

Abstract
A key goal of pharmacogenetics — the use of genetic variation to elucidate inter-individual variation in drug treatment response — is to aid the development of predictive genetic tests that could maximize drug efficacy and minimize drug toxicity. The completion of the Human Genome Project and the associated HapMap Project, together with advances in technologies for investigating genetic variation, have greatly advanced the potential to develop such tests; however, many challenges remain. With the aim of helping to address some of these challenges, this article discusses the steps that are involved in the development of predictive tests for drug treatment response based on genetic variation, and factors that influence the development and performance of these tests.
The clinical premise of pharmacogenetics is based on more accurate, and hence more useful, predictive medicine. The underlying assumption is that a person's genotype will determine whether they will respond to, or have toxicity from, a given medication. It appears that genetic variation, along with clinical and behavioural information, will be useful in this regard. However, the relative importance of genetic versus clinical factors over the broad range of medications currently in clinical use is yet to be determined.
Existing pharmacogenetic tests can be broadly divided into two categories: identifying responders or non-responders to a given drug and identifying those individuals at risk of serious adverse events.
Pharmacogenetics has benefited from the rapid advances associated with the Human Genome Project and its derivative the HapMap Project (see Further information). In 2000, an overall map of the human genome was released1,2, and in the past 5 years, the HapMap Project has catalogued human genetic variation, which is defined as “genetic variation with the allele frequency of more than 10%”, in three ethnic groups: Caucasians, Africans and Asians (both Han Chinese and Japanese). This has allowed researchers to use linkage disequilibrium to find genetic variation related to common complex traits, such as drug treatment response3. As can be seen from FIG. 1, there are two broad types of variation in the human genome: common genetic variation and rare genetic variation. Common genetic variation occurs with an allele frequency of more than 1%, and tends to be evolutionary older and to affect gene regulatory function4. Rare genetic variation occurs with an allele frequency of less than 1%, and tends to arise more recently in human evolution and, when present at non-synonymous sites in coding regions, it is more likely to alter protein structure4. The relationship of these population genetic principles to pharmacokinetic and pharmacodynamic responses are unknown at present but worthy of further investigation.
Figure 1. Common genetic variation and rare genetic variation with allele frequencies.
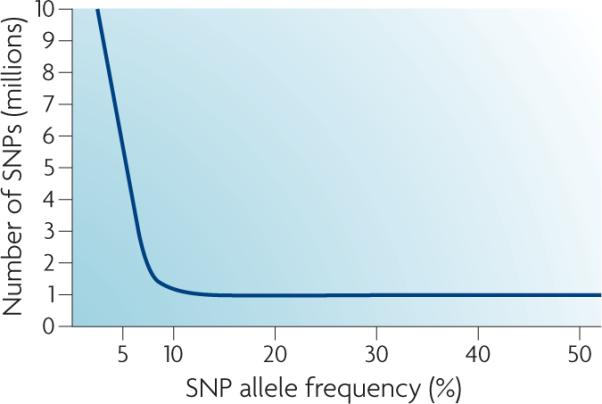
Genetic variants that occur with an allele frequency of less than 1% are more likely, by definition, to be rare. They are more likely to be non-synonymous coding variants, which will influence pharmacokinetic responses, and to be under natural selection. Variants that occur with an allele frequency of more than 1% are more likely, by definition, to be common. They are more likely to influence gene regulation, which is important in determining pharmacodynamic responses, and are unlikely to be under significant natural selection pressure. SNP, single nucleotide polymorphism.
With the rapid expansion of knowledge and tools that enable the study of complex human genetic traits, and with genome-wide data rapidly accumulating, now is an opportune time to focus on the factors affecting the development of pharmacogenetic tests. In this article, we discuss key factors in the development of pharmacogenetic tests. These include those that influence the rationale for developing such tests for particular purposes, those that affect test performance in general, and those relating to test regulation and uptake by clinicians. We also propose an approach to predict treatment responses in complex clinical drug-response phenotypes. We are focusing this manuscript on actual genetic variation and its influence on drug treatment response, recognizing that transcriptomic and proteomic approaches to predict drug response are also possible and desirable (for a recent review see Ross et al.5). Our focus is also on the identification and application of knowledge of the influence of genetic variation on the effects of approved drugs, rather than on the application of pharmacogenetic tests in the development of investigational drugs, although many of the points highlighted could also be relevant in this case. For a discussion of this topic, see Jain6.
Although the use of pharmacogenetic testing to guide therapy is a new approach, the use of laboratory testing to guide therapy is not novel. For instance, serum creatinine levels are routinely used to guide drug dosage, and antibiotic sensitivity testing is used to select drugs that work for specific conditions. Other examples include following the international normalized ratio after warfarin treatment, and monitoring the concentrations of some drugs such as phenytoin, lithium and gentamicin to guide dose selection and the timing of dosing. Pharmacogenetic testing can be seen as an addition to the treatment algorithm that should not be difficult to implement, in theory. There are various issues including education and cost that could have an important impact in this respect in practice, as discussed later in this article.
