

Abstract
Ure2 is the protein determinant of the Saccharomyces cerevisiae prion [URE3]. Ure2 has structural similarity to glutathione transferases, protects cells against heavy metal and oxidant toxicity in vivo, and shows glutathione-dependent peroxidase activity in vitro. Here we report that Ure2 (which has no cysteine residues) also shows thiol-disulfide oxidoreductase activity similar to that of glutaredoxin enzymes. This demonstrates that disulfide reductase activity can be independent of the classical glutaredoxin CXXC/CXXS motif or indeed an intrinsic catalytic cysteine residue. The kinetics of the glutaredoxin activity of Ure2 showed positive cooperativity for the substrate glutathione in both the soluble native state and in amyloid-like fibrils, indicating native-like dimeric structure within Ure2 fibrils. Characterization of the glutaredoxin activity of Ure2 sheds light on its ability to protect yeast from heavy metal ion and oxidant toxicity and suggests a role in reversible protein glutathionylation signal transduction. Observation of allosteric enzyme behavior within amyloid-like Ure2 fibrils not only provides insight into the molecular structure of the fibrils but also has implications for the mechanism of [URE3] prion formation.
The tripeptide glutathione (GSH)2 is abundant in the cell. It plays an important role as a reducing agent in vivo, such as in endogenous free radical scavenging, reversible protein S-glutathionylation, and the reduction of the active sites of enzymes. One major class of enzyme that uses GSH as a reductant is glutaredoxin (GRX), which is a small protein involved in reduction of ribonucleotide reductase for the formation of deoxyribonucleotides for DNA synthesis (1), reduction of 3′-phosphoadenylylsulfate reductase (2) for generation of sulfite, signal transduction, and protection against oxidative stress (3). GRXs are ubiquitous thiol-disulfide oxidoreductases that belong to the thioredoxin superfamily (4). GRXs also show dehydroascorbic acid (DHA) reductase (DHAR) activity (5). Yeast Saccharomyces cerevisiae has at least seven GRXs, which can be divided into two classes according to the number of cysteines in their active site motif: dithiol GRXs with the active site motif CXXC and monothiol GRXs with the motif CXXS (6–9). The dithiol GRXs catalyze protein disulfide reduction using a dithiol mechanism for which both the active site cysteines are essential. On the other hand, both the dithiol and monothiol GRXs can catalyze the reduction of GSH-protein mixed disulfides using a monothiol mechanism that only requires the N-terminal active site cysteine. This reaction and mechanism is important for reversible protein glutathionylation in redox signaling and oxidative stress (10).
Glutathione S-transferases (GSTs) are a large versatile family of enzymes with multiple functions, particularly associated with cellular detoxification (11). In terms of overall structure, they belong to the thioredoxin superfamily, like GRX (4). In general, GSTs catalyze the conjugation of reduced GSH to hydrophobic substrates containing an electrophilic atom. In addition, GSTs bind a broad spectrum of ligands and show many other functions. For example, some GSTs show overlapping functions with glutathione-dependent peroxidases (GPxs), which use GSH to reduce hydrogen peroxide and/or organic hydroperoxides and thus are responsible for protection against both endogenous and exogenous oxidant toxicity (11). Interestingly Omega class and Beta class GSTs (such as Escherichia coli GST (EGST)) possess typical GRX activity toward widely used substrates, such as 2-hydroxyethyl disulfide (HEDS) (12–16). These GSTs have an active site cysteine, which is indispensable for GRX activity but not GST activity.
The yeast prion protein Ure2 is composed of a disordered protease-sensitive N-terminal prion domain and a compact globular C-terminal domain, which shows high structural similarity to EGST (17). The C-terminal domain of Ure2 can be further structurally divided into two subdomains, the all-α-helix subdomain and the thioredoxin fold subdomain, which shows high structural homology to GRX. Ure2 is involved in the regulation of nitrogen metabolism and resistance to heavy metal ion toxicity (especially cadmium) and oxidative stress in S. cerevisiae (18, 19). In addition, Ure2 shows GPx activity toward both hydrogen peroxide and organic hydroperoxides such as cumene hydroperoxide and tert-butyl hydroperoxide (20). The discovery of the GPx activity of Ure2 (20) provides an explanation for its ability to protect yeast cells from oxidant toxicity (18). However, the reason that ure2Δ yeast cells are hypersensitive to cadmium remains unclear. In general, cadmium ions have a drastic effect on yeast cell growth, and the reasons are complicated. One possible reason for cadmium ion toxicity is that thioltransferases or GRXs can be inhibited by direct binding of cadmium to the two essential cysteine residues present in the thioltransferase active site (21). The inhibition of GRXs leads to complex effects on cell growth. Therefore, we used an in vitro assay to provide a system that allows detailed analysis of the activity of Ure2 and its relationship to that of GRX enzymes. Characterization of the allosteric behavior of the GRX activity of Ure2 revealed that Ure2 forms an active dimer within fibrils. In addition to providing information about the molecular structure of Ure2 fibrils, this also has implications for the molecular mechanism of Ure2 prion formation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—Ure2 and mutants (42Ure2, 90Ure2, or Ure2-CTD with and without point mutations) were produced in E. coli with an N-terminal His6 tag and purified by nickel chromatography as described previously (22, 23). 42Ure2 and 90Ure2 lack the N-terminal 1–41 and 1–89 residues, respectively. Ure2-CTD lacks the entire N-terminal unstructured region (residues 1–104) and contains only the C-terminal domain of Ure2 (residues 105–354). Each protein was judged by SDS-PAGE to be at least 98% pure and was stored at –80 °C. All proteins were defrosted in a 25 °C water bath immediately prior to use. The concentration of the protein was determined by the absorbance at 280 nm using a calculated extinction coefficient of 48,200 m–1 cm–1 (22, 24), and all protein concentrations are expressed in terms of monomer. EGST (16), S. cerevisiae yeast GRX1 (yGRX1) (25), and E. coli thioredoxin (TRX) (26) were expressed and purified as described previously.
Hydrogen Peroxide Treatment of yGRX1 and Ure2—Typically 300 μm yGRX1 or 80 μm Ure2-CTD was incubated with ∼10 mm H2O2 at 4 °C for 2 h. The incubation mixture was then dialyzed thoroughly against 20 mm Tris-HCl, pH 8.4. The number of free thiol groups in each molecule of yGRX1 was determined using 5,5′-dithiobis(2-nitrobenzoic acid) as described previously (27).
