

Abstract
Acetylcholine (ACh), the first neurotransmitter to be identified, regulate the activities of central and peripheral functions through interactions with muscarinic receptors. Changes in muscarinic acetylcholine receptor (mAChR) have been implicated in the pathophysiology of many major diseases of the central nervous system (CNS). Previous reports from our laboratory on streptozotocin (STZ) induced diabetic rats showed down regulation of muscarinic M1 receptors in the brainstem, hypothalamus, cerebral cortex and pancreatic islets. In this study, we have investigated the changes of acetylcholine esterase (AChE) enzyme activity, total muscarinic and muscarinic M1 receptor binding and gene expression in the corpus striatum of STZ – diabetic rats and the insulin treated diabetic rats. The striatum, a neuronal nucleus intimately involved in motor behaviour, is one of the brain regions with the highest acetylcholine content. ACh has complex and clinically important actions in the striatum that are mediated predominantly by muscarinic receptors. We observed that insulin treatment brought back the decreased maximal velocity (Vmax) of acetylcholine esterase in the corpus striatum during diabetes to near control state. In diabetic rats there was a decrease in maximal number (Bmax) and affinity (Kd) of total muscarinic receptors whereas muscarinic M1 receptors were increased with decrease in affinity in diabetic rats. We observed that, in all cases, the binding parameters were reversed to near control by the treatment of diabetic rats with insulin. Real-time PCR experiment confirmed the increase in muscarinic M1 receptor gene expression and a similar reversal with insulin treatment. These results suggest the diabetes-induced changes of the cholinergic activity in the corpus striatum and the regulatory role of insulin on binding parameters and gene expression of total and muscarinic M1 receptors.
Background
The most and well known effects of diabetes mellitus on CNS is dysfunction of neurotransmitters, which is secondary to the metabolic disorders such as hyperglycemia and acidosis. It has been proposed that an unbalanced autonomic nervous system may be a major cause of the metabolic syndrome [1] Diabetes mellitus have also been reported to be accompanied by a number of behavioral and hormonal abnormalities, including hyperphagia, reduced motor activity [2,3]. CNS abnormalities including neuronal atrophy and axonal degenerations [4,5] are also associated with diabetes. The altered levels of neurotransmitter in specific brain areas in patients with diabetes mellitus [6] and in animals with experimental diabetes [7-12] have been documented and implicated in the CNS disorders. ACh, a major neurotransmitter from autonomic nervous system, regulates the cholinergic stimulation of insulin secretion, through interactions with muscarinic receptors. Recently we have reported that muscarinic M1 receptor gene expressions were decreased in the cerebral cortex, brainstem, hypothalamus and pancreatic islets of STZ induced diabetic rats and insulin modulates the binding parameters and gene expression [13,14]. Acetylcholine muscarinic receptors, members of the superfamily of G protein-coupled receptors, are classified pharmacologically into M1 to M5 subtypes, with M1, M3 and M5 receptors preferentially coupling to Gαq/11 proteins and M2 and M4 receptors to Gαi/o proteins [9]. All five muscarinic receptors are expressed by striatal neurones, with M1 and M4 receptors as the predominant subtypes, conforming together nearly 80% of the receptor population in the rat as shown by immunodetection [15,16] The main objective of the present study was to determine whether uncontrolled hyperglycemia, as a consequence of diabetes, altered the acetylcholine esterase enzyme activity, total and muscarinic M1 receptor binding parameters and muscarinic M1 receptor gene expression and the regulatory role of insulin in the rat corpus striatum.
Materials and methods
Biochemicals used in the present study were purchased from Sigma Chemical Co., St. Louis, USA. All other reagents were of analytical grade purchased locally. Quinuclidinyl benzilate, L- [Benzilic-4,4'-3H], ([3H] QNB)(Sp. Activity 42 Ci/mmol) was purchased from NEN life sciences products Inc., Boston, U.S.A. Tri-reagent kit was purchased from MRC, USA. Real Time PCR Taqman probe assays on demand were purchased from Applied Biosystems, Foster City, CA, USA.
Male adult Wistar rats of 180–240 g body weight were used for all experiments. They were housed in separate cages under 12 hour light and 12 hour dark periods. Rats have free access to standard food and water ad libitum. All animal care and procedures were in accordance with the Institutional and National Institute of Health guidelines. Diabetes was induced in rats by single intrafemoral injection of streptozotocin freshly dissolved in 0.1 M citrate buffer, pH 4.5, under anesthesia [17]. Streptozotocin was given at a dose of 55 mg/Kg body weight [18,19]. Animals were divided into the following groups: i) Control ii) diabetic iii) insulin-treated diabetic rats. Each group consisted of 6–8 animals. The insulin-treated diabetic group received subcutaneous injections (1 Unit/kg body weight) of Lente and Plain insulin (Boots India) daily during the entire period of the experiment. The last injection was given 24 hrs before sacrificing the rats. Rats were sacrificed on 15th day by decapitation. The corpus striatum was dissected out quickly over ice according to the procedure of Glowinski and Iversen 1966[20], the tissues were stored at -70°C until assayed.