Currently, pharmacogenetic tests are used for assigning the dose of 6-mercaptopurine based on the thiopurine S-methyltransferase (TPMT) genotype7-9; to decide whether or not to use codeine or tamoxifen based on the cytochrome P450 family 2, subfamily D, polypeptide 6 (CYP2D6) genotype10; and to design optimal treatment regimens for colon cancer using irinotecan with the UDP glucuronosyltransferase 1 family, polypep-tide A1 (UGT1A1) genotype. In addition, pharmacogenetic tests have recently been developed to define warfarin dosage more accurately on the basis of vitamin K epoxide reductase complex, subunit 1 (VKORC1) and CYP2C9 genotypes (TABLE 1).
Table 1.
Examples of recently approved or emerging pharmacogenetic tests
| Gene | Drug | Consequence |
|---|---|---|
| KIT mutations* | Imatinib mesylate‡ | Altered survival |
| TPMT | Mercaptopurine and azathioprine* | Neutropaenia |
| UGT1A1* | Irinotecan‡ | Neutropaenia |
| CYP2C9/VKORC1 | Warfarin | International normalized ratio |
| CYP2D6* | 5-HT3 receptor antagonists | Inadequate anti-emetic control |
| Antidepressants | Inadequate benefit | |
| ADHD drugs | Inadequate benefit | |
| Codeine derivatives* | Inadequate benefit |
US Food and Drug Administration (FDA)-approved device.
FDA package insert information. 5-HT3, 5-hydroxytryptamine (serotonin) 3; ADHD, attention-deficit/hyperactivity disorder; CYP, cytochrome P450; TPMT, thiopurine S-methyltransferase; UGT1A1, UDP glucuronosyltransferase 1 family, polypeptide A1; VKORC1, vitamin K epoxide reductase complex, subunit 1.
Factors affecting test development
The clinical consequences of pharmacogenetic tests differ substantially depending on the setting. The utility of a clinical test is dependent on the clinical options available. If there are no clinical options then the test only provides a warning of potential events to come. If there are clinical options then the clinician can err on the side of either false positives or false negatives depending on the clinical risk of making an erroneous decision. The clinical context, the availability of other tests in the clinical environment, as well as the frequency and severity of the drug-response phenotype, all have important roles in determining how useful, and widely applied, a given predictive test will be. This therefore underlies whether a test for a particular drug response should be developed in the first place. For instance, the use of the UGT1A1 test to prevent irinotecan toxicity represents a different clinical context compared with the use of the VKORC1 test to predict optimal warfarin dosage.
Just as there are different clinical models for drug treatment response, there are different genetic models as well. Some drug effects behave in a Mendelian fashion and are due to a single coding variant in a single gene. An example of this would be the common genetic variant in the CyP2C9 enzyme — a C to T nucleotide substitution at codon 430, which results in a change from arginine to cysteine at amino-acid 144 (see Further information). This mutation in CYP2C9 causes a decrease in the metabolism of warfarin and thus, an increased risk of bleeding compared with a person without this variant receiving the same dose11. In other cases, the drug effect might be due to multiple single nucleotide polymorphisms (SNPs), some coding and some regulatory, in a single gene. An example of this would be variants in the β2 adrenergic receptor (ADRB2), which influences the response to inhaled short-acting β-agonists12. These multiple variants could be combined to create a haplotype or combination of variants.
In contrast to these single gene effects, many drug treatment response phenotypes are complex, produced by multiple coding and regulatory variants in multiple genes that often interact in a signalling pathway. In these cases, each variant could contribute to the variance in the phenotype and there is no clear model of genetic inheritance. Genetic factors that influence whether a drug treatment response is complex include mode of inheritance (recessive versus dominant or additive); pleiotropy; incomplete penetrance; and epistasis, due to gene–environment interactions and environmental phenocopies. All of these factors contribute to the complexity of the response phenotypes. As of yet we have no good examples of such complex drug treatment response tests.
The clinical phenotype itself may be difficult and elusive. In some cases the definition of a responder or a non-responder is clear, whereas in others it can be arbitrary. As shown in FIG. 2, the response to inhaled corticosteroids, as measured by the forced expiratory volume in 1 second (FEV1), can be considered a linear relationship. One might also categorize the negatives or non-responders (the bottom 30% of the distribution), as individuals who have a non-response, or a clinically poor response phenotype, to inhaled corticosteroids as characterized by FEV1. This example is emblematic of a certain class of genetic tests in which a continuous phenotype, here FEV1, is dichotomized to create a binary variable with a discrete response. In many clinical situations, the response might involve a biomarker (for example, the cholesterol level in response to a statin medication) rather than a specific disease phenotype. linear modelling can also be used to model continuous drug-response phenotypes such as those already noted. Thus, there are multiple models for the separation of drug-response phenotypes. However, if the purpose of the test is to separate groups of patients according to response phenotypes (for example, responders or nonresponders) then the separation of these two groups should be made using well-recognized clinical response measures when they are available. For instance, we know that the degree of change in bone density affects the risk of bone fracture, and that the magnitude of change in low-density lipoprotein or blood pressure levels is associated with cardiovascular risk, and so these can be assessed continuously or as dichotomous phenotypes.