Assay of GRX Activity—GSH-dependent disulfide reductase activity of Ure2 and its mutants was measured at 25 °C using GSH and HEDS as substrates in a spectrometric coupled assay as described previously (28). Typically a 1-ml reaction volume in a cuvette containing 100 mm Tris-HCl or sodium phosphate buffer, pH 7.5, 0.2–5 mm GSH, 0.2–5 mm HEDS or 5 mm trans-4,5-dihydroxy-1,2-dithiane (DTTox), 0.25–0.8 units/ml glutathione reductase, 0.2 mm NADPH, and 1 mm EDTA was preincubated at 25 °C for 2 min. The enzyme was then added to the cuvette to trigger the reaction. (The final enzyme concentration was 0.1–1.0 μm when using HEDS as a substrate and 4.0–7.0 μm when using DTTox as a substrate.) The activity was measured from the continuous decrease of NADPH absorption at 340 nm for 3 min. All initial velocities were corrected by subtraction of the non-enzymatic reaction measured using an equivalent volume of buffer in place of the protein solution.
To examine the effect of substrate concentration, the initial velocity was measured over a wide range of concentrations of one substrate and a fixed concentration of the other substrate. When the concentration of GSH was fixed, the data were fitted to the Michaelis-Menten and Lineweaver-Burk equations. Values obtained from these plots were the same within experimental error. When the concentration of HEDS was fixed, the kinetic behavior of Ure2 and its mutants followed a sigmoidal curve. Therefore, the data were fitted to the Hill equations (Equations 1 and 2) describing the kinetic behavior of an allosteric enzyme,
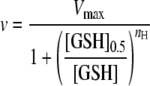 |
(Eq. 1) |
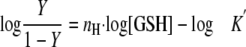 |
(Eq. 2) |
where nH is the Hill coefficient and Y is the ratio of the initial rate to the maximum rate. The nH values obtained from fitting to the two equations were the same within error. The steadystate kinetic mechanism for Ure2-CTD was determined from a family of double reciprocal plots, which was obtained by varying both HEDS and GSH concentrations.
Assay of Dehydroascorbic Acid Reductase Activity—GSH-dependent dehydroascorbic acid reductase activity was determined as described previously (29). Briefly a 1-ml reaction volume in a cuvette containing 100 mm sodium phosphate buffer, pH 7.5, 1 mm GSH, 1 mm DHA, 0.4 units/ml glutathione reductase, 0.2 mm NADPH, 1 mm EDTA, and 4–5 μm Ure2 protein or mutants was incubated at 25 °C. The decrease in absorbance at 340 nm was recorded spectrophotometrically. DHAR activity was measured after subtracting the spontaneous reaction rate between DHA and GSH in the absence of Ure2. DHA was used within 2 h of preparation.
Turbidimetric Assay of Insulin Disulfide Reductase Activity—The reduction of insulin by Ure2 was assayed by monitoring turbidity as described previously (30). Briefly a 0.5-ml cuvette containing 100 mm Tris-HCl buffer, pH 7.5, 1 μm enzyme, 1 mm EDTA, and reductant (0.8 mm DTT or 8 mm GSH) was incubated at 25 °C. Then 80 μm bovine insulin was added to start the reaction. Increasing turbidity from the precipitation of the insulin B chain was monitored by the absorbance at 650 nm. TRX and yGRX1 were used as controls. The nonenzymatic reduction of insulin by DTT or GSH was also monitored.
GRX Activity within Amyloid-like Fibrils of Ure2—Assay of GRX activity during the time course of Ure2 fibril formation was performed essentially in the same way as for GPx activity (20). Briefly the 50-μl total reaction mixture (30 μm Ure2 in 50 mm Tris-HCl buffer, pH 8.4, and 0.2 m NaCl), 50-μl supernatant, and 50-μl resuspended pellet were assayed with 1.5 mm GSH, 3 mm HEDS, 0.25 units/ml glutathione reductase, and 0.2 mm NADPH in 100 mm Tris-HCl buffer, pH 7.5, and 1 mm EDTA at 25 °C.
Once the plateau phase of the time course of Ure2 fibril formation had been reached, the total reaction mixture was centrifuged at 100,000 × g for 20 min after which the pellet was resuspended using the same volume of distilled deionized water and stored at 4 °C. The resuspended pellet was visualized using electron microscopy. Aliquots of resuspended pellet were assayed with 100 mm sodium phosphate buffer, pH 7.5, 0.1–5 mm GSH when the HEDS concentration was fixed at 3 mm, and 0.3–5 mm HEDS when the GSH concentration was fixed at 1 mm at 25 °C.
Steady-state Kinetic Analysis of the GRX Activity of Ure2, Its Prion Domain Mutants, and Its Fibrils—Given the relatively low affinity of Ure2 for GSH (20, 23, 31), it is therefore reasonable to assume that rapid equilibrium conditions apply to this reaction. Given this condition and the fact that the crystal structure of Ure2 shows that there is only one binding site for GSH within each Ure2 monomer (31), we can therefore apply the graphical method of Wang and Srivastava (32) to determine the number of essential sites within the Ure2 homodimer in solution and also the number of active sites in each catalytic unit within fibrils of Ure2. The data were analyzed according to this method (32) as follows. The initial velocity for an enzyme bearing n binding sites for the substrate GSH of which k sites are essential for catalytic activity is given as Equation 3.
 |
(Eq. 3) |
Ai and Bi are constants if the change in reactant concentration is negligible (i.e. less than 5%) as is the case here. Ai[GSH]i in the denominator is related to the total enzyme species bound to i GSH molecules, and Bi[GSH]i in the numerator is associated with functional enzyme species bound to i GSH molecules. If 1/[GSH] = x, Equation 3 can be rearranged to give Equation 4.
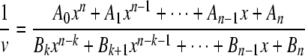 |
(Eq. 4) |
After multiplying both sides by 1/xp or [GSH]p (where p is an integer 0, 1, 2, 3,...), Equation 4 can be further rearranged to Equation 5, the limits of which are described by Equation 6.
 |
(Eq. 5) |
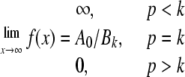 |
(Eq. 6) |
By plotting [GSH]p/v0 versus 1/[GSH] at different values of p (0, 1, 2, 3,... etc.), the appearance of a horizontal asymptote therefore identifies the condition where p = k (i.e. the number of essential binding sites in the catalytic unit) and the appearance of a zero asymptote for p = k + 1 confirms the validity of the result. In this case we found that k = 2 for both soluble dimer and fibrils of Ure2 (see “Results”). Given the presence of one GSH binding site per subunit (17, 31), this means that n = 2 for the soluble dimer. For the fibrils, given that k = 2 and the Hill coefficient approaches 2, then n must be ≥2. When n = 2 and k = 2, Equation 3 becomes Equation 7.
