-C. However, the low metabolic capacity could not account exclusively for the weak response of p53 in MCF-10A cells (Figure 4B) as we observed that treatments of MCF-10A cells with 1, 2-DHD-6-NHOH did not result in a distinct increase of p53. To determine whether the observed differences in p53 responses were due to the nature of the DNA damaging species or to differences in cell types, we examined the effects of BPDE, the ultimate carcinogenic form of BaP. Both 1, 2-DHD-6-NHOH-C and BPDE induced stronger p53 responses in MCF-7 cells than those obtained from MCF-10A cells (Figure 4). Thus, the differential p53 responses observed between MCF-7 and MCF-10A cells following treatments with ultimate carcinogens are due primarily to the differences in cell types.
Several studies have shown that p21Cip1 expression can be induced in both p53-dependent and p53-independent manners [26–29]. In this study, we also demonstrated that p21Cip1 protein was increased by 6-NC in a p53-independent manner and this effect appeared to depend on cell types since it was only observed in MCF-10A cells but not in MCF-7 cells. To clarify if the differences in p21Cip1 response were due to the carcinogen itself or to the differences in cell types, we conducted a parallel experiment using BaP. We found that both carcinogens induced a significant increase of p21Cip1 in MCF-10A cells after 24 h (Figure 2B). Although the increased expression of p21Cip1 in MCF-10A cells by BaP may be through both p53-dependent and independent pathways, these results suggest that a p53-independent p21Cip1 regulation pathway was involved in the up-regulation of p21Cip1 by 6-NC in MCF-10A cells but not in MCF-7 cells.
Cell cycle progression is controlled by a highly complex network. The roles of p53 and p21Cip1 in cell cycle regulation have been demonstrated [19, 30]; in addition, literature data suggest the involvement of AhR and ER in the cell cycle progression [31–33]. As described above, 6-NC induced p21Cip1 expression that is independent of p53 and only in MCF-10A cells. Therefore, to link the effect of p21Cip1 expression to changes in the cell cycle distribution induced by 6-NC, additional experiments were conducted using p21Cip1 siRNA. Although the results are considered preliminary and expected based on literature data, we demonstrate the linkage between p21Cip1 expression and G1 accumulation in MCF-10A cells treated with 6-NC. However, future studies are required to support this observation.
The differential responses of p53 and p21Cip1 observed between MCF-7 and MCF-10A cells may be due, in part, to the different genetic make-up of the two cell lines used in this study. Estrogen receptor alpha has been correlated with the response of DNA damage as well as cell cycle progression [33–35]. MCF-7 cell line which is derived from ductal adenocarcinoma expresses ERα; however, the immortalized mammary epithelial cell line MCF-10A is ERα-negative. It is likely that the different status of ERα may contribute, in part, to the varied responses of p53, p21Cip1 and cell cycle regulation observed between the two cell lines in this study. In addition to ER status, many of the biological actions of PAH or NO2-PAH, such as the induction of cytochrome p450s, are believed to be mediated through the action of aryl hydrocarbon receptor (AhR) [3, 31]. AhR is also known to be involved in regulation of cell cycle progression [31, 32] as well as ER-mediated cell signaling pathway [36]. Earlier studies have shown that 6-NC can induce the expression of cytochrome P450 1A1 [37, 38]; our preliminary studies (unpublished data) using a luciferase assay also show that 6-NC is a potent AhR agonist. Clearly, the dysregulation of cell cycle induced by 6-NC can be due to the interactions of factors described above.
In summary, our studies demonstrate that by contrast to numerous chemical carcinogens such as BaP which stimulates p53 expression, the lack of p53 response by 6-NC imply the lack of protective functions mediated by p53 (e.g. DNA repair machinery) and this may contribute to its remarkable carcinogenicity in the mammary tissue.
Acknowledgments
This work was supported by the National Institutes of Health Grant, No CA 35519. Carcinogens and metabolites used in this study were provided by Penn State Cancer Institute-Organic Synthesis Facility. We thank the members in flow cytometry facility at College of Medicine Penn State University for their technical assistance.