Figure 2. response to inhaled corticosteroids, as measured by FEV1.

This shows the distribution of improvement in forced expiratory volume in 1 second (FEV1), a measure of lung function, in response to inhaled corticosteroids taken over a 6−8 week period. The x axis is change in FEV1 and the y axis is the number of subjects having that level of change in FEV1 in three different studies: an adult clinical trial (Adult study), the childhood asthma management program (CAMP) and one of the Asthma Clinical Research Network (ACRN) clinical trials. The histogram shows a wide range of response between individuals to the inhaled corticosteroid medication. This figure is modified with permission from REF. 32 © (2004) Oxford University Press.
It is important to recognize that while pharmacogenetic tests are intended to be applied to individual patients, their predictive power is based on population samples of patients and their drug responses. This means that pharmacogenetic testing can be used for predictive medicine but not for truly personalized medicine, as the testing is based on how a group of individuals with the same polymorphisms and the same drug-response phenotype would behave in a similar clinical situation.
Because pharmacogenetic testing can be predictive of treatment outcome, its use differs from other clinical tests. Clinical tests (such as serum drug levels) are less specifically diagnostic of disease and drug response, and often require integration with other clinical variables to have any clinical utility. However, similar to other clinical tests (such as serum creatinine, serum cholesterol, serum drug concentrations of gentamicin or phenytoin, or fasting glucose level), the predictive capabilities of pharmacogenetic testing are population based.
The expectation is that genetics will allow more precise prediction than clinical variables alone. This is partly because of the stability of DNA in the laboratory compared with other biological samples such as RNA or protein. This is in addition to the fact that germline DNA does not change in an individual's lifetime and the robust nature of genetic tests, which provide clear data on DNA sequence as opposed to a measurement of a linear variable such as the density of an RNA species on an array or microchip, or the presence of a protein on a Western blot. However, more important than these biochemical properties, the accuracy of a given prediction depends on whether the full set of relevant genes (and their genetic variants) that are related to the phenotype of interest has been delineated. Furthermore, accuracy will also depend on clinical and environmental factors that influence the drug response, and on the genetic and predictive models that relate genetic factors to drug response. It remains unknown whether transcriptomic, or proteomic predictive tests are superior to genetic tests. Measurement of protein levels will probably provide the most specific model for drug treatment response, but few comparisons between these approaches and a genetic one are available.
Predictive genetic tests could also be of value in the drug development process, rescuing drugs that would not pass approval by the US Food and Drug Administration (FDA) in Phase III trials because of the susceptibility of a subset of patients to an adverse event. Tests of high predictive accuracy that are able to identify patients who are susceptible to serious adverse events, or to treatment non-response, could help salvage drug approval. An example of this is the use of major histocompatibility complex, class I, B (HlA-B)*5701 testing in patients with HIv who are receiving abacavir13.
Finally, drug safety and drug efficacy are central to developing clinical pharmacogenetic tests. In general, drugs are approved because they have been shown in Phase III clinical trials to benefit individuals treated with the medication relative to placebo, or relative to the standard of care. However, there are still individuals at the extreme of the drug-response distribution who do not respond, as well as some individuals who have adverse responses. Although adverse responses might be rare, and might only come to light in the post-marketing surveillance of a medication (or when Phase III trials are initiated), non-responders are often ignored unless they represent a significant proportion of the patient population. Pharmacogenetic testing can be used to identify both those who are unlikely to respond and those that are most likely to have an adverse reaction, thus maximizing the clinical utility of a drug.
Factors influencing test performance
Although many technical and logistical hurdles must be overcome before a test can be implemented in a clinical laboratory, several additional factors can also influence the clinical impact of a pharmacogenetic test. Many of these factors cannot be controlled or manipulated, but are clinically important. Among the genetic factors that influence test performance are allele frequency, linkage disequilibrium, epistasis and the number of functional gene variants.
Allele frequency has important practical implications for pharmacogenetic testing. variants with a low allele frequency might have substantial influence on toxicity risk or effectiveness, but the small number of patients with the genotype of interest could make it difficult to establish the clinical and/ or economic value of prospective testing. An example is the use of CYP2C19 genotype for predicting clinical resistance to proton pump inhibitors in gastric ulcer therapy. The low frequency of a homozygous variant genotype in European-derived white populations is largely responsible for the low uptake in CYP2C19 testing in the Western world. The opposite is true in East Asia where the at-risk genotype occurs in ∼20% of patients14,15.
Linkage disequilibrium also has an influence on pharmacogenetic testing. The power of linkage disequilibrium for discovering genes of interest in the entire genome is clear, but problematic. The variant that is associated with the clinical phenotype of interest is not necessarily causative. This was seen with the discovery of VKORC1 haplotypes, which are associated with greater sensitivity to the anticoagulant effects of warfarin16. It now appears that the original predictive SNPs are actually SNPs in linkage disequilibrium for a promoter region SNP that alters VKORC1 transcription and thereby warfarin dose requirements.
