

Abstract
The immunoglobulin (Ig) constant CH2 domain is critical for antibody effector functions. Isolated CH2 domains are promising as scaffolds for construction of libraries containing diverse binders that could also confer some effector functions. However, previous work has shown that an isolated murine CH2 domain is relatively unstable to thermally induced unfolding. To explore unfolding mechanisms of isolated human CH2 and increase its stability γ1 CH2 was cloned and a panel of cysteine mutants was constructed. Human γ1 CH2 unfolded at a higher temperature (Tm = 54.1 °C, as measured by circular dichroism) than that previously reported for a mouse CH2 (41 °C). One mutant (m01) was remarkably stable (Tm = 73.8 °C). Similar results were obtained by differential scanning calorimetry. This mutant was also significantly more stable than the wild-type CH2 against urea induced unfolding (50% unfolding at urea concentration of 6.8 m versus 4.2 m). The m01 was highly soluble and monomeric. The existence of the second disulfide bond in m01 and its correct position were demonstrated by mass spectrometry and nuclear magnetic resonance spectroscopy, respectively. The loops were on average more flexible than the framework in both CH2 and m01, and the overall secondary structure was not affected by the additional disulfide bond. These data suggest that a human CH2 domain is relatively stable to unfolding at physiological temperature, and that both CH2 and the highly stable mutant m01 are promising new scaffolds for the development of therapeutics against human diseases.
Monoclonal antibodies (mAbs)2 with high affinity and specificity are now well established therapeutics and invaluable tools for biological research. It appears that their use will continue to expand in both targets and disease indications. However, a fundamental problem for full-size mAbs that limits their applications is their poor penetration into tissues (e.g. solid tumors) and poor or absent binding to regions on the surface of some molecules (e.g. on the HIV envelope glycoprotein) that are accessible by molecules of smaller size. Antibody fragments, e.g. Fabs (∼60 kDa) or single chain Fv fragments (scFvs) (20∼30 kDa), are significantly smaller than full-size antibodies (∼150 kDa), and have been used as imaging reagents and candidate therapeutics. Even smaller fragments of antibodies are of great interest and advantageous for pharmaceutical applications including cancer targeting and imaging.
During the last decade a large amount of work has been aimed at developing of small size binders with scaffolds based on various highly stable human and non-human molecules (1–8). A promising direction is the development of binders based on the heavy or light chain variable region of an antibody; these fragments ranging in size from 11 kDa to 15 kDa were called “domain antibodies” or “dAbs” (7, 9). A unique kind of antibodies composed only of heavy chains are naturally formed in camels, dromedaries, and llamas, and their variable regions can also recognize antigens as single domain fragments (10). Not only is the overall size of the dAbs much smaller than that of full-size antibodies but also their paratopes are concentrated over a smaller area so that the dAbs provide the capability of interacting with novel epitopes that are inaccessible to conventional antibodies or antibody fragments with paired light and heavy chain variable domains.
The structure of the constant antibody domains is similar to that of the variable domains consisting of β-strands connected mostly with loops or short helices. The second domain of the α, δ, and γ heavy chain constant regions, CH2, is unique in that it exhibits very weak carbohydrate-mediated interchain protein-protein interactions in contrast to the extensive interchain interactions that occur between the other domains. The expression of murine CH2 in bacteria, which does not support glycosylation, results in a monomeric domain (11). It has been hypothesized that the CH2 domain (CH2 of IgG, IgA, and IgD, and CH3 of IgE and IgM) could be used as a scaffold and could offer additional advantages compared with those of dAbs because it contains binding sites or portions of binding sites conferring effector and stability functions (12).