Estimation of blood glucose
Blood glucose was estimated by the spectrophotometric method using glucose oxidase-peroxidase reactions. Blood samples were collected from the tail vein at 0 hours (Before the start of the experiment), 3rd day, 6th day, 10th day and 14th day and the glucose levels were estimated. Blood samples were collected 3 hrs after the administration of morning dose. The results were expressed in terms of milligram per deciliter of blood.
Acetylcholine Esterase Assay
Acetylcholine esterase asssay was done using the spectrophotometric method of Ellman et al, (1961) [21]. The corpus striatum homogenate (10%) was prepared in sodium phosphate buffer (30 mM, pH-7). One ml of 1% Triton × 100 was added to the homogenate to release the membrane bound enzyme and centrifuged at 10,000 × g for 30 minutes at 4°C. Different concentrations of acetylthiocholine iodide were used as substrate. The mercaptan formed as a result of the hydrolysis of the ester reacts with an oxidising agent 5,5'-dithiobis (2-Nitrobenzoate) absorbs at 412 nm.
Total Muscarinic and muscarinic M1 receptor binding studies in the corpus striatum
[3H]QNB binding assay in corpus striatum was done according to the modified procedure of Yamamura and Snyder (1981) [22]. Corpus striatum was homogenised in a polytron homogeniser with 20 volumes of cold 50 mM Tris-HCl buffer, containing 1 mM EDTA, pH.7.4. The supernatant was then centrifuged at 30,000 × g for 30 minutes and the pellets were resuspended in appropriate volume of Tris-HCl-EDTA buffer.
Total muscarinic receptor binding parameter assays were done using different concentrations i.e., 0.1–2.5 nM of [3H] QNB in the incubation buffer, pH 7.4 in a total incubation volume of 250 μl containing appropriate protein concentrations (200–250 μg). Non-specific binding was determined using 100 μM atropine. Competition studies were carried out with 1 nM [3H]QNB in each tube with atropine concentrations varying from 10-9 – 10-4 M atropine. Tubes were incubated at 22°C for 60 minutes and filtered rapidly through GF/C filters (Whatman). The filters were washed quickly by three successive washing with 5.0 ml of ice cold 50 mM Tris-HCl buffer, pH 7.4. Bound radioactivity was counted with cocktail-T in a Wallac 1409 liquid scintillation counter. The non-specific binding determined showed 10% in all our experiments.
Muscarinic M1 receptor binding assays were done using different concentrations i.e., 0.1–2.5 nM of [3H]QNB in the incubation buffer, pH 7.4 in a total incubation volume of 250 μl containing appropriate protein concentrations (200–250 μg). Non-specific binding was determined using 100 μM pirenzepine. Competition studies were carried out with 1 nM [3H]QNB in each tube with pirenzepine concentrations varying from 10-9 – 10-4 M. Tubes were incubated at 22°C for 60 minutes and filtered rapidly through GF/C filters (Whatman). The filters were washed quickly by three successive washing with 5.0 ml of ice cold 50 mM Tris-HCl buffer, pH 7.4. Bound radioactivity was counted with cocktail-T in a Wallac 1409 liquid scintillation counter. The non-specific binding determined showed 10% in all our experiments.
Protein determination
Protein was measured by the method of Lowry et al, (1951) [23] using bovine serum albumin as standard. The intensity of the purple blue colour formed was proportional to the amount of protein, which was read in a spectrophotometer at 660 nm
Receptor data analysis
The receptor binding parameters were determined using Scatchard analysis [24]. The specific binding was determined by subtracting non-specific binding from the total. The binding parameters, maximal binding (Bmax) and equilibrium dissociation constant (Kd), were derived by linear regression analysis by plotting the specific binding of the radioligand on X-axis and bound/free on Y-axis using Sigma plot software (version 2.0, Jandel GmbH, Erkrath, Germany). The maximal binding is a measure of the total number of receptors present in the tissue and the equilibrium dissociation constant is the measure of the affinity of the receptors for the radioligand. The Kd is inversely related to receptor affinity.
Displacement curve analysis
The displacement data were analysed by nonlinear regression using GraphPad PRISM™ software, GraphPad Inc., San Diego, USA. The concentration of the competing drug that competes for half the specific binding was defined as EC50 which is same as IC50. The affinity of the receptor for the competing drug is designated as Ki and is defined as the concentration of the competing ligand that will bind to half the binding sites at equilibrium in the absence of radioligand or other competitors. The Hill slope was used to indicate a one or two-sited model of curve-fitting.
Analysis of gene expression by Real-Time PCR
RNA was isolated from the corpus striatum of experimental rats using the Tri reagent (MRC, USA). Total cDNA synthesis was performed using ABI PRISM cDNA arhive kit in 0.2 ml microfuge tubes. The reaction mixture of 20 μl contained 0.2 μg total RNA, 10 × RT buffer, 25 × dNTP mixture, 10 × random primers, MultiScribe RT (50 U/μl) and RNase free water. The cDNA synthesis reactions were carried out at 25°C for 10 minutes and 37°C for 2 hours using an Eppendorf Personal Cycler. Real-time PCR assays were performed in 96-well plates in ABI 7300 real-time PCR instrument (Applied Biosystems). The primers and probes were purchased from Applied Biosystems, Foster City, California, USA. The TaqMan reaction mixture of 20 μl contained 25 ng of total RNA-derived cDNAs, 200 nM each of the forward primer, reverse primer, and TaqMan probe for Muscarinic M1 receptor gene and endogenous control (β-actin) and 12.5 μl of Taqman 2× Universal PCR Master Mix (Applied Biosystems) and the volume was made up with RNAse free water. The following thermal cycling profile was used (40 cycles): 50°C for 2 min, 95°C for 10 min, 95°C for 15 sec and 60°C for 1 min.