Linkage disequilibrium occurs for most of the world's population, but for many genes there are substantial differences in the degree of linkage disequilibrium observed across populations. Furthermore, there are often multiple genotype explanations for an altered phenotype, making linkage disequilibrium inadequate to detect the causative variants. This is the case for TPMT and thiopurine sensitivity, for which 85−90% of at-risk patients will be detected using only three non-synonymous SNPs, but over 20 causative variants have been observed across the world17,18. Allele heterogeneity, the concept that all causative SNPs do not produce identical phenotypes, is also important in the interpretation of pharmacogenetic tests. For example, genetic variants in CYP2D6 may range from variants that are non-functional, giving rise to poor metabolizers, to variants that are intermediate in function.
Epistasis or gene–gene interactions may also affect the outcome of pharmacogenetic tests. Although looking at several genes together makes for complex analysis, by analysing one gene at a time we might miss a clinically important interaction between variants within a gene or between genes.
Recently, there has been evidence for the need to consider pharmacogenetics in a quantitative rather than qualitative manner. A hallmark of cancer cells is aneuploidy (an abnormal number of chromosomes) primarily at the level of large segments of genomic loss or amplification. The altered regions often contain genes of biological importance, sparing genes whose function might not be directly related to tumour biology but may nevertheless have some pharmacogenetic function19,20. However, recent studies in leukaemia have demonstrated that the interaction between gene polymorphism and gene amplification can have a quantitative functional impact21. A patient with a TPMT heterozygous genotype (for example, G/A) and three copies of TPMT in their leukaemia blast cells actually has a G/G/A or G/A/A genotype, which affects the enzyme's activity. This phenomenon cannot be readily detected by the genotyping technologies that are used in clinical laboratories, thus illustrating the need for diagnostic approaches that provide the necessary information to integrate such data into practice22. A similar distinction has also been observed in the context of solid organ transplantation, in which the genotype of the donor organ was found to be more predictive of drug-associated nephrotoxicity rather than the recipient's genome23.
Clinical or biological factors that influence the performance of a pharmacogenetic test are not necessarily related to genetic variation. Indeed, drug–drug and nutrient–drug interactions are important sources of discrepancy between genotype and phenotype. For example, the CYP2D6 genotype of patients with breast cancer appears to influence the metabolism of tamoxifen24. Nutritional factors, such as folate, have also influenced pharmacogenetic tests for drugs such as warfarin. In the case of warfarin, folate intake also influences the predictive value of genetic tests for VKORC1 as a predictor of anticoagulant response25. These findings indicate that, in addition to clinical information, pharmacogenetic tests will provide added information about the pharmacological milieu of a patient. However, these tests will not replace good clinical management of a patient's treatment regimen, and, in order to be clinically valuable in treatment decisions, they must be interpreted by professionals that are capable of taking the clinical context into account.
Expertise in the interpretation of pharmacogenetic tests is particularly challenging. By definition, a pharmacogenetic test involves a therapy and the condition it is designed to treat, and it cannot be of value if it is considered in an isolated manner. The test should predict a clinically meaningful difference in drug effect or it will have little value, even if its analytical specifications are robust. Interpretation of a clinically meaningful difference inevitably depends on both the disease and the context, and this decision is best made by clinical experts. When pharmacogenetic tests have been used in the treatment of osteoporosis, endocrinologists and rheumatologists have defined the degree of change in biomarkers for bone strength that affect the actual risk of bone fracture. In some areas of medicine — such as the treatment of hypertension, hyperlipidaemia, osteoporosis, depression or some cancers — clearly defined clinically relevant changes due to drug therapy have been defined by consensus panels, and these should be used in the interpretation of tests aimed at improving therapy. That said, in many areas of pharmacotherapy, such clear guidelines do not exist and the response to treatment remains frustratingly variable. In these cases, we must await the development of meaningful biomarkers of treatment response before the value of some tests can be assessed.
Physicians remain the most important medical decision makers and their involvement in the development and use of pharmacogenetic tests is crucial. However, given that pharmacogenetics is a rapidly evolving field, ideally, a multidisciplinary team approach to pharmacogenetic test interpretation, involving physicians, pharmacists, geneticists, clinical chemists and nurses, should evolve. Without better physician education, there is a real danger that pharmacogenetic tests will be used in ways that do not optimize health care, or may actually harm patients by resulting in the use of the wrong drug at the wrong time in the wrong patient.
Finally, all clinical tests, including genetic tests, are judged by their intrinsic test characteristics; that is, their sensitivity, specificity, reproducibility and precision (BOX 1).
Factors influencing test approval/uptake
Most clinical tests have been developed and released into clinical practice with limited attention to their characteristics and performance. Although tests for research purposes require no approval, any laboratory test that will be used for patient care, including any pharmacogenetic test, must comply with the Clinical laboratory Improvement Amendment of the United States or comply with the In Vitro Diagnostic Directive of the European Union; that is, the test is able to pass sufficient laboratory standards to receive a Committee Europe mark. Equivalent standards exist in Japan and other Asian countries.
Box 1| Clinical test characteristics.