It was found previously that an isolated murine CH2 is relatively unstable at physiological temperature with a temperature of 50% unfolding (Tm) slightly higher than 37 °C (11). We have hypothesized that human CH2 would exhibit different stability because of significant differences in the sequence compared with its murine counterpart. Therefore, we have extensively characterized the stability of an isolated unglycosylated single CH2 domain. We found that its stability is significantly higher than the previously reported for the murine CH2. We further increased the stability of the human CH2 by engineering an additional disulfide bond between the A and G strands. One of the newly developed mutants, denoted as m01, exhibited significantly higher stability (Tm = 73.8 °C) than that of wild type CH2. We suggest that both the wild type CH2 and the newly developed mutant, m01, could be used as scaffolds for binders. These results also demonstrate for the first time that the stability of constant antibody domains can be dramatically increased by engineering of an additional disulfide bond. The increase in stability of isolated domains may result in an increase in stability of larger antibody fragments, e.g. Fc, and therefore could have implications as a general method for increasing antibody stability. Thus, it appears that further development of CH2 and its more stable variants as scaffolds could provide new opportunities for identification of potentially useful therapeutics.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of CH2 Domains—Human γ1 CH2 was cloned in bacterial expression vectors and used for transformation of Escherichia coli strain HB2151 cells, which were grown at 37 °C in SB medium to an optical density of A600 ∼0.6–0.8. Expression was induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside at 37 °C for 12–16 h. Bacterial cells were harvested and resuspended in Buffer A (50 mm Tris·Cl, 450 mm NaCl, pH 8.0) at 1:10 (volume of Buffer A:culture volume). Polymyxin B sulfate (Sigma-Aldrich) (0.5 mu/ml) was added to the suspension (1:1000, volume of polymyxin B sulfate: culture volume). The cell lysate was subsequently clarified by centrifugation at 15,000 rpm for 45 min at 4 °C and tested for expression by SDS-PAGE and Western. The clarified supernatant was purified by using 1 ml of HiTrap Chelating HP Ni-NTA column (GE Healthcare). After elution with Buffer B (50 mm Tris·Cl, 450 mm NaCl, 200 mm imidazole, pH 8.0), the imidazole was removed by Amicon Ultra-15 Centrifugal Filter Devices (Millipore), and the purified proteins were kept in Buffer A or PBS (9.0 g/liter NaCl, 144 mg/liter KH2PO4, 795 mg/liter Na2HPO4, pH 7.4). The proteins were checked for purity by SDS-PAGE, and their concentrations were determined by measuring the UV absorbance (13).
CH2 Mutant Design and Plasmid Construction—To design the CH2 mutants we used the IgG1 b12 crystal structure (14) and the crystal structure of an isolated unglycosylated human CH2 (15). Five mutants, V10/E103 to C10/C103, F11/K104 to C11/C104, L12/T105 to C11/C105, L12/K104 to C12/C104, and V10/K104 to C10/C104,were selected for characterization by analyzing the structure with the computer program VMD 1.8.6 (16). They were made by PCR-based site-directed mutagenesis and cloned into bacterial expression vectors. The clones were verified by direct sequencing and used for transformation of the E. coli strain HB2151. The mutants were expressed and purified similarly to the wild type CH2.
Size Exclusion Chromatography—The purified CH2, m01, and m02 were loaded into the Hiload 26/60 Superdex 75 HR 10/30 column (GE Healthcare) running onÄKTA BASIC pH/C chromatography system (GE Healthcare) to assess possible oligomer formation. Buffer A was selected as mobile phase. A gel filtration of standards consisting of aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (44 kDa), chymotrypsinogen A (25 kDa), and ribonuclease A (13.7 kDa) was used to define the molecular weight of CH2, m01, and m02.
Determination of Disulfide Bonds by Mass Spectrometry—The total number of disulfide bonds in purified CH2, m01, and m02 were determined using a Voyager 4700 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Framingham, MA) by comparing the molecular masses after (A) reduction and alkylation of all SH groups and (B) alkylation of the original free SH groups without reduction of disulfide bonds. Reduction was carried out using TCEP, and alkylation was performed using iodoacetamide.
Circular Dichroism—The secondary structures of CH2, m01, and m02 were determined by CD spectroscopy. The purified proteins were dissolved in PBS at the final concentration of 0.49 mg/ml, and the CD spectra were recorded on AVIV Model 202 CD Spectrometer (Aviv Biomedical). Wavelength spectra were recorded at 25 °C using a 0.1-cm path-length cuvette for native structure measurements. Thermal stability was measured at 216 nm by recording the CD signal in the temperature range of 25–90 °C with heating rate 1 °C/min. The temperature was recorded with an external probe sensor and the temperature inside the microcuvette was calculated by calibration; it was about 2–3 °C (range from 1.9 to 3.8 °C for temperatures from 20 to 80 °C) lower than the one measured by the external sensor. After heating, wavelength spectra were recorded at 90 °C. For evaluation of the refolding, all the samples were kept at 4 °C overnight and measured at 25 °C again.
Differential Scanning Calorimetry (DSC)—The thermal stabilities of CH2, m01, and m02 were further monitored with a VP-DSC MicroCalorimeter (MicroCal, Northampton, MA). The concentrations of the three proteins were 1.5 mg/ml in PBS (pH 7.4). The heating rate used was 1 °C/min, and the scanning was performed from 25 to 100 °C.
Spectrofluorometry—The intrinsic fluorescence of CH2, m01, and m02 were recorded on a Fluorometer Fluoromax-3 (HORIBA Jobin Yvon, NJ). Intrinsic fluorescence measurements were performed using a protein concentration of 10 μg/ml with excitation wavelength at 280 nm, and emission spectra recorded from 320 to 370 nm at 25 °C. Buffer A in the presence of urea from 0 to 8 mm was used. With all samples, fluorescence spectra were corrected for the background fluorescence of the solution (buffer + denaturant). Fluorescence intensity at 340 nm was used for unfolding evaluation.