Fluorescence signals measured during amplification were considered positive if the fluorescence intensity was 20-fold greater than the standard deviation of the baseline fluorescence. The ΔΔCT method of relative quantification was used to determine the fold change in expression. This was done by first normalizing the resulting threshold cycle (CT) values of the target mRNAs to the CT values of the internal control β-actin in the same samples (ΔCT = CTTarget – CTβ-actin). It was further normalize with the control (ΔΔCT = ΔCT – CTControl). The fold change in expression was then obtained as (2-ΔΔCT) and the graph was plotted using log 2-ΔΔCT.
Statistics
Statistical evaluations were done by ANOVA, expressed as mean ± S.E.M using InStat (Ver.2.04a) computer programme.
Results
Blood glucose level of all rats before streptozotocin administration was within the normal range. Streptozotocin administration led to a significant increase (p < 0.001) in blood glucose level of diabetic rats when compared to control rats. Insulin treatment was able to significantly reduce (p < 0.001) the increased blood glucose level to near the control value when compared to diabetic group (Table 1)
Table 1.
Blood glucose (mg/dl) level in Experimental rats
| Animal status | 0 day (Before STZ injection) | 3rd day (Initial) | 6th day | 10th day | 14th day (Final) |
| Control | 86.2 ± 1.4 | 93.5 ± 1.6 | 89.4 ± 0.8 | 101.2 ± 2.2 | 97.7 ± 1.21 |
| Diabetic | 79.4 ± 1.5 | 253.1 ± 0.5 | 303.1 ± 0.8 | 309.7 ± 0.6 | 311.9 ± 1.4*** |
| D + I | 85.2 ± 0.8 | 256.8 ± 0.5 | 303.6 ± 0.7 | 190.9 ± 1.5 | 137.0 ± 1.3ψψψφφφ |
Values are mean ± S.E.M of 4–6 rats in each group. Each group consist of 6–8 rats
*** P < 0.001 when compared to control, ψψψ P < 0.001 when compared to diabetic group, φφφ p < 0.001 when compared with initial reading
Acetylcholine esterase activity in the Corpus striatum of experimental rats
Acetylcholine esterase kinetics studies showed that Vmax was significantly decreased (p < 0.001) in the corpus striatum of diabetic group with no significant change in Km. Insulin treatment significantly reversed the Vmax (p < 0.001) to near control value when compared to diabetic group (Table 2).
Table 2.
Acetylcholine esterase activity in the corpus striatum of Control, Diabetic and D+I group rats
| Animal status | Vmax (μmoles/min/mg protein) |
Km (μM) |
| Control | 2420.0 ± 11.5 | 51.0 ± 0.5 |
| Diabetic | 1631.3 ± 16.1*** | 51.3 ± 0.8 |
| Diabetic + Insulin treated (D+I) | 2647.0 ± 26.5ψψψ | 51.0 ± 0.5 |
Values are mean ± S.E.M of 4–6 separate experiments. Each group consist of 6–8 rats
*** P < 0.001 when compared to control, ψψψ P < 0.001 when compared to diabetic group
Total Muscarinic receptor analysis
Scatchard analysis of [3H] QNB binding against atropine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated diabetic rats
The Scatchard analysis showed that the Bmax and Kd of the [3H]QNB receptor binding decreased significantly (p < 0.001) in the corpus striatum of diabetic rats when compared to control group. In insulin treated diabetic group Bmax and Kd were significantly (p < 0.001) reversed back to near control value when compared to diabetic group. (Fig 1 & Table 3)
Figure 1.

Representative graph showing Scatchard analysis of [3H]QNB binding against atropine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats. Control (black circle), Diabetic (open circle), Insulin treated diabetic rats (black triangle). Total muscarinic receptor binding parameter assays were done using different concentrations i.e., 0.1–2.5 nM of [3H]QNB in the incubation buffer, pH 7.4 in a total incubation volume of 250 μl containing appropriate protein concentrations (200–250 μg). Non-specific binding was determined using 100 μM atropine. Tubes were incubated at 22°C for 60 minutes and filtered rapidly through GF/C filters (Whatman). The filters were washed quickly by three successive washing with 5.0 ml of ice cold 50 mM Tris-HCl buffer, pH 7.4. Bound radioactivity was counted with cocktail-T in a Wallac 1409 liquid scintillation counter. The non-specific binding determined showed 10% in all our experiments.
Table 3.