Sensitivity and specificity
Sensitivity is defined as the number of subjects in the test that test positive and have a positive drug response divided by the total number of subjects who have a positive drug response. Specificity is defined as the number of subjects in the test that test negative and have no drug response divided by the total number of subjects who have no drug response31. Sensitivity and specificity are intrinsic characteristics of a test. However, a test's predictive value depends not only on sensitivity and specificity, but also on response prevalence; that is, how well the drug works in how many people. A high response prevalence is associated with more false positives. A low response prevalence is associated with more false negatives. One of the ways of quantifying the clinical performance of a given predictive test is to construct a receiver operating characteristic (ROC) curve31.
Reproducibility and precision
Additional concepts that are crucial to interpreting diagnostic or prognostic tests are reproducibility and precision. Reproducibility is simply the capability of a test to give the same results repeatedly in the same individuals. Characteristics that can influence reproducibility include the testing method (for example, sequencing of both DNA strands (the gold standard) or genotyping by Taqman or by oligonucleotide array), and the quality of the DNA sample. A test should be robust, that is, it should function well in all types of clinical situations. It should also be highly reproducible when tested on individuals under standard conditions. In the case of tests for genetic variants that are relevant to therapeutic response, the stability of DNA lends itself to clear and reproducible results, but every test must be validated in the clinical environment for which it is intended.
These standards include reproducibility and precision — parameters that validate that the test actually measures what it is intended to measure. Tests that are not highly reproducible and precise have little chance of being clinically useful. It is of note that the US FDA also has a process for approving clinical tests, including genetic tests, and has recently established guidelines for this process that include the demonstration of clinical utility, a requirement that is not imposed by the regulatory agencies in other countries26. In addition, the FDA requires studies that determine test performance, describe the rationale for the cut-off used for the test, precision and reproducibility data, analytic specificity data (indicating no interference or cross-reactivity), as well as details about the analysis and conditions of the test27. Although FDA approval is not currently a requirement for all genetic tests, this regulatory question remains open. The FDA has approved a small number of pharmacogenetic tests, including the Amplichip CyP450 Test (Roche), which analyses CYP2D6 and CYP2C19, and the Invader UGT1A1 Molecular Assay (Third Wave Technologies/Genzyme), which is used in guiding irinotecan dosage. The FDA-approved labels for azathioprine, 6-mercaptopurine, atomoxetine, irinotecan and warfarin all include references to the availability of a pharmacogenetic test that will aid health-care professionals make therapeutic decisions.
Medical science has already provided an array of clinical variables with which to predict response to drug therapy. Physicians use these routinely, and they are well integrated into the practice of medicine. Thus, the clinical value of any new test depends, in part, on what is currently available and routinely used. For example, the effectiveness of statins is usually followed by the measurement of cholesterol and lipoprotein subfractions. In the cancer field, tumour stage and grade are used as measures of disease severity, and oncologists routinely use them to predict how patients will respond to treatment. In breast cancer, for example, oestrogen and progesterone receptor status, HER2 status, and in some patients a 21-gene expression panel can be measured, which, together with the tumour grade and stage are used to determine the most appropriate therapy28. A woman with an oestrogen receptor-positive tumour is most likely to receive an endocrine treatment, such as tamoxifen or an aromatase inhibitor, to target that receptor.
In the case of antihypertensive agents, their effectiveness is monitored by the measurement of blood pressure, and changes in dose or drug are frequently made as a result. Similarly, the extent of β-blockade is assessed by measurement of the heart rate; the adverse effects from dopaminergic blockade by watching for extrapyramidal signs; and the adverse effects of anxiolytic therapy by monitoring patients for excessive sedation or memory loss. For a new pharmacogenetic test to be useful in these situations, it should not only be robust in a clinical environment (involving real patients taking multiple drugs and with a range of disease severity), but it should also be more predictive than current approaches. The field of breast-cancer treatment is particularly rich in genomic predictive tests and similar tests are becoming available in the treatment of other cancers, in which both the somatic DNA, present in the tumour itself, and the patient's germline DNA are used29. Both may be useful, and the testing characteristics and performance for each should be similar.
An approach to prediction
Pharmacogenetics is still struggling to identify clinically meaningful treatment responses that can be predicted by genetic testing. Here, we describe one of the approaches that is being used by investigators in the Pharmacogenetics Research Network (PGRN) to predict treatment response in complex clinical drug-response phenotypes (determined by multiple clinical variables and multiple genes that interact in a network or pathway) (FIG. 3). Currently, this approach is being used by the Pharmacogenetics of Asthma Treatment Group of the PGRN to develop genetic tests that predict asthma exacerbations, response to inhaled corticosteroids and response to inhaled β-agonists.
Figure 3. Proposed methodology for developing pharmacogenetic predictive tests.