Abbreviations
- PAH
polycyclic aromatic hydrocarbons
- NO2-PAH
nitropolycyclic aromatic hydrocarbons
- BaP
benzo[a]pyrene
- BPDE
anti-7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- 6-NC
6-nitrochrysene
- 1,2-DHD-6-NHOH-C
1,2-dihydroxy-1,2-dihydro-6-hydroxylaminochrysene
- 6-AC
6-aminochrysene
- N-OH-6-AC
N-hydroxy-6-aminochrysene
- 1,2-DHD-6-NC
1,2-dihydroxy-6-nitrochrysene
- 1,2-DHD-6-AC
1,2-dihydroxy-6-aminochrysene
- PMSF
phenylmethylsulfonyl fluoride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.ACS. Cancer Facts and Figures 2006. American Cancer Society; Atlanta, GA: [Google Scholar]
- 2.Amin S, El-Bayoumy K. Tumorigenicity of Polycyclic Aromatic Hydrocarbons. On the possible contribution of PAHs and their Nitro-Derivatives to the Development of Human Breast Cancer. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons Imperial College Press Chapter. 2005;9:p315–351. [Google Scholar]
- 3.Jeffy BD, Chirnomas RB, Romagnolo DF. Epigenetics of breast cancer: polycyclic aromatic hydrocarbons as risk factors. Environ Mol Mutagen. 2002;39:235–244. doi: 10.1002/em.10051. [DOI] [PubMed] [Google Scholar]
- 4.El-Bayoumy K, Sharma AK, Lin JM, Krzeminski J, Boyiri T, King LC, Lambert G, Padgett W, Nesnow S, Amin S. Identification of 5-(deoxyguanosin-N2-yl)-1,2-dihydroxy-1,2-dihydro-6-aminochrysene as the major DNA lesion in the mammary gland of rats treated with the environmental pollutant 6-nitrochrysene. Chem Res Toxicol. 2004;17:1591–1599. doi: 10.1021/tx049849+. [DOI] [PubMed] [Google Scholar]
- 5.IARC monographs on the evaluation of carcinogenic risks to humans. Diesel and gasoline engine exhausts and some nitroarenes. International Agency for Research on Cancer. IARC Monogr Eval Carcinog Risks Hum. 1989;46:1–458. [PMC free article] [PubMed] [Google Scholar]
- 6.Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14:473–486. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 8.Rundle A, Tang D, Hibshoosh H, Schnabel F, Kelly A, Levine R, Zhou J, Link B, Perera F. Molecular epidemiologic studies of polycyclic aromatic hydrocarbon-DNA adducts and breast cancer. Environ Mol Mutagen. 2002;39:201–207. doi: 10.1002/em.10048. [DOI] [PubMed] [Google Scholar]
- 9.Hecht SS, El-Bayoumy K, Rivenson A, Amin S. Potent mammary carcinogenicity in female CD rats of a fjord region diol-epoxide of benzo[c]phenanthrene compared to a bay region diol-epoxide of benzo[a]pyrene. Cancer Res. 1994;54:21–24. [PubMed] [Google Scholar]
- 10.El-Bayoumy K, Chae YH, Upadhyaya P, Rivenson A, Kurtzke C, Reddy B, Hecht SS. Comparative tumorigenicity of benzo[a]pyrene, 1-nitropyrene and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine administered by gavage to female CD rats. Carcinogenesis. 1995;16:431–434. doi: 10.1093/carcin/16.2.431. [DOI] [PubMed] [Google Scholar]
- 11.El-Bayoumy K, Desai D, Boyiri T, Rosa J, Krzeminski J, Sharma AK, Pittman B, Amin S. Comparative tumorigenicity of the environmental pollutant 6-nitrochrysene and its metabolites in the rat mammary gland. Chem Res Toxicol. 2002;15:972–978. doi: 10.1021/tx020019a. [DOI] [PubMed] [Google Scholar]
- 12.Zwirner-Baier I, Neumann HG. Polycyclic nitroarenes (nitro-PAHs) as biomarkers of exposure to diesel exhaust. Mutat Res. 1999;441:135–144. doi: 10.1016/s1383-5718(99)00041-8. [DOI] [PubMed] [Google Scholar]
- 13.Chae YH, Yun CH, Guengerich FP, Kadlubar FF, El-Bayoumy K. Roles of human hepatic and pulmonary cytochrome P450 enzymes in the metabolism of the environmental carcinogen 6-nitrochrysene. Cancer Res. 1993;53:2028–2034. [PubMed] [Google Scholar]
- 14.Boyiri T, Leszczynska J, Desai D, Amin S, Nixon DW, El-Bayoumy K. Metabolism and DNA binding of the environmental pollutant 6-nitrochrysene in primary culture of human breast cells and in cultured MCF-10A, MCF-7 and MDA-MB-435s cell lines. Int J Cancer. 2002;100:395–400. doi: 10.1002/ijc.10505. [DOI] [PubMed] [Google Scholar]
- 15.Majumder B, Wahle KW, Moir S, Schofield A, Choe SN, Farquharson A, Grant I, Heys SD. Conjugated linoleic acids (CLAs) regulate the expression of key apoptotic genes in human breast cancer cells. Faseb J. 2002;16:1447–1449. doi: 10.1096/fj.01-0720fje. [DOI] [PubMed] [Google Scholar]
- 16.Cox R. The multi-step nature of carcinogenesis and the implications for risk analysis. Int J Radiat Biol. 1998;73:373–376. doi: 10.1080/095530098142194. [DOI] [PubMed] [Google Scholar]