Scatchard analysis of [3H] QNB binding against atropine in the corpus striatum of Control, Diabetic, and Diabetic+Insulin treated group rats
| Animal status | Bmax (fmoles/mg protein) |
Kd (nM) |
| Control | 214.00 ± 3.05 | 0.19 ± 0.01 |
| Diabetic | 150.00 ± 5.77*** | 0.27 ± 0.05*** |
| Diabetic + Insulin treated | 184.00 ± 3.05ψψψ | 0.17 ± 0.05ψψψ |
Values are mean ± S.E.M of 4–6 separate experiments. Each group consist of 6–8 rats
*** P < 0.001 when compared to control, ψψψ P < 0.001 when compared to diabetic group
Displacement analysis of [3H]QNB using Atropine
In the displacement analysis, the competitive curve fitted to a one-sited model in all groups with Hill slope values were near to unity. The log (EC50) did not alter in all the experimental groups. The Ki decreased in diabetic condition (Fig 2 & Table 4).
Figure 2.

Representative graph showing displacement analysis of [3H]QNB binding against atropine in the hypothalamus of Control, Diabetic and Diabetic+Insulin treated group rats. Control (black circle), Diabetic (upward pointing black triangle), Insulin treated diabetic rats (downward pointing black triangle). Competition studies were carried out with 1 nM [3H]QNB in each tube with atropine concentrations varying from 10-9, 10-4 M. Data were fitted with iterative nonlinear regression software (Prism, GraphPad, San Diego, CA). Ki The affinity of the receptor for the competing drug. EC50 is the concentration of the competitor that competes for half the specific binding.
Table 4.
Binding parameters of [3H] QNB against atropine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats
| Experimental Group | Best-fit model | Log (EC50) | Ki | Hill slope |
| Control | One-site | -7.076 | 2.10 × 10-8 | 0.9832 |
| Diabetic | One-site | -6.818 | 3.80 × 10-8 | 0.9883 |
| Insulin treated diabetic | One-site | -6.913 | 3.05 × 10-8 | 0.9889 |
Values are mean of 4–6 separate experiments. Each group consist of 6–8 rats
Muscarinic M1 receptor analysis
Scatchard analysis of [3H]QNB binding against pirenzepine in the corpus striatum of Control, Diabetic, and Diabetic+Insulin treated diabetic rats
The Scatchard analysis showed that the Bmax of muscarinic M1 receptors of corpus striatum was increased significantly (p < 0.001) in diabetic condition when compared to control group while the Kd was decreased significantly when compared to control group (p < 0.001). In insulin treated diabetic rats Bmax was significantly (p < 0.001) reversed back to near control value when compared to diabetic group but Kd was not reversed back to near control value when compared to diabetic group (Fig 3 & Table 5).
Figure 3.
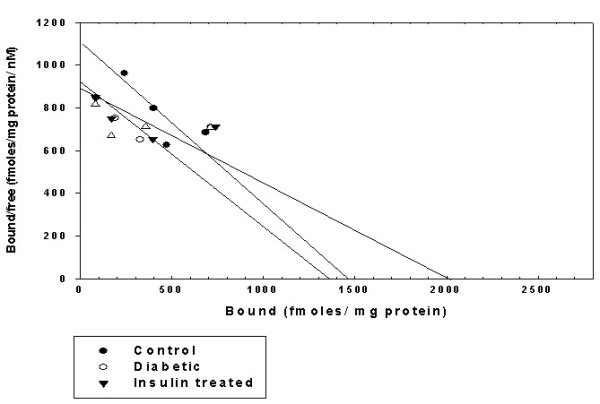
Representative graph showing Scatchard analysis of [3H]QNB binding against pirenzepine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats. Control (black circle), Diabetic (open circle), Insulin treated diabetic rats (black triangle). Muscarinic M1 receptor binding parameter assays were done using different concentrations i.e., 0.1–2.5 nM of [3H]QNB in the incubation buffer, pH 7.4 in a total incubation volume of 250 μl containing appropriate protein concentrations (200–250 μg). Non-specific binding was determined using 100 μM pirenzepine. Tubes were incubated at 22°C for 60 minutes and filtered rapidly through GF/C filters (Whatman). The filters were washed quickly by three successive washing with 5.0 ml of ice cold 50 mM Tris-HCl buffer, pH 7.4. Bound radioactivity was counted with cocktail-T in a Wallac 1409 liquid scintillation counter. The non-specific binding determined showed 10% in all our experiments.
Table 5.
Scatchard analysis of [3H] QNB binding against pirenzepine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats
| Animal status | Bmax (fmoles/mg protein) | Kd (nM) |
| Control | 1460.00 ± 30.55 | 1.34 ± 0.02 |
| Diabetic | 2060.00 ± 30.55*** | 0.45 ± 0.02*** |
| Diabetic + Insulin treated | 1550.00 ± 28.86ψψψ | 0.56 ± 0.01 |
Values are mean ± S.E.M of 4–6 separate experiments. Each group consist of 6–8 rats
*** P < 0.001 when compared to control, ψψψ P < 0.001 when compared to diabetic group
Displacement analysis of [3H]QNB using pirenzepine
In the displacement analysis, the competitive curve fitted to a one-site model in all the experimental conditions. Hill slopes were near unity confirming the one-site model. There were no changes in the log (EC50) values. The Ki value was decreased in diabetic condition (Fig 4 & Table 6).