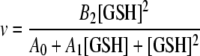 |
(Eq. 7) |
The initial velocity data were fit to Equation 7 to obtain the parameters Ai and Bi, which were then used to calculate the theoretical curves according to Equation 5 at different p values (p = 0, 1, 2, 3,...). We also attempted to fit the fibril data using values of n > 2, including 3, 4, 5, 6, 7, and 8, but no reasonable fit could be obtained, whereas using n = 2 gave excellent agreement between the experimental data and theoretical curves (see “Results”). This implies that the functional catalytic unit within fibrils is indeed a dimer rather than a higher order oligomer.
RESULTS
Ure2 Shows GRX Activity—The ability of Ure2 and its mutants to reduce the disulfide of HEDS was monitored by recording the continuous decrease in absorbance at 340 nm due to the oxidization of NADPH by the product GSSG (oxidized glutathione) under steady-state conditions. When Ure2 protein was added to the cuvette containing preincubated GSH and HEDS, a significant decrease in absorbance at 340 nm was observed. This indicates that Ure2 has GSH-dependent disulfide reductase activity toward the disulfide substrate HEDS (Fig. 1). The initial velocity for reaction of HEDS was found to be proportional to the concentration of Ure2 protein. The effect of the prion domain on the GRX activity of Ure2 was examined by comparing a series of mutants with increasing truncation in the prion domain. The truncated mutants 42Ure2, 90Ure2, and Ure2-CTD showed increasing levels of enzyme activity correlating with the increasing extent of the truncation of the prion domain (Fig. 1).
FIGURE 1.

Glutathione-dependent disulfide reduction of HEDS catalyzed by Ure2. The initial velocities of the Ure2 catalyzed reaction are plotted as a function of concentration for full-length Ure2 (•) and its N-terminally truncated mutants 42Ure2 (▴), 90Ure2 (○), and Ure2-CTD (▪). The reaction conditions were 100 mm sodium phosphate buffer, pH 7.5, 1 mm GSH, and 3 mm HEDS at 25 °C. Other details are as described under “Experimental Procedures.” The initial velocities were proportional to the concentration of Ure2 protein. No activity was observed when GSH was omitted from the reaction system.
Effect of Hydrogen Peroxide and Cadmium Chloride on the GRX Activity of Yeast GRX1 and Ure2—It is reported that Ure2 protects yeast cells from oxidant and heavy metal ion toxicity, such as against hydrogen peroxide and cadmium (18). We therefore measured the effect of hydrogen peroxide and cadmium on the enzyme activity of yGRX1 and Ure2.
After H2O2 treatment (and subsequent removal of residual H2O2; see “Experimental Procedures”), the GRX activity of yGRX1 decreased significantly, whereas Ure2 retained the same level of activity (Fig. 2A). The number of free thiol groups determined using 5,5′-dithiobis(2-nitrobenzoic acid) (see “Experimental Procedures”) was 1.8 per yGRX1 molecule before H2O2 treatment and 0.0 after treatment. (Ure2 contains no free thiol groups.) The sensitivity of yGRX1 to H2O2 treatment is consistent with the fact that yGRX1 has two active site cysteines, which become oxidized after treatment with H2O2, whereas Ure2 has no cysteine residues and remains fully active after treatment with H2O2.
FIGURE 2.

Effect of oxidation and cadmium ions on the GRX activity of yGRX1 and Ure2. The GRX activity of yGRX1 (filled bars) and Ure2-CTD (empty bars) are shown. The GRX activity determined before hydrogen peroxide treatment or in the absence of cadmium ions was set as 100% for each protein (shown as “Control”). The reaction conditions were as described in the legend to Fig. 1 unless otherwise indicated. The concentration of yGRX1 was 0.1 μm. The error bars shown represent the S.E. of at least three independent measurements. A, effect of treatment with 10 mm hydrogen peroxide prior to assay of GRX activity. The concentration of Ure2-CTD was 0.5 μm. B, effect of the presence of 10 μm cadmium chloride on GRX activity. The concentration of HEDS was 2 mm, and the concentration of Ure2-CTD was 0.4 μm. In the presence of cadmium ions, 10 units/ml glutathione reductase was used.
To measure the effects of cadmium ions, we had to make an adjustment to the coupled assay system: because the activity of glutathione reductase (which is part of the coupled enzyme assay system) can also be inhibited up to 5-fold by 10 μm cadmium chloride (data not shown), we used 5–10-fold more glutathione reductase in the assay in the presence of 10 μm cadmium chloride under which conditions the total glutathione reductase activity was at least as high as in the conditions without cadmium. As reported for human GRX previously (21), the activity of yGRX1 was impaired by cadmium (Fig. 2B). Interestingly Ure2 shows slightly higher sensitivity to cadmium than yGRX1. A possible reason is that binding of metal ions within the Ure2 active site may inhibit catalysis. These results indicate that Ure2 may play a complementary or backup role to yGRX1 in protection of yeast cells against oxidant and heavy metal toxicity.
Comparison of GRX Activity of Ure2 and Its Mutants—The observation of disulfide reductase or thioltransferase activity for a protein without any cysteine residues is extremely unexpected. Therefore, to investigate the catalytic mechanism, we examined the thioltransferase activity of active site mutants of Ure2. Given the higher activity of Ure2 when the N-terminal 104 residues are truncated (Ure2-CTD; Fig. 1), we used the active site mutants of Ure2-CTD here. The widely used substrate HEDS as well as DTTox were adopted as substrates. The bacterial thioltransferase EGST (16), which contains one active cysteine, was used as a positive control. Interestingly Ure2 mutants N124A and N124V showed negligible activity with either substrate, indicating the importance of Asn-124 for GRX activity (Fig. 3, A and B). A122S showed lower activity than Ure2-CTD toward both HEDS and DTTox, suggesting impairment of the active site by the introduction of a hydroxyl group to the side chain of residue 122. However, the level of GRX activity toward HEDS for A122C was even lower than A122S, whereas A122C showed similar or even higher activity than Ure2-CTD toward DTTox. This may be because the cysteine in A122C can be covalently modified by the disulfide substrate HEDS, whereas DTTox, an extremely weak oxidant, cannot oxidize free thiols.
FIGURE 3.

Comparison of GSH-dependent disulfide reductase activity for Ure2 and mutants. Each protein contains only the C-terminal domain of Ure2 (Ure2-CTD) with or without point mutations (as indicated). The reaction conditions were 100 mm sodium phosphate buffer, pH 7.5, at 25 °C. The error bars shown represent the S.E. of at least three independent measurements. A, GRX activity assayed with 1 mm GSH and 5 mm HEDS. The structure of HEDS is shown. The activity of EGST is shown as a control. B, GRX activity assayed with 5 mm GSH and 5 mm DTTox. The structure of DTTox is shown. EGST and bovine serum albumin (BSA) are shown as controls.