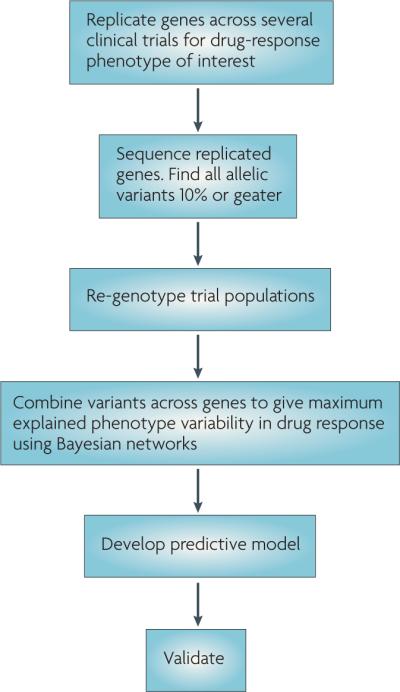
This flow diagram describes the steps to take in the development of a hypothetical predictive pharmacogenetic test. The cut-off of 10% in step two is arbitrary. The methodology for combining variants in step four could be any multivariate approach not just Bayesian networks. Validation (step six) will probably occur in samples from the general population rather than in randomized controlled trials.
The first step is to use linkage dis-equilibrium within genes to identify a set of genes that replicate an association with a drug-response phenotype across several clinical trials in a single ethnic group. The critical question at this stage is what constitutes adequate sample sizes for prediction. This is dependent on the SNP set that is being used for prediction: is it a subset of a whole genome (300,000−1,000,000,000 SNPs) or just a subset of SNPs in candidate genes? The problem of too many predictors for a small sample of cases and controls is well known in bioinformatics: the developed prediction model is not generalizable and hence not reproducible in a second population. Initial predictive models for pharmacogenetics were built with as few as 200 cases and controls, with candidate gene sets of several thousand SNPs. Now, they are utilizing a similar set of cases and between 500,000−1,000,000 SNPs.
The bioinformatics approach used to investigate the epistatic interactions between these predictive gene sets relies on software that utilizes Bayesian networks, a form of statistical modelling, that takes into account the dependencies between the outcome (in this case drug treatment response as a dichotomous variable, that is, responder/ non-responder) and multiple SNPs. The software (Bayes’ Discoverer) evaluates each individual SNP as a potential predictor node of an epistatic network based on the ratio of two probabilities (Bayes factors) that quantitatively assess the marginal contribution of the SNPs to the overall predictive model30. The resulting network models seem to have reasonable predictive power based on the receiver operating characteristic (ROC) curve area and have been shown to replicate across trials of one ethnic group.
Box 2 | Ethical issues.
Given the recent increased focus on the privacy of health-care data, and the high likelihood that all individuals in the population harbour susceptibility alleles for some pharmacogenetic phenotype, as well as the goals of genetic non-discrimination, the possibility of discrimination on the basis of pharmacogenetic test results seems limited. Despite the limited possibility of stigmatization based on disease susceptibility, there remains concern about the differential application of these tests in specific ethnic groups. The possibility that results from a predictive test will differ between racial and ethnic groups is a concern. The genetic context here refers explicitly to varying linkage disequilibrium patterns and differing allele frequencies based on the evolutionary history of different populations. This may be an important concern particularly in the first generation of predictive tests, which will primarily be based on linkage disequilibrium rather than on functional variation. This could lead to varying predictive performance in different racial and ethnic groups. In the future development and use of pharmacogenetic tests, it will be important that all communities become active participants and that patients, physicians and health-care providers are made aware of their existence. Ancestral informative markers, that is, genetic variation that is associated with human populations based on their evolutionary history, will aid in understanding linkage disequilibrium patterns among racial and ethnic groups.
Developing the initial model is done with linkage disequilibrium tagging SNPs, but it is unlikely that these will be generalizable to other racial groups. Although currently untested, it stands to reason that generalizability can be achieved with linkage disequilibrium tagging SNPs if these SNPs are known for other ethnic groups besides the group that was initially chosen. Thanks to data from the HapMap Project, linkage disequilibrium tagging SNPs for the whole genome have been identified for several racial groups. However, some addition and substitution may need to be done to use a linkage disequilibrium tagging approach for epistatic prediction before all functional variants are defined.
The second step is to try to replicate the epistatic interaction model in an independent clinical trial population and explain a significant portion of the area under the ROC curve. Once replication is achieved, then the third step is to define the functional variants in the genes identified using the Bayesian network modelling approach as described above. In this step the functional variants rather than the variants based on linkage disequilibrium are used in an iterative fashion to find the epistatic interactions between the functional variants leading to the clinical phenotype. Then, one could build a new model based on these functional variants as predictors of the clinical phenotype. It is likely that this will have broader generalizability than a model built using linkage disequilibrium SNPs. After the model is validated, either in an observational study or in a clinical trial, these variants can be put on a chip to determine whether the test is adequately sensitive and specific for predicting clinical outcomes in a variety of clinical populations.
Alternative or complimentary approaches to the one outlined here would include use of gene-expression signatures to model response, and using linear or logistic regression to focus on main effects rather than epistasis. It is likely that all of these models will be used by different groups of investigators.
It is also important to develop interpretation guidelines for the test, especially regarding how it might influence drug-prescribing patterns. The guidelines would describe either a change in dose or selection of drug types to use as a result of the test. Finally, it would be desirable to validate such a predictive model through testing in the general population, as prospective randomized genotype-guided dosing trials will take too long and be too costly to perform routinely and thus, validation is likely to be performed in observational cohorts.