Figure 4.

Representative graph showing displacement analysis of [3H]QNB binding against pirenzepine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats. Control (black circle), Diabetic (upward pointing black triangle), Insulin treated diabetic rats (downward pointing black triangle). Competition studies were carried out with 1 nM [3H]QNB in each tube with pirenzepine concentrations varying from 10-9 to 10-4 M. Data were fitted with iterative nonlinear regression software (Prism, GraphPad, San Diego, CA). Ki – The affinity of the receptor for the competing drug. EC50 is the concentration of the competitor that competes for half the specific binding.
Table 6.
Binding parameters of [3H]QNB against pirenzepine in the corpus striatum of Control, Diabetic and Diabetic+Insulin treated group rats
| Experimental Group | Best-fit model | Log (EC50) | Ki | Hill slope |
| Control | One-site | -7.107 | 2.23 × 10-8 | 0.7224 |
| Diabetic | One-site | -6.459 | 9.92 × 10-8 | 0.7444 |
| Insulin treated diabetic | One-site | -7.322 | 4.76 × 10-8 | 0.6826 |
Values are mean of 4–6 separate experiments. Each group consist of 6–8 rats
Real Time-PCR analysis
Real Time-PCR analysis showed that the muscarinic M1 receptor gene expression was increased significantly (p < 0.01) in diabetic condition and it reversed to near control value in insulin treated diabetic rats (Fig 5 & Table 7).
Figure 5.

Representative graph showing Real-Time amplification of muscarinic M1 mRNA from the corpus striatum of control, diabetic and insulin treated diabetic rats. Control, Diabetic, Insulin treated diabetic rats. The ΔΔCT method of relative quantification was used to determine the fold change in expression. This was done by first normalizing the resulting threshold cycle (CT) values of the target mRNAs to the CT values of the internal control β-actin in the same samples (ΔCT = CTTarget – CT β-actin). It was further normalize with the control (ΔΔCT = ΔCT – CTControl). The fold change in expression was then obtained (2-ΔΔCT). The graph was plotted using log 2-ΔΔCT. Values are mean ± S.D of 4–6 separate experiments. Relative Quantification values and standard deviations are shown in the table. The relative ratios of mRNA levels were calculated using the ΔΔCT method normalized with β-actin CT value as the internal control and Control CT value as the calibrator.
Table 7.
Real Time amplification of Muscarinic M1 receptor mRNA from the Corpus striatum of Control, Diabetic, and Diabetic+Insulin treated group rats
| Experimental group | RQ Value |
| Control | 0 |
| Diabetic | 3.14 ± 0.25** |
| Diabetic + Insulin treated | 0.86 ± -0.66ψψ |
Values are mean ± S.D of 4–6 separate experiments. Each group consist of 6–8 rats Relative Quantification values and standard deviations are shown in the table. The relative ratios of mRNA levels were calculated using the ΔΔCT method normalized with β-actin CT value as the internal control and Control CT value as the calibrator.
** p < 0.01 when compared with control
ψψp < 0.01 when compared with diabetic group
Discussion
The brain neurotransmitters receptor activity and hormonal pathways control many physiological functions in the body. The present study analyzed the changes of acetylcholine esterase enzyme activity, total muscarinic and muscarinic M1 receptors in the corpus striatum of STZ diabetic and insulin-treated diabetic rats. CNS mAChRs regulate a large number of important central functions including cognitive, behavioural, sensory, motor and autonomic processes [25-27]. A key feature of type 2 diabetes is that pancreatic β-cells fail to release sufficient amounts of insulin despite elevated blood glucose levels [28]. Glucose-stimulated insulin secretion (GSIS) is regulated by numerous hormones and neurotransmitters most of which act on specific G-protein-coupled receptors (GPCRs) expressed by pancreatic β-cells [29,30]. Many studies have shown that ACh, following its release from intra-pancreatic parasympathetic nerve endings, can stimulate β-cell mAChRs, leading to enhanced GSIS in a strictly glucose-dependent fashion [29,30]. mAChRs are members of the superfamily of GPCRs. Molecular-cloning studies have revealed the existence of five molecularly distinct mammalian mAChR subtypes, M1-M5 [31,9]. Earlier studies from our laboratory have established the central neurotransmitter receptor subtypes functional regulation during diabetes, pancreatic regeneration and cell proliferation [32-37]. M1 mAChRs are abundantly expressed in all major regions of the forebrain, including striatum, hippocampus, and cerebral cortex [38-40,15]. It is therefore likely that M1 mAChRs play a role in the many central actions of ACh that involve the activity of forebrain mAChRs. Pharmacological evidence suggests that M1 receptors are involved in mediating higher cognitive processes, such as learning and memory [41,42,28]. The striatum, a neuronal nucleus intimately involved