Steady-state Kinetic Analysis of Ure2 and Its Mutants—To further investigate the GRX activity of Ure2, we obtained apparent steady-state kinetic parameters for the enzymatic reaction. The results for the GRX activity of Ure2 and its mutants toward HEDS and GSH are shown in Fig. 4 and Table 1. When the GSH concentration was fixed at 1.0 mm, GRX activity was hyperbolic over a certain range of HEDS concentrations (Fig. 4A). However, at HEDS concentrations greater than 3 mm, inhibition of GRX activity was observed. When the HEDS concentration was fixed at 3.0 mm, GRX activity was sigmoidal with respect to GSH concentration (Fig. 4B). The sigmoidal kinetic behavior toward GSH remained when the HEDS concentration was varied from 0.3 to 3.0 mm, and the shape of the sigmoidal curve was independent of the Ure2 concentration (data not shown). In general, the observation of a sigmoidal curve for the GRX activity of Ure2 means that it shows positive cooperativity toward the binding of GSH, indicating that there is an allosteric interaction between the two GSH binding sites within the dimer. The Hill coefficients (nH) are shown in Table 1. All four soluble proteins show a similar nH value of around 2, consistent with the presence of two active sites within the Ure2 homodimer and indicating a high degree of cooperativity. The binding of GSH to the G-site in one of the subunits of the Ure2 dimer may result in a change in overall dimer conformation and thus may help the subsequent binding of GSH to the G-site in the other subunit of the dimer. The positive cooperative behavior of Ure2 GRX activity toward the substrate GSH is consistent with the behavior of yeast Omega GST (15) and E. coli GRX1 when assayed with substrate peptide and GSH (33), suggesting that the cooperativity provides a regulatory function in their redox roles in vivo.
FIGURE 4.

Kinetic behavior of GRX activity for Ure2 and its N-terminally truncated mutants. The parameters obtained from the fits are shown in Table 1 or in the legend to Fig. 8. A, Michaelis-Menten plots of GRX activity measured at various concentrations of HEDS with 1.0 mm GSH. The concentrations were 0.8 μm for Ure2 (•), 42Ure2 (▴), or 90Ure2 (○) and 0.5 μm for Ure2-CTD (▪). At concentrations of HEDS greater than 3 mm, inhibition of activity is observed. B, GRX activity measured at various concentrations of GSH with 3.0 mm HEDS. The concentrations were 0.5μm for Ure2 (•) or 42Ure2 (▴), 0.4 μm for 90Ure2 (○), and 0.2 μm for Ure2-CTD (▪). The fit to the Hill equation (Equation 1) is shown (solid lines). For wild-type Ure2, the fit to Equation 7 is also displayed (dashed line) and overlays with the fit to Equation 1. C, double reciprocal plots of the initial velocity data for the GRX activity of Ure2-CTD. The HEDS concentration was varied from 0.25 to 2 mm, while the GSH concentration was fixed at 0.33, 0.5, 1, or 2 mm as indicated in the figure. A group of converging lines intersecting on the x axis indicates a sequential mechanism and also indicates that the apparent Km(HEDS) value is independent of GSH concentration.
TABLE 1.
Apparent steady-state kinetic parameters and Hill coefficients for the GRX activity of soluble Ure2, its prion domain mutants, and Ure2 fibrils
|
Protein
|
Fixed [GSH] at 1
mma
|
Fixed [HEDS] at 3
mmb
|
|||
|---|---|---|---|---|---|
| Km(HEDS)(app) | Vmax(app)/[E]0 | nH | [GSH]0.5 | Vmax(app)/[E]0 | |
| mm | s-1 | mm | s-1 | ||
| Ure2 | 2.4 ± 0.1 | 0.32 ± 0.02 | 2.0 ± 0.1 | 2.3 ± 0.1 | 1.0 ± 0.1 |
| 42Ure2 | 2.4 ± 0.2 | 0.35 ± 0.03 | 2.1 ± 0.1 | 2.2 ± 0.1 | 1.2 ± 0.1 |
| 90Ure2 | 2.0 ± 0.2 | 0.52 ± 0.02 | 1.9 ± 0.1 | 1.9 ± 0.1 | 1.4 ± 0.1 |
| Ure2-CTD | 1.9 ± 0.2 | 1.2 ± 0.1 | 1.7 ± 0.2 | 1.6 ± 0.2 | 1.8 ± 0.1 |
| Ure2 fibrils | 2.2 ± 0.2 | ≥0.16 ± 0.01c | 1.7 ± 0.1 | 2.3 ± 0.1 | ≥0.40 ± 0.02c |
The errors shown are the S.E. of at least three independent measurements.
The errors shown are the S.E. of the fit to Equation 1.
The values were calculated according to the maximum Ure2 concentration in fibrils on the assumption that all soluble Ure2 was converted into amyloid-like fibrils. These are therefore minimum values for Vmax/[E]0 as the actual concentration of Ure2 in fibrils maybe lower due to formation of amorphous aggregates.
To investigate the kinetic mechanism for the two substrates, we performed the GRX activity assay for Ure2-CTD by varying the concentration of both GSH and HEDS over a 4 × 5 matrix of concentrations. Fig. 4C shows the double reciprocal plot of the data. A group of converging lines intersecting on the x axis indicates a sequential mechanism and also indicates that Km(HEDS) is independent of GSH concentration. Given that the uncatalyzed reaction rate for HEDS and GSH is very low and is proportional to the HEDS concentration when the GSH concentration is fixed (data not shown), we can assume that the total GSH concentration remains approximately constant and that the total concentration of GS-SCH2CH2OH is proportional to the HEDS concentration. Therefore, the apparent Km(HEDS) of Ure2-CTD has the same value under different GSH concentrations, suggesting that 1) the binding of GSH at one active site does not influence the binding of HEDS (or GS-SCH2CH2OH) on either the same subunit or a different subunit and that 2) the binding of HEDS (or GS-SCH2CH2OH) at one subunit does not influence the binding of a second HEDS (or GS-SCH2CH2OH) molecule on the adjacent subunit. The observation of a sequential mechanism for the GRX activity of Ure2-CTD is consistent with the mechanism observed for yGRX1 (34) and yeast monothiol ScGRX7 (8), suggesting that Ure2 follows either a steady-state ordered Bi Bi or a rapid equilibrium random Bi Bi mechanism.
Influence of the Prion Domain on GRX Activity of Ure2—From the results of the standard activity assay (Fig. 1), we found that the level of GRX activity differs depending on the extent of truncation of the prion domain with activity varying in the order Ure2-CTD > 90Ure2 > 42Ure2 ≥ full-length Ure2. We therefore compared the apparent kinetic behavior and parameters of these mutants (Table 1 and Fig. 4, A and B). When the concentration of GSH was fixed at 1.0 mm, the apparent Km(HEDS) values for Ure2 and its three mutants were approximately the same within error, suggesting the same ability to bind substrate. However, the apparent Vmax values varied in the order mentioned above. This indicates that the presence of the N-terminal domain, especially residues 90–104, interferes with the catalytic activity of the C-terminal domain as was also observed for GST activity of Ure2 point mutants (23). Given the similar apparent Km(HEDS) values, this interference effect is unlikely to involve any significant conformational change at the active site.