Conclusions
Expectations are high regarding the application of the results of the Human Genome Project to clinical medicine; one of the most direct applications of the data will be in the area of prediction, including pharmacogenetics. With the recent enactment of the Genetic Information Nondiscrimination Act in the United States this year, discrimination concerns would appear to be less of an issue, although ethical concerns in genetic testing are still relevant (BOX 2).
Questions remain as to the realistic time frame within which to expect the development of pharmacogenetic tests and how the field will move forward. The trend is to use ever larger sets of SNPs, iterative multivariate modelling and replication with ROC area as the best approach to evaluate the results. We propose that developmentally useful tests in pharmacogenetics for complex traits will become increasingly available at an exponential rate over the next 10 years. Existing clinical pharmacogenetic tests and prototypes have already been developed, or are under development by research groups within the PGRN (see Further information). The next tests are likely to take advantage of whole-genome data and to include multiple gene variants. It will take time to integrate these emerging tests into daily practice given the need for iterative development, validation and ultimately FDA approval. Even currently available tests approved by the FDA are not used routinely in all clinical practices and many physicians feel that they can predict drug response just as well with their clinical experience as they can with a genetic test. However, within 10 years, clinically useful predictive tests with reasonable sensitivity and specificity are likely to be available for many drug-response phenotypes. It is likely that these will cost under US$100 per test and that they will be interpreted by trained clinicians. Despite this, these tests will not substitute for good clinical judgment and will not replace careful thought by the practitioner about the dose and choice of drug. Predictive medicine will be one of the first major applications of the human genome to clinical medicine, and more precise prediction of drug response will improve drug prescribing for all patients.
Glossary.
Environmental phenocopy
A clinical case of a complex trait due solely to environmental factors.
Epistasis
The interaction or interdependence of two or more genes.
Incomplete penetrance
Occurs when less than 100% of a population with an identical mutant genotype display the associated phenotype.
Linkage disequilibrium
The nonrandom association of alleles in the genome.
Mode of inheritance
Dominant mode of inheritance occurs when only one copy of the allele is necessary to produce the phenotype. Recessive mode of inheritance occurs when both copies of the allele are necessary to produce the phenotype.
Pleiotropy
A single mutation that has more than one biological effect or phenotype.
Receiver operating characteristic (ROC) curve
A curve that plots I-sensitivity on the y axis and specificity on the x axis. The area under this curve is a measure of test performance.
Severe adverse event
An event that occurs less than 1 in 10,000 administrations of the medication and is life threatening.
Acknowledgements
We would like to thank Richard M. Weinshilboum, MD and the White Paper subcommittee of the PGRN network for valuable suggestions. Financial support is also acknowledged: U01 HL065899 (S.T.W.); U01 GM63340 (H.L.M.); UO1 GM061373 (D.A.F.); DA 20830 (N.L.B.); U01 GM074492 (J.A.J.); GM61393 (M.J.R. and M.E.D.); and GM61390 (K.M.G.).
Footnotes
The authors have written this article on behalf of the Pharmacogenetics Research Network (PGRN) investigators.
DATABASES
OMIM: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
ADRB2 | CYP2C19 | CYP2C9 | CYP2D6 | TPMT | UGT1A1 | VKORC1
FURTHER INFORMATION
CYP2C9 allele nomenclature: http://www.cypalleles.ki.se/cyp2c9.htm
HapMap Project: http://www.hapmap.org
Human Genome Project: http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml
Pharmacogenetics research Network: http://www.nigms.nih.gov/Initiatives/PGRN
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Scott T. Weiss, Channing Laboratory, Brigham and Women's Hospital, 181 Longwood Ave, Boston, Massachusetts 02115, USA.
Howard L. McLeod, University of North Carolina, Chapel Hill, Campus BOX 7360, 3203 Kerr Hall, Chapel Hill, North Carolina 27599, USA.
David A. Flockhart, Indiana University School of Medicine, Wishard Hospital, WD OPW 320, 1001 West 10th Street, Indianapolis, Indianapolis 46202, USA.
M. Eileen Dolan, Department of Medicine, Committee on Clinical Pharmacology and Pharmacogenomics, 5841 South Maryland Avenue, Box MC2115, University of Chicago, Chicago, Illinois 60637−1470, USA..
Neal L. Benowitz, Division of Clinical Pharmacology and Experimental Therapeutics, University of California, San Francisco, BOX 1220, San Francisco, California 94143−1220, USA.
Julie A. Johnson, University of Florida College of Pharmacy, Department of Pharmacy Practice, P.O. BOX 100486, Gainesville, Florida 32610−0486, USA.
Mark J. Ratain, University of Chicago, 5841 South Maryland Avenue, MC 2115, Chicago, Illinois 60637−1470, USA.
Kathleen M. Giacomini, University of California, San Francisco, School of Pharmacy, Box 0446 513 Parnassus Avenue, San Francisco, California 94143−0446, USA.