in motor behaviour, is one of the brain regions with the highest acetylcholine content [43]. The mRNA for M1 is present in more than 80% of striatal neurons [44] including cholinergic neurons, substance P neurons, enkephalin neurons, and somatostatin neurons [45]. Recent studies from our laboratory have showed the significance of muscarinic and muscarinic M1 receptors in the cerebral cortex, hypothalamus, brainstem, and pancreatic islets of STZ induced diabetic rats and its functional regulation in insulin secretion. [13,14]. ACh, through vagal muscarinic and non-vagal muscarnic pathways [46] increases insulin secretion [47]. They function through muscarinic receptors present on pancreatic islet cells. Receptor localization studies suggest that multiple muscarinic receptors (M1, M3, M4, and M5) are expressed in pancreatic islets/β-cells [48]. From our previous studies it was observed that muscarinic M1 receptors were down-regulated during STZ diabetes [13]. The enzyme AChE indirectly plays an important role in transmission of nerve impulse. It hydrolyses the ACh released at the cholinergic synapse and thus terminates the action of this neurotransmitter. In addition to their role in cholinergic transmission, cholinesterases may also play a role during morphogenesis and neurodegenerative diseases [49,50]. We observed a significant decrease in Vmax of acetylcholine esterase in the striatum of diabetic rats which was reversed to near control level by insulin treatment. Akmayev et al (1978) [51] showed that there is a difference in distribution of enzyme in the neurons of the central vagal nuclei in normal and adult male rats. It is suggested that the changes in the plasma glucose or insulin may be that stimulus that influence the activity of cholinergic neurons. Insulin treatment reversed the altered maximum velocity toward the control level. Corpus striatum is best known for its role in the planning and modulation movement pathways but also involved in a variety of cognitive process involving executive function. In corpus striatum total muscarinic receptor numbers and affinity were decreased during diabetes, whereas muscarinic M1 receptors number was increased in STZ diabetic rats with decrease in affinity. The changes in the receptor number and affinity observed are due to the alterations of receptor protein and synthesis. Real-time PCR analysis showed an up-regulation of the muscarinic M1 receptor mRNA level in the striatum of diabetic rats, whereas it reversed to near control when treated with insulin. This is in accordance with our receptor binding studies. ACh has complex and clinically important actions in the striaturn that are mediated predominantly by muscarinic receptors. Based on physiological and pharmacological studies, several specific actions of ACh in the striatum have been suggested. ACh regulates its own release from cholinergic interneurons through presynaptic autoreceptors. Noncholinergic striatal neurons are directly affected by ACh through postsynaptic receptors and presynaptic heteroceptors and the release of excitatory amino acids and dopamine by extrinsic striatal afferents may be under presynaptic control of ACh through presynaptic heteroceptors [52-54]. In diabetic corpus striatum total muscarinic receptors activities were decreased. The insulin treatment reversed these altered parameters to near control level. Muscarinic receptor subtypes other than M1 may also be affected by the diabetic condition. Further studies have to be carried out to elucidate the role of other subtypes. The present study suggests that drugs that can selectively activate muscarinic receptors may be of significant therapeutic benefit in the diabetes management. Thus our results revealed the significance of central muscarinic receptor changes during diabetes and the regulatory role of insulin on muscarinic receptors.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GG and CSP designed research; GG performed experiments; PKT and JM helped GG in experiments; GG and CSP analyzed the data; GG and CSP wrote the paper. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by a research grant from KSCSTE, Govt. of Kerala, DST, and ICMR, Govt. of India to Dr. C.S.Paulose. Gireesh.G thanks KSCSTE for JRF
Contributor Information
G Gireesh, Email: giri_ganga2000@yahoo.co.in.
T Peeyush Kumar, Email: peeyushchembio@yahoo.co.in.
Jobin Mathew, Email: eattathottu@gmail.com.
CS Paulose, Email: cspaulose@cusat.ac.in.
References
- Kreier F, Yilmaz A, Kalsbeek A, Romijn JA, Sauerwein HP, Fliers E, Buijs RM. Hypothesis: shifting the equilibrium from activity to food leads to autonomic unbalance and the metabolic syndrome. Diabetes. 2003;52:2652–2656. doi: 10.2337/diabetes.52.11.2652. [DOI] [PubMed] [Google Scholar]
- Marchall JF, Friedman TG, Heffner MI. Reduced anorexic and locomotor – stimulant action of d-amphetamine in alloxan – diabetic rats. Brain Research. 1976;111:428–432. doi: 10.1016/0006-8993(76)90789-7. [DOI] [PubMed] [Google Scholar]