Dehydroascorbic Acid Reductase Activity—Because typical GRXs are found to have intrinsic DHAR activity (29), we tested whether Ure2 was able to increase the reaction rate for GSH and DHA. Ure2-CTD and its point mutants were used. Ure2-CTD shows a level of DHAR activity that is around two-thirds of the level observed for A122C, whereas mutants N124A and N124V do not show DHAR activity at all (Fig. 5). The relative level of DHAR activity of these Ure2 mutants, especially A122C, was consistent with their thioltransferase activity toward the substrate DTTox. Because Ure2-CTD precipitates at pH 7.0, we can only determine DHAR activity at around pH 7.5 or above under which conditions the spontaneous reaction rate between GSH and DHA is high. It was therefore not feasible to perform a more detailed kinetic study of DHAR activity for Ure2.
FIGURE 5.

Ure2 shows DHAR activity. DHAR activity of Ure2 and its mutants is shown. Ure2-CTD with or without point mutations (as indicated) was used. The structure of DHA is shown. The reaction conditions were 100 mm sodium phosphate buffer, pH 7.5, 1 mm EDTA, 1 mm GSH, and 1 mm DHA at 25 °C. The error bars shown represent the S.E. of at least three independent measurements.
Insulin Reduction Catalyzed by Ure2 Using GSH as Reductant—Insulin reduction is a classic method to detect disulfide reductase activity (30). This assay was used to determine whether Ure2 can reduce the disulfide bond between the A and B chains of insulin using yGRX1 and TRX as controls. Ure2-CTD and its mutants were used to perform this assay. TRX is very effective, whereas under the same conditions yGRX1 and Ure2 showed little ability to reduce the disulfide bridge of insulin when DTT was used as the reductant (Fig. 6A). For TRX, the two free thiols in the active site (-CGPC-) can directly reduce the disulfide bridge in insulin and then become an intramolecular disulfide bond that can be reduced by DTT in the assay solution or by thioredoxin reductase and NADPH in vivo. The absence of insulin reductase activity for Ure2 in the presence of DTT is consistent with the lack of a cysteine residue in the Ure2 protein. Interestingly however, yGRX1, which like TRX has a two-thiol CPYC active site motif, could not increase insulin B chain aggregation in the presence of DTT (Fig. 6A). These results are consistent with those reported for bovine heart glutaredoxin (35). However, we found that both yGRX1 and Ure2-CTD could reduce the disulfide bond of insulin in the presence of GSH (Fig. 6B). Furthermore in the presence of GSH, Ure2-CTD showed a level of insulin reductase activity similar to that of TRX, whereas the N124A mutant showed lower activity, and yGRX1 showed higher activity (Fig. 6B). This activity of Ure2 and yGRX1 using GSH as a reductant is consistent with that observed for Chlamydomonas reinhardtii cytosolic GRX1 (36), which likewise can use GSH as a reductant to reduce insulin. Presumably Ure2 can bind GSH, and then the Ure2-GSH complex can function in a way similar to a monothiol GRX. As in the GPx (20, 23) and GRX activities of Ure2, it is still unknown how Ure2 activates GSH, but it may involve residue Asn-124 and is clearly independent of any intrinsic cysteine residue.
FIGURE 6.

Ure2 shows insulin reductase activity. The reaction conditions were 100 mm Tris-HCl, pH 7.5, 80 μm insulin, and 1 μm enzyme (Ure2-CTD, yGRX1, or TRX) at 25 °C. A, insulin reductase activity of Ure2-CTD and yGRX1 using 0.8 mm DTT as reductant and TRX as a positive control. Ure2 does not have significant activity when DTT is used as reductant, consistent with the fact that Ure2 has no cysteine residues. B, insulin reductase activity of Ure2-CTD with and without N124A mutation using 8 mm GSH as reductant. TRX and yGRX1 are shown as controls.
GRX Activity Measured during the Time Course of Fibril Formation—The sigmoidal time course for formation of amyloid-like fibrils of Ure2 measured using the fluorescent dye thioflavin T under the GRX assay conditions is shown in Fig. 7A. Concomitant with the increase in thioflavin T fluorescence, the GRX activity of both the total reaction mixture and the supernatant fraction decreased dramatically. The GRX activity of the total mixture plateaued at a level significantly lower than that of the native protein, whereas the activity in the supernatant fraction almost disappeared. The GRX activity of the pellet fraction showed a time course similar to that of thioflavin T fluorescence and plateaued at a level similar to that observed for the complete sample.
FIGURE 7.

GRX activity of Ure2 fibrils. A, relationship between GRX activity and the time course of fibril formation. Incubation was in 50 mm Tris-HCl, pH 8.4, and 0.2 mm NaCl at 37 °C with shaking. Fibril formation was monitored by assaying changes in thioflavin T (ThT) binding (upper panel; ▴). In parallel (lower panel), the GRX activity in the total reaction mixture (•), supernatant fraction (○), and resuspended pellet fraction (▵) were assayed. The initial velocity is shown for a final protein concentration in the GRX assay of 1.5 μm for the total reaction mixture and a maximum of 1.5 μm in either the pellet or supernatant fraction. A.U., arbitrary units. B, initial velocity of Ure2 fibril-catalyzed GRX activity as a function of GSH concentration at a fixed level of HEDS. The reaction conditions were 100 mm sodium phosphate buffer, pH 7.5, 3 mm HEDS, and 1 mm EDTA at 25 °C. The velocity is normalized by maximum fibril concentration of the aliquots used in the assay (2 μm). Other details are as described under “Experimental Procedures.” The continuous line shows the best fit to the Hill equation (Equation 1), which gave the parameters shown in Table 1. The fit to Equation 7, which gave the parameters shown in the legend to Fig. 8, is displayed as a dashed line (and overlays with the fit to Equation 1). The upper inset is the Hill plot of the data according to Equation 2 where Y is the ratio of the initial rate to the maximum rate (0.40 s–1). The lower inset shows the electron microscopy image of Ure2 fibrils used in the enzymatic assay.
These results indicate that mature fibrils retain GRX activity, but the level of GRX activity in fibrils is significantly lower than in the native dimeric form. This is in contrast to the results for the GPx activity of Ure2 and the GST activity of Ure2 point mutants, which showed a similar level before and after fibril formation (20, 23). However, the significantly greater sensitivity of the GRX activity assay compared with the previously described GPx and GST assays for Ure2 meant that we could apply the GRX assay in further characterization of the enzymatic behavior of Ure2 even in its fibrillar form.