- 17.Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004;3:1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 18.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–508. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 19.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 20.Ramet M, Castren K, Jarvinen K, Pekkala K, Turpeenniemi-Hujanen T, Soini Y, Paakko P, Vahakangas K. p53 protein expression is correlated with benzo[a]pyrene-DNA adducts in carcinoma cell lines. Carcinogenesis. 1995;16:2117–2124. doi: 10.1093/carcin/16.9.2117. [DOI] [PubMed] [Google Scholar]
- 21.Bjelogrlic NM, Makinen M, Stenback F, Vahakangas K. Benzo[a]pyrene-7,8-diol-9,10-epoxide-DNA adducts and increased p53 protein in mouse skin. Carcinogenesis. 1994;15:771–774. doi: 10.1093/carcin/15.4.771. [DOI] [PubMed] [Google Scholar]
- 22.Sadikovic B, Rodenhiser DI. Benzopyrene exposure disrupts DNA methylation and growth dynamics in breast cancer cells. Toxicol Appl Pharmacol. 2006;216:458–468. doi: 10.1016/j.taap.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 23.Wang A, Gu J, Judson-Kremer K, Powell KL, Mistry H, Simhambhatla P, Aldaz CM, Gaddis S, MacLeod MC. Response of human mammary epithelial cells to DNA damage induced by BPDE: involvement of novel regulatory pathways. Carcinogenesis. 2003;24:225–234. doi: 10.1093/carcin/24.2.225. [DOI] [PubMed] [Google Scholar]
- 24.Jeffy BD, Chen EJ, Gudas JM, Romagnolo DF. Disruption of cell cycle kinetics by benzo[a]pyrene: inverse expression patterns of BRCA-1 and p53 in MCF-7 cells arrested in S and G2. Neoplasia. 2000;2:460–470. doi: 10.1038/sj.neo.7900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basile JR, Eichten A, Zacny V, Munger K. NF-kappaB-mediated induction of p21(Cip1/Waf1) by tumor necrosis factor alpha induces growth arrest and cytoprotection in normal human keratinocytes. Mol Cancer Res. 2003;1:262–270. [PubMed] [Google Scholar]
- 26.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U S A. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alpan RS, Pardee AB. p21WAF1/CIP1/SDI1 is elevated through a p53-independent pathway by mimosine. Cell Growth Differ. 1996;7:893–901. [PubMed] [Google Scholar]
- 28.Gorospe M, Liu Y, Xu Q, Chrest FJ, Holbrook NJ. Inhibition of G1 cyclin-dependent kinase activity during growth arrest of human breast carcinoma cells by prostaglandin A2. Mol Cell Biol. 1996;16:762–770. doi: 10.1128/mcb.16.3.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane (DIM) induces a G(1) cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis. 2002;23:1297–1305. doi: 10.1093/carcin/23.8.1297. [DOI] [PubMed] [Google Scholar]
- 30.Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, Ding W, Hutchins B, Hockenberry T, Kirschmeier P, Greene JR, Liu S. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–3654. doi: 10.1038/sj.onc.1206477. [DOI] [PubMed] [Google Scholar]
- 31.Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- 32.Huang G, Elferink CJ. Multiple mechanisms are involved in Ah receptor-mediated cell cycle arrest. Mol Pharmacol. 2005;67:88–96. doi: 10.1124/mol.104.002410. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, Rajasekaran AK, Das GM. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006;281:9837–9840. doi: 10.1074/jbc.C600001200. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Yao J, Pisha E, Yang Y, Hua Y, van Breemen RB, Bolton JL. Oxidative DNA damage induced by equine estrogen metabolites: role of estrogen receptor alpha. Chem Res Toxicol. 2002;15:512–519. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- 35.Chen X, Danes C, Lowe M, Herliczek TW, Keyomarsi K. Activation of the estrogen-signaling pathway by p21(WAF1/CIP1) in estrogen receptor-negative breast cancer cells. Journal of the National Cancer Institute. 2000;92:1403–1413. doi: 10.1093/jnci/92.17.1403. [DOI] [PubMed] [Google Scholar]
- 36.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 37.Chen RM, Chou MW, Ueng TH. Induction of cytochrome P450 1A in hamster liver and lung by 6-nitrochrysene. Arch Toxicol. 1998;72:395–401. doi: 10.1007/s002040050519. [DOI] [PubMed] [Google Scholar]
- 38.Chen RM, Chou MW, Ueng TH. Induction of cytochrome P450 1A1 in human hepatoma HepG2 cells by 6-nitrochrysene. Toxicol Lett. 2000;117:69–77. doi: 10.1016/s0378-4274(00)00242-3. [DOI] [PubMed] [Google Scholar]