- Marchall JF. Further analysis of the resistance of the diabetic rat to d-amphetamine. Pharmacol Biochem Behav . 1978;8(3):281–286. doi: 10.1016/0091-3057(78)90317-9. [DOI] [PubMed] [Google Scholar]
- Rossi GL, Bestetti G. Morphological changes in the hypothalamic-hypophyseal-gonodal axis of male rats after twelve months of streptozotocin induced diabetes. Diabetologia. 1981;21:476–481. doi: 10.1007/BF00257789. [DOI] [PubMed] [Google Scholar]
- Reske-Nielsen E, Lunbeak K. Diabetic encephalopathy. Diffuse and focal lesions of the brain in long-term diabetes. Acta Neurol Scand Suppl . 1963;39(4 Suppl 4):273–293. [PubMed] [Google Scholar]
- Lackovic Z, Salkovic M, Kuci Z, Relja M. Dopamine, norepinephrine and serotonin in the brain of diabetic rats and man. Journal of Neurochemistry. 1985;44:94–98. [Google Scholar]
- Bitar M, Koulu M, Rapoport SI, Linnoila M. Dibetes induced alterations in brain monoamine metabolism in rats. Journal of Pharmacology and Experimental Therapeutics. 1986;236:432–437. [PubMed] [Google Scholar]
- Saller CF. Dopaminergic activity is reduced in diabetic rats. Neuroscience Letters. 1984;49:301–306. doi: 10.1016/0304-3940(84)90306-9. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJM. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Kwok RPS, Walls EK, Juoria AV. The concentrations of dopamine 5 Hydroxytryptamine and some of their acid metabolites in the brain of genetically diabetic rats. Neurochemical Research. 1985;10:611–616. doi: 10.1007/BF00964400. [DOI] [PubMed] [Google Scholar]
- Chen CC, Yang JC. Effects of short and long-lasting diabetes mellitus on mouse brain monoamines. Brain Research. 1991;552:175–179. doi: 10.1016/0006-8993(91)90677-n. [DOI] [PubMed] [Google Scholar]
- Tasaka Y, Matsumoto H, Inove Y, Hirata Y. Brain catecholamine concentrations in hyperosmolar diabetic and diabetic rats. Diabetes Research. 1992;19:1–7. [PubMed] [Google Scholar]
- Gireesh G, Kaimal SB, Kumar TP, Paulose CS. Decreased muscarinic M1 receptor gene expression in the hypothalamus, brainstem, and pancreatic islets of streptozotocin-induced diabetic rats. J Neurosci Res. 2007. [DOI] [PubMed]
- Gireesh G, Reas SK, Jobin M, Paulose CS. Decreased muscarinic M1 receptor gene expression in the cerebral cortex of streptozotocin-induced diabetic rats and Aegle marmelose leaf extract's therapeutic function J. Ethnopharmacol. 2007. [DOI] [PubMed]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Development of antisera selective for m4 and m5 muscarinic cholinergic receptors: distributions of m4 and m5 receptors in rat brain. 1993;43:149–157. [PubMed] [Google Scholar]
- Junod A, Lambert AE, Staufferacher W, Renold AE. Diabetogenic action of Streptozotocin: Relationship of dose to metabolic response. J Clin Invest. 1969;48:2129–2139. doi: 10.1172/JCI106180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenegger M, Rudas B. Kidney functions in experimental diabetic ketosis. Diabetologia. 1971;17:334–338. doi: 10.1007/BF01219467. [DOI] [PubMed] [Google Scholar]
- Arison RN, Ciaccio EI, Glitzer MS, Cassaro AV, Pruss M. Light and electron microscopy of lesions in rats rendered diabetic with streptozotocin. Diabetes. 1967;16:51–56. doi: 10.2337/diab.16.1.51. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain: the disposition of [3H] norepinephrine, [3H] dopa in various regions of brain. J Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- Yamamura HI, Synder G. Binding of [3H] QNB in rat brain. Proc. Natl Acad Sci. 1981;71:1725–1729. doi: 10.1073/pnas.71.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Roserbrough NJ, Farr AL, Randall RJ. Protein measurement with Folin Phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Scatchard G. The attraction of proteins for small molecules and ions. Ann N Y Acad Sci. 1949;51:660–672. [Google Scholar]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol. 1996;10:69–99. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- Eglen RM. Muscarinic receptor subtype pharmacology and physiology. Prog Med Chem. 2005;43:105–136. doi: 10.1016/S0079-6468(05)43004-0. [DOI] [PubMed] [Google Scholar]
- Kahn SE. The importance of βcell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. doi: 10.1210/jcem.86.9.7713. [DOI] [PubMed] [Google Scholar]
- Ahren B. Autonomic regulation of islet hormone secretion – implications for health and disease. Diabetologia. 2000;43:393–410. doi: 10.1007/s001250051322. [DOI] [PubMed] [Google Scholar]
- Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic βcell function. Endocr Rev. 2001;22:565–604. doi: 10.1210/edrv.22.5.0440. [DOI] [PubMed] [Google Scholar]
- Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- Sudha B, Paulose CS. Induction of DNA synthesis in primary culture of rat hepatocyte by serotonin: Possible involvement of serotonin S2 receptor. Hepatology. 1998;27:62–66. doi: 10.1002/hep.510270111. [DOI] [PubMed] [Google Scholar]