Use of GRX Activity to Characterize the Number of GSH Binding Sites within the Catalytic Unit of Ure2 in Soluble and Fibrillar Forms—The GRX activity of Ure2 fibrils toward HEDS follows the Michaelis-Menten equation and shows hyperbolic curves similar to those of the soluble protein (data not shown). The apparent values of Km(HEDS) and Vmax/[E]0 for Ure2 fibrils are shown in Table 1. Notably the apparent Km(HEDS) value is the same as that of soluble Ure2 within error, suggesting that the binding capacity of soluble Ure2 to HEDS or GS-SCH2CH2OH is not affected by fibril formation. The maximum possible Ure2 concentration within the fibrils (2 μm) was used to calculated Vmax/[E]0. However, because of the presence of amorphous aggregates in the fibril pellet, which may be denatured or blocked from substrate binding, the Vmax/[E]0 shown in Table 1 is a minimum value.
The GSH dependence of the GRX activity of Ure2 amyloid-like fibrils shows a sigmoidal curve indicating positive cooperativity (Fig. 7B). This is very similar to the behavior of soluble Ure2 and its mutants (Fig. 4B). The Hill coefficient nH for Ure2 fibrils is close to 2 as for the soluble dimer (Table 1). These results strongly suggest that the catalytic unit in Ure2 fibrils contains at least two subunits that interact with each other during catalysis.
To investigate this further, we used the graphical method developed by Wang and Srivastava (32) to determine the number of essential sites for substrate binding in one catalytic unit of the oligomeric enzyme (see “Experimental Procedures”). Fig. 8A shows plots of [GSH]p/v0 versus 1/[GSH] at different values of p. When p = 2, the plot approaches a horizontal asymptote, and when p = 3, a zero asymptote is observed. These results indicate that the parameter k in Equation 3, representing the number of essential substrate binding sites in the catalytic unit in the soluble Ure2 homodimer, is equal to 2. The same result was obtained for Ure2 fibrils (Fig. 8B). This indicates that the Ure2 dimer (which contains one GSH binding site per monomer) must bind two GSH molecules to catalyze the GRX reaction. In other words, monomeric Ure2 or an oligomer binding only one GSH molecule does not show GRX activity. These results indicate that the dimer represents the minimum catalytic unit for the GRX activity of Ure2 in both soluble and fibrillar forms.
FIGURE 8.

Analysis of the number of essential binding sites within the catalytic unit of soluble and fibrillar Ure2. The data for the GSH dependence of GRX activity for wild-type soluble Ure2 (as shown in Fig. 4B) and Ure2 fibrils (as shown in Fig. 7B) were replotted in the form [GSH]p/v0 versus 1/[GSH] for different integer values of p. According to the Wang and Srivastava (32) graphical method, the observation of a horizontal asymptote indicates that p is equal to the number of essential binding sites (k) within the catalytic unit of the enzyme (see “Experimental Procedures”). The continuous lines represent the theoretical curves according to Equation 5 using n = 2, k = 2, and the values of A0, A1, and B2 derived from fitting the original plots of the data (dashed lines in Figs. 4B and 7B) to Equation 7. A, plots for soluble dimeric Ure2. The parameters used in Equation 5 were A0 = 4.7 ± 0.3 mm2, A1 = 0.6 ± 0.5 mm, and B2 = 1.0 ± 0.1 s–1 (where the error indicates the S.E. of the fit). B, plots for Ure2 fibrils. The parameters used in Equation 5 were A0 = 3.3 ± 0.3 mm2, A1 = 1.6 ± 0.6 mm, and B2 = 0.45 ± 0.04 s–1. The initial velocity was normalized using the maximum fibril concentration of 2μm in a final reaction volume of 1 ml.
DISCUSSION
The yeast prion protein Ure2, which shows structural similarity to GST and GRX, protects yeast cells from heavy metal ion and oxidant toxicity. Although its low level of GPx activity (20) explains the in vivo role of Ure2 to a certain extent, it is still hard to understand the detoxification activity of Ure2 toward heavy metal ions, especially cadmium. Cadmium ions not only cause elevated reactive oxygen species levels in the cell but also directly inactivate glutaredoxin and thioredoxin, which bear a CXXC active site motif (21). The N-terminal subdomain of the Ure2 globular region (residues 105–196) adopts a canonical thioredoxin fold, which is found widely in nature. This subdomain of Ure2 shows high homology to glutaredoxin by Dali structural comparison search, especially E. coli GRX2. For these reasons, we decided to test whether Ure2 can function as a glutaredoxin.
Here observation of a significant level of GRX-like activity for Ure2 indicates that it can directly participate in catalysis of disulfide and dehydroascorbic acid reduction. The apparent kcat measured for the GRX activity of Ure2-CTD toward HEDS was 1.2 s–1 (Table 1), which is similar to the value of 2.2 s–1 reported for yGRX1 (34). Classical GRX enzymes have an active site CXXC or CXXS motif in which the most N-terminal cysteine is essential for activity. The free cysteine, the thiol of which has a low pKa value, is able to attack the sulfur atom in disulfide bridges, especially GSH mixed disulfide bonds, and thus promote thiol transfer. In fact, Omega class (12, 14, 15) and Beta class (13, 16) GST members have been found to show GRX activity toward HEDS. Similar to GRX family members, the active site cysteine of these GST proteins is essential for their GRX-like activity. However, the Ure2 protein does not have any cysteine residues. Site-directed mutagenesis of Ure2 indicates that the important residue for this redox activity may be Asn-124 (Figs. 3, 5, and 6B), which is also essential for the GPx activity of Ure2 (23). How Asn-124 activates GSH and facilitates the thiol transfer reaction remains to be elucidated. The structurally equivalent position in Ure2 to the yeast GRX1 and GRX2 active site -CPYC- motif is -APNG- (residues 122–125), which contains a conserved Pro like most GRXs and many GSTs. The introduction of a free thiol group at site 122 before this Pro increases the enzyme activity toward DTTox and DHA (Figs. 3B and 5), suggesting that Ure2 still retains some characteristics of an ancient GRX.
Thiol-independent GRX activity may be physiologically important for yeast cells especially under stress conditions. Many proteins related to oxidative and heavy metal ion stress resistance or redox signaling, such as yGRX1, contain active site cysteines, which may be easily oxidized when acute stress occurs (Fig. 2A). Ure2, a protein containing no cysteine residues, still possesses a significant level of GRX activity toward small molecule disulfides (or GSH mixed disulfide bonds) and protein disulfides, but because of its lack of cysteines, it is immune to direct oxidative damage (Fig. 2A). These findings broaden the known range of catalytic mechanisms for thiol transfer and help us to understand the in vivo role of Ure2 in oxidative stress resistance. In fact, the GRX activity of both yGRX1 and Ure2 can be inhibited by cadmium ions to a similar level (Fig. 2B). This provides an explanation for the sensitivity of wild-type yeast cells toward cadmium and the increased sensitivity of ure2Δ cells (18).