References
- 1.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Altshuler D, et al. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urban TJ, et al. Functional genomics of membrane transporters in human populations. Genome Res. 2006;16:223–230. doi: 10.1101/gr.4356206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross JS, Symmans WF, Pusztai L, Hortobagyi GN. Pharmacogenomics and clinical biomarkers in drug discovery and development. Am. J. Clin. Pathol. 2005;124:S29–S41. doi: 10.1309/XYQAFANAPYNC6X59. [DOI] [PubMed] [Google Scholar]
- 6.Jain KK. Challenges of drug discovery for personalized medicine. Curr. Opin. Mol. Ther. 2006;8:487–492. [PubMed] [Google Scholar]
- 7.Yates CR, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann. Intern. Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 8.Relling MV, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J. Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 9.Schaeffeler E, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype–genetype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004;14:407–417. doi: 10.1097/01.fpc.0000114745.08559.db. [DOI] [PubMed] [Google Scholar]
- 10.Caraco Y, Sheller J, Wood AJ. Pharmacogenetic determination of the effects of codeine and prediction of drug interactions. J. Pharmacol. Exp. Ther. 1996;278:1165–1174. [PubMed] [Google Scholar]
- 11.Higashi MK, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287:1690–1698. doi: 10.1001/jama.287.13.1690. [DOI] [PubMed] [Google Scholar]
- 12.Hawkins GA, et al. Sequence, haplotype and association analysis of ADRβ2 in multi-ethnic asthma case–control subjects. Am. J. Respir. Crit. Care Med. 2006;174:1101–1109. doi: 10.1164/rccm.200509-1405OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallal S, et al. for the PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 14.Furuta T, et al. Influence of CYP2C19 pharmacogenetic polymorphism on proton pump inhibitor-based therapies. Drug Metab. Pharmacokinet. 2005;20:153–167. doi: 10.2133/dmpk.20.153. [DOI] [PubMed] [Google Scholar]
- 15.Desta Z, Zhao X, Shin JG, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 2002;41:913–958. doi: 10.2165/00003088-200241120-00002. [DOI] [PubMed] [Google Scholar]
- 16.Rieder MJ, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005;352:2285–2293. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- 17.Relling MV, Pui CH, Cheng C, Evans WE. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107:843–844. doi: 10.1182/blood-2005-08-3379. [DOI] [PubMed] [Google Scholar]
- 18.Marsh S, McLeod HL. Cancer pharmacogenetics. Br. J. Cancer. 2004;90:8–11. doi: 10.1038/sj.bjc.6601487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rae JM, et al. Genotyping for polymorphic drug metabolizing enzymes from paraffinembedded and immunohistochemically stained tumor samples. Pharmacogenetics. 2003;13:501–507. doi: 10.1097/00008571-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Marsh S, Mallon MA, Goodfellow P, McLeod HL. Concordance of pharmacogenetic markers in germline and colorectal tumor DNA. Pharmacogenomics. 2005;6:873–877. doi: 10.2217/14622416.6.8.873. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Q, et al. Karyotypic abnormalities create discordance of germline genotype and cancer cell phenotypes. Nature Genet. 2005;37:878–882. doi: 10.1038/ng1612. [DOI] [PubMed] [Google Scholar]
- 22.McLeod HL, Marsh S. Pharmacogenetics goes 3D. Nature Genet. 2005;37:794–795. doi: 10.1038/ng0805-794. [DOI] [PubMed] [Google Scholar]
- 23.Hauser IA, et al. ABCB1 genotype of the donor but not of the recipient is a major risk factor for cyclosporine-related nephrotoxicity after renal transplantation. J. Am. Soc. Nephrol. 2005;16:1501–1511. doi: 10.1681/ASN.2004100882. [DOI] [PubMed] [Google Scholar]
- 24.Jin Y, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl Cancer Inst. 2005;97:30–39. doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 25.Custodio das Dores SM, et al. Relationship between diet and anticoagulant response to warfarin: a factor analysis. Eur. J. Nutr. 2007;46:147–154. doi: 10.1007/s00394-007-0645-z. [DOI] [PubMed] [Google Scholar]
- 26.Evans BJ, Flockhart FD. The unfinished business of US drug regulation. Food Drug Law J. 2006;61:45–64. [PubMed] [Google Scholar]
- 27.Lesko LJ, et al. Pharmacogenetics and pharmacogenomics in drug development and regulatory decision making: report of the first FDA–PWG–PhRMA–DruSafe Workshop. J. Clin. Pharmacol. 2003;43:342–358. doi: 10.1177/0091270003252244. [DOI] [PubMed] [Google Scholar]
- 28.Reis-Filho JS, Westbury C, Pierga JY. The impact of expression profiling on prognostic and predictive testing in breast cancer. J. Clin. Pathol. 2006;59:225–231. doi: 10.1136/jcp.2005.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan C, et al. Concordance among gene-expression-based predictors for breast cancer. N. Engl. J. Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 30.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. NatureGenet. 2005;37:435–440. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sackett DL, Hayes RB, Guyatt GH, Tugwell P. Clinical Epidemiology. 2nd Edn Little Brown; Boston, Massachusetts: 1991. [Google Scholar]
- 32.Tantisira KG, et al. Corticosteroid pharmacogenetics: association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum. Mol. Genet. 2004;13:1353–1359. doi: 10.1093/hmg/ddh149. [DOI] [PubMed] [Google Scholar]