- Biju MP, Pyroja S, Rajesh NV, Paulose CS. Hepatic GABA A receptor functional regulation during liver cell proliferation. Hepatol Res. 2001;21:136–146. doi: 10.1016/s1386-6346(01)00092-4. [DOI] [PubMed] [Google Scholar]
- Renuka TR, Ani DV, Paulose CS. Alterations in the muscarinic M1 and M3 receptor gene expression in the brain stem during pancreatic regeneration and insulin secretion in weanling rat. Life Sci. 2004;75:2269–2280. doi: 10.1016/j.lfs.2004.03.034. [DOI] [PubMed] [Google Scholar]
- Mohanan VV, Kaimal SB, Paulose CS. Decreased 5-HT1A receptor gene expression and 5HT1A receptor protein in the cerebral cortex and brain stem during pancreatic regeneration in rats. Neurochem Res. 2005;30:25–32. doi: 10.1007/s11064-004-9682-7. [DOI] [PubMed] [Google Scholar]
- Kaimal SB, Gireesh G, Paulose CS. Decreased GABAA receptor function in the brain stem during pancreatic regeneration in rats. Neurochem Res. 2007 doi: 10.1007/s11064-007-9283-3. [DOI] [PubMed] [Google Scholar]
- Abraham A, Paulose CS. Age related alterations in noradrenergic function in brain stem of streptozotocin-diabetic rats. J Biochem Mol Biol Biophys. 1999;31:171–176. [Google Scholar]
- Levey AI. Immunological localization of M1–M5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci. 1993;52:441–48. doi: 10.1016/0024-3205(93)90300-r. [DOI] [PubMed] [Google Scholar]
- Vilaró MT, Mengod G, Palacios JM. Advances and limitations of the molecular neuroanatomy of cholinergic receptors: the example of multiple muscarinic receptors. Prog Brain Res . 1993;(98):95–101. doi: 10.1016/s0079-6123(08)62385-7. [DOI] [PubMed] [Google Scholar]
- Wolfe BB, Yasuda RP. Development of selective antisera for muscarinic cholinergic receptor subtypes. Ann NY Acad Sci. 1995;757:186–93. doi: 10.1111/j.1749-6632.1995.tb17474.x. [DOI] [PubMed] [Google Scholar]
- Quirion R, Aubert I, Lapchak PA, Schaum RP, Teolis S. Muscarinic receptor subtypes in human neurodegenerative disorders: focus on Alzheimer's disease. Trends Pharmacol Sci . 1989;10:80–84. [PubMed] [Google Scholar]
- Fisher A, Heldman E, Gurwitz D, Haring R, Karton Y. M1 agonists for the treatment of Alzheimer's disease. Novel properties and clinical update. Ann NY Acad Sci. 1996;177:189–196. doi: 10.1111/j.1749-6632.1996.tb34418.x. [DOI] [PubMed] [Google Scholar]
- Hoover DB, Muth EA, Jacobowitz DM. A mapping of the distribution of acetylcholine, choline acetyltransferase and acetylcholinesterase in discrete areas of rat brain. Brain Res. 1978;153:295–306. doi: 10.1016/0006-8993(78)90408-0. [DOI] [PubMed] [Google Scholar]
- Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci USA. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12:3591–600. doi: 10.1523/JNEUROSCI.12-09-03591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg GR, Pokol-Daniel S. Neural modulation of glucose-dependent insulinotropic peptide (GIP) and insulin secretion in conscious dogs. Pancreas. 1994;9:531–535. doi: 10.1097/00006676-199407000-00018. [DOI] [PubMed] [Google Scholar]
- Tassava MT, Okuda T, Romsos DR. Insulin secretion from ob/ob mouse pancreatic islets: effects of neurotransmitters. Am J Physiol. 1992. p. 262. [DOI] [PubMed]
- Iismaa TP, Kerr EA, Wilson JR, Carpenter L, Sims N, Biden TJ. Quantitative and functional characterization of muscarinic receptor subtypes in insulin-secreting cell lines and rat pancreatic islets. Diabetes. 2000;493:92–398. doi: 10.2337/diabetes.49.3.392. [DOI] [PubMed] [Google Scholar]
- Reyes AE, Perez DR, Alvarez A, Garrido J, Gentry MK, Doctor BP, Inestrosa NC. A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem Biophys Res Commun. 1997;232:652–655. doi: 10.1006/bbrc.1997.6357. [DOI] [PubMed] [Google Scholar]
- Layer PG, Alber R, Sporns O. Quantitative development and molecular forms of acetylcholinesterase and butyrylcholinesterase during morphogenesi s and synaptogenesi s of chick brain and retina. J Neurochem. 1987;49:175–182. doi: 10.1111/j.1471-4159.1987.tb03411.x. [DOI] [PubMed] [Google Scholar]
- Akmayev IG, Rubberia E, Fiddina OV. CNS-endocrine pancreas system. IV. Evidence for the existence of a direct hypothalamic-vagal descending pathway. Endokrinologie. 1978;71:169–174. [PubMed] [Google Scholar]
- Waelbroeck M, Tastenoy M, Camus J, Christophe J. Bindingof selective antagonists to four muscarinic receptors (M1 – M4) in rat forebrain. Mol Pharmacol. 1990;38:267–273. [PubMed] [Google Scholar]
- Ehlert F, Tran LLP. Regional distribution of M 1, M2 and non- Ml, non-M2 subtypes of muscarinic binding sites in rat brain. J Pharmacol Exp Ther. 1990;255:1148–1157. [PubMed] [Google Scholar]
- Sugita S, Chimura NU, Jiang ZG, North R. Distinct muscarinic receptors inhibit release of gamma-aminobutyric acid and excitatory amino acids in mammalian brain. Proc Natl Acad Sci USA. 1991;88:2608–2611. doi: 10.1073/pnas.88.6.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]