Many enzymes show cross-reactivity with related substrates, termed “enzyme promiscuity.” The observation that yeast GRX1 and GRX2 show GRX activity toward HEDS, GPx activity toward organic hydroperoxides, and GST activity toward 1-chloro-2,4-dinitrobenzene (37, 38) would seem to be a good example of this phenomenon. Ure2, harboring GRX, GPx, DHAR, and insulin reductase activity, is also a good example of a promiscuous enzyme. In addition, Ure2 shows GST activity after single site mutation (23). The glutathione-dependent peroxidase activity (20) and disulfide reductase activity of Ure2 identified in this study suggest that it is an atypical glutaredoxin without a CXX(C/S) motif and may play an important role under stress conditions in vivo, complementary to that of GRX family members.
It is reported that thioredoxin and glutaredoxin reduce protein disulfide bonds using a dithiol mechanism. However, the inability of yGRX1 to reduce insulin disulfide using DTT shows that either DTT cannot reduce the intramolecular disulfide at the yGRX1 active site or another catalytic mechanism is used instead of the dithiol mechanism. GSH alone can reduce the insulin interchain disulfide bridge at a very low rate. We found that Ure2 is able to accelerate the reduction of the insulin disulfide bond by GSH. Although the precise physiological role of Ure2 as an antioxidant in yeast cells has not yet been defined, the insulin reductase activity clearly shows that Ure2 harbors the ability to reduce protein disulfides, such as resulting from oxidative or heavy metal ion stress, and like members of the GRX family (10) may fulfill some functions related to reversible protein glutathionylation signal transduction.
The structure of Ure2 amyloid-like fibrils is an intriguing problem, and there is no widely accepted model at present. To date, two basic models have been proposed for the structure of Ure2 fibrils. The first one is the so-called “amyloid backbone” model (39, 40) in which the N-terminal domain of Ure2 forms an amyloid core surrounded by C-terminal domains in a largely native conformation (41–43). Another model (44, 45) suggests that soluble Ure2 dimers must disassociate to monomer and then rearrange to assemble into fibrils. Although both models are compatible with near-native conformation of the C-terminal domain in fibrils, they do not provide details about the relative arrangement of the monomers (43, 45). Melki and co-workers (44, 45) proposed that Ure2 fibrils might assemble through intermolecular interaction between the N-terminal domain and the hydrophobic region at the dimer interface of the C-terminal domain. However, the observation here of positive cooperativity toward GSH for the GRX activity of Ure2 fibrils, as seen also for soluble dimeric Ure2, indicates that the minimum catalytic unit of the C-terminal domains of Ure2 within the fibrils is a dimer with native-like interaction occurring between adjacent subunits. The Hill coefficient for the GRX activity of Ure2 in both soluble and dimeric forms was close to 2 (Table 1), indicating a high degree of cooperativity between the subunits within each dimer. The application of the Wang and Srivastava (32) graphical method shows that the number of essential GSH binding sites in the catalytic unit within Ure2 fibrils is two (Fig. 8B) as in the soluble dimer (Fig. 8A). Thus, the dimer represents the minimum catalytic unit for the GRX activity of Ure2 in both soluble and fibrillar forms. According to these results, we can rule out interaction between the hydrophobic surface area of the dimer interface with the α-cap region (44) or N-terminal domain (45) in Ure2 fibrils. It is possible that some non-native arrangement of the Ure2 dimer might show the same interaction or cooperativity between the two GSH binding sites as the native dimer, but this explanation seems unlikely. Thus, definition of the relative molecular arrangement of Ure2 subunits within amyloid-like fibrils as similar to that in the native dimer not only has implications for architectural models but also has implications for the molecular mechanism of prion formation.
Acknowledgments
We thank Prof. Congzhao Zhou, University of Science and Technology of China, for kindly providing the yGRX1 plasmid. We thank Prof. Zhixin Wang, Tsinghua University, for helpful discussions, suggestions, and comments on the manuscript. We thank Dr. Xi Wang, Institute of Biophysics, for advice and for providing purified TRX protein. We also thank Prof. Junmei Zhou and Prof. Chih-chen Wang for continued encouragement and support.
This work was supported by National Natural Science Foundation of China Grants 30620130109, 30670428, and 30870482; Chinese Ministry of Science and Technology Grants 2006CB500703 and 2006CB910903; and Chinese Academy of Sciences Grant KSCX2-YW-R-119.
Footnotes
The abbreviations used are: GSH, reduced glutathione; GPx, glutathione-dependent peroxidase; GRX, glutathione-dependent reductase or glutaredoxin; DHA, dehydroascorbic acid; DHAR, dehydroascorbic acid reductase; GST, glutathione S-transferase; HEDS, 2-hydroxyethyl disulfide; Ure2-CTD, Ure2 C-terminal domain (residues 105–354); DTTox, trans-4,5-dihydroxy-1,2-dithiane; TRX, E. coli thioredoxin; yGRX1, yeast glutaredoxin 1; EGST, E. coli GST; DTT, dithiothreitol.
References
- 1.Holmgren, A. (1976) Proc. Natl. Acad. Sci. U. S. A. 73 2275–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lillig, C. H., Potamitou, A., Schwenn, J. D., Vlamis-Gardikas, A., and Holmgren, A. (2003) J. Biol. Chem. 278 22325–22330 [DOI] [PubMed] [Google Scholar]
- 3.Fernandes, A. P., and Holmgren, A. (2004) Antioxid. Redox Signal. 6 63–74 [DOI] [PubMed] [Google Scholar]
- 4.Martin, J. L. (1995) Structure (Lond.) 3 245–250 [DOI] [PubMed] [Google Scholar]
- 5.Wells, W. W., Xu, D. P., Yang, Y. F., and Rocque, P. A. (1990) J. Biol. Chem. 265 15361–15364 [PubMed] [Google Scholar]
- 6.Herrero, E., and de la Torre-Ruiz, M. A. (2007) CMLS Cell. Mol. Life Sci. 64 1518–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lillig, C. H., Berndt, C., and Holmgren, A. (2008) Biochim. Biophys. Acta 1780 1304–1317 [DOI] [PubMed] [Google Scholar]
- 8.Mesecke, N., Mittler, S., Eckers, E., Herrmann, J. M., and Deponte, M. (2008) Biochemistry 47 1452–1463 [DOI] [PubMed] [Google Scholar]
- 9.Mesecke, N., Spang, A., Deponte, M., and Herrmann, J. M. (2008) Mol. Biol. Cell 19 2673–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallogly, M. M., and Mieyal, J. J. (2007) Curr. Opin. Pharmacol. 7 381–391 [DOI] [PubMed] [Google Scholar]
- 11.Sheehan, D., Meade, G., Foley, V. M., and Dowd, C. A. (2001) Biochem. J. 360 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Board, P. G., Coggan, M., Chelvanayagam, G., Easteal, S., Jermiin, L. S., Schulte, G. K., Danley, D. E., Hoth, L. R., Griffor, M. C., Kamath, A. V., Rosner, M. H., Chrunyk, B. A., Perregaux, D. E., Gabel, C. A., Geoghegan, K. F., and Pandit, J. (2000) J. Biol. Chem. 275 24798–24806 [DOI] [PubMed] [Google Scholar]
- 13.Caccuri, A. M., Antonini, G., Allocati, N., Di Ilio, C., De Maria, F., Innocenti, F., Parker, M. W., Masulli, M., Lo Bello, M., Turella, P., Federici, G., and Ricci, G. (2002) J. Biol. Chem. 277 18777–18784 [DOI] [PubMed] [Google Scholar]
- 14.Whitbread, A. K., Masoumi, A., Tetlow, N., Schmuck, E., Coggan, M., and Board, P. G. (2005) Methods Enzymol. 401 78–99 [DOI] [PubMed] [Google Scholar]
- 15.Garcera, A., Barreto, L., Piedrafita, L., Tamarit, J., and Herrero, E. (2006) Biochem. J. 398 187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang, X. Y., Zhang, Z. R., and Perrett, S. (2009) Biochem. J. 417 55–64 [DOI] [PubMed] [Google Scholar]
- 17.Bousset, L., Belrhali, H., Janin, J., Melki, R., and Morera, S. (2001) Structure (Lond.) 9 39–46 [DOI] [PubMed] [Google Scholar]
- 18.Rai, R., Tate, J. J., and Cooper, T. G. (2003) J. Biol. Chem. 278 12826–12833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rai, R., and Cooper, T. G. (2005) Yeast 22 343–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai, M., Zhou, J. M., and Perrett, S. (2004) J. Biol. Chem. 279 50025–50030 [DOI] [PubMed] [Google Scholar]
- 21.Chrestensen, C. A., Starke, D. W., and Mieyal, J. J. (2000) J. Biol. Chem. 275 26556–26565 [DOI] [PubMed] [Google Scholar]
- 22.Perrett, S., Freeman, S. J., Butler, P. J., and Fersht, A. R. (1999) J. Mol. Biol. 290 331–345 [DOI] [PubMed] [Google Scholar]
- 23.Zhang, Z. R., Bai, M., Wang, X. Y., Zhou, J. M., and Perrett, S. (2008) J. Mol. Biol. 384 641–651 [DOI] [PubMed] [Google Scholar]
- 24.Gill, S. C., and von Hippel, P. H. (1989) Anal. Biochem. 182 319–326 [DOI] [PubMed] [Google Scholar]
- 25.Yu, J., Zhang, N. N., Yin, P. D., Cui, P. X., and Zhou, C. Z. (2008) Proteins 72 1077–1083 [DOI] [PubMed] [Google Scholar]
- 26.Zhao, Z., Peng, Y., Hao, S. F., Zeng, Z. H., and Wang, C. C. (2003) J. Biol. Chem. 278 43292–43298 [DOI] [PubMed] [Google Scholar]
- 27.Ellman, G. L. (1959) Arch. Biochem. Biophys. 82 70–77 [DOI] [PubMed] [Google Scholar]
- 28.Holmgren, A., and Aslund, F. (1995) Methods Enzymol. 252 283–292 [DOI] [PubMed] [Google Scholar]
- 29.Wells, W. W., Xu, D. P., and Washburn, M. P. (1995) Methods Enzymol. 252 30–38 [DOI] [PubMed] [Google Scholar]
- 30.Holmgren, A. (1979) J. Biol. Chem. 254 9627–9632 [PubMed] [Google Scholar]
- 31.Bousset, L., Belrhali, H., Melki, R., and Morera, S. (2001) Biochemistry 40 13564–13573 [DOI] [PubMed] [Google Scholar]
- 32.Wang, Z. X., and Srivastava, D. K. (1994) Anal. Biochem. 216 15–26 [DOI] [PubMed] [Google Scholar]
- 33.Peltoniemi, M. J., Karala, A. R., Jurvansuu, J. K., Kinnula, V. L., and Ruddock, L. W. (2006) J. Biol. Chem. 281 33107–33114 [DOI] [PubMed] [Google Scholar]
- 34.Discola, K. F., de Oliveira, M. A., Rosa Cussiol, J. R., Monteiro, G., Barcena, J. A., Porras, P., Padilla, C. A., Guimaraes, B. G., and Netto, L. E. (2009) J. Mol. Biol. 385 889–901 [DOI] [PubMed] [Google Scholar]
- 35.Jung, C. H., and Thomas, J. A. (1996) Arch. Biochem. Biophys. 335 61–72 [DOI] [PubMed] [Google Scholar]
- 36.Zaffagnini, M., Michelet, L., Massot, V., Trost, P., and Lemaire, S. D. (2008) J. Biol. Chem. 283 8868–8876 [DOI] [PubMed] [Google Scholar]
- 37.Collinson, E. J., Wheeler, G. L., Garrido, E. O., Avery, A. M., Avery, S. V., and Grant, C. M. (2002) J. Biol. Chem. 277 16712–16717 [DOI] [PubMed] [Google Scholar]
- 38.Collinson, E. J., and Grant, C. M. (2003) J. Biol. Chem. 278 22492–22497 [DOI] [PubMed] [Google Scholar]
- 39.Taylor, K. L., Cheng, N., Williams, R. W., Steven, A. C., and Wickner, R. B. (1999) Science 283 1339–1343 [DOI] [PubMed] [Google Scholar]
- 40.Speransky, V. V., Taylor, K. L., Edskes, H. K., Wickner, R. B., and Steven, A. C. (2001) J. Cell Biol. 153 1327–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baxa, U., Speransky, V., Steven, A. C., and Wickner, R. B. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5253–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baxa, U., Taylor, K. L., Wall, J. S., Simon, M. N., Cheng, N., Wickner, R. B., and Steven, A. C. (2003) J. Biol. Chem. 278 43717–43727 [DOI] [PubMed] [Google Scholar]
- 43.Kajava, A. V., Baxa, U., Wickner, R. B., and Steven, A. C. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 7885–7890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bousset, L., Thomson, N. H., Radford, S. E., and Melki, R. (2002) EMBO J. 21 2903–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fay, N., Redeker, V., Savistchenko, J., Dubois, S., Bousset, L., and Melki, R. (2005) J. Biol. Chem. 280 37149–37158 [DOI] [PubMed] [Google Scholar]