Nuclear Magnetic Resonance (NMR)—For the NMR experiments E. coli was first grown in 2×YT medium. Single colony was inoculated in 3 ml of 2×YT medium for about 3 h, then turbidity was checked and bacteria were transferred to 1 liter of 2×YT medium for further growth at 37 °C until A600 ∼0.8–0.9 was reached. The cell culture was then centrifuged to remove the 2×YT medium and replaced with a M9 minimum medium with [15N]NH4Cl and [13C]glucose as sole 15N and 13C sources, respectively (17). The cells were incubated at 30 °C overnight, and induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside. Harvested cells were suspended in TES buffer (10 ml of buffer for 1 liter of culture) for 1 h on ice. Osmotic shock to release periplasmic proteins was induced by adding 1.5 volume TES/5 on ice for 4 h. The supernatant was then dialyzed in a dialysis buffer (50 mm Tris·Cl, 0.5 m NaCl) overnight at 4 °C. The protein was purified by the method described above for an initial purification. Fractions containing a significant amount of the protein were then loaded on Sephacryl S-200 column (GE Healthcare) for further purification. The separated fractions samples were collected in Buffer A.
NMR experiments were performed in 40 mm Tris·Cl buffer at pH 7.8 containing 64 mm NaCl in 95% H2O, 5% D2O and a sample volume of ∼300 μl in a 5-mm Shigemi tube (Shigemi Inc) with a protein concentration of 0.5–0.8 mm at 25 °C. NMR experiments were conducted using Bruker Avance 600 MHz spectrometer, which is equipped with a cryogenic probe (Bruker Instruments). Water-flip back sequences were used for 1H-15N HSQC and 1H-15N NOE experiments to minimize exchange between amide protons and water protons (18). 1H-15N HSQC spectra were recorded with 1024 complex points for an acquisition dimension with a spectral width of 8012 Hz, and 256 complex points for an indirect (t1) dimension. 1H-15N NOE experiments were conducted with similar number of points by recording two sets of spectra, with and without proton saturation at 3 and 4 s repetition delays, respectively (19). Uncertainties of the NOE values were estimated from root mean square deviation (RMSD) noise of the two spectra and peak heights.
Signal assignments were performed based on HNCA, CBCACONH, CCONH experiments for the CH2 domain, and HNCACB and CBCACONH and 13C, 15N simultaneous evolution NOESY for m01 (20, 21). NMR data were processed and analyzed using the nmrPipe and CARA (22, 23). To color significance of chemical shift changes on the CH2 backbone structure, a normalized chemical shift changes (Equation 1),
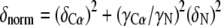 |
(Eq. 1) |
its average, and S.D. were calculated, and are grouped to four classes: δnorm > 3.0 (red), 3.0 > δnorm > 2.0 (orange), 2.0 > δnorm > 1.0 (yellow), and (4) δnorm < 1 (blue).
RESULTS
Isolated, Unglycosylated Human γ1 CH2 Domain Is Relatively Stable—Human γ1 heavy chain CH2 (Fig. 1A) was cloned in a bacterial expression vector, expressed and purified as described under “Experimental Procedures.” It expresses at high levels as soluble protein (more than 10 mg per liter of bacterial culture) and is highly soluble (more than 10 mg/ml). It is monomeric in PBS at pH 7.4 as determined by size exclusion chromatography (Fig. 1B) (15), and runs on SDS-PAGE gels with an apparent molecular mass (MW) of about 14–15 kDa, which is close to the calculated MW (14.7 kDa, including the His and FLAG tags) and as expected is much smaller than the MWs of scFv, Fab, and IgG1 (Fig. 1C).
FIGURE 1.

A, amino acid sequence alignment of human CH2 (NCB Accession J00228) and mouse CH2 (NCB Accession J00453). Identical and similar residues were 67 and 92%, respectively. B, size exclusion chromatogram of human CH2. The inset is a standard curve. C, molecular size of CH2 compared with scFv, Fab, and IgG1. SDS-PAGE gel of purified CH2 (lane 1,10 μg; lane 2,5 μg), scFv m9 (lane 3), Fab X5 (lane 4), and IgG X5 (lane 5).
Previously, it has been found that an isolated unglycosylated murine CH2 domain is relatively unstable at physiologically relevant temperatures (Tm = 41 °C as measured by CD (11). The sequence of human CH2 differs from its murine counterpart (Fig. 1A), which could lead to different stabilities. To test the thermal stability of human γ1 CH2 we used both CD and DSC. As measured by CD the secondary structure of CH2 consisted primarily of β strands at 25 °C (Fig. 2A). The CH2 unfolding started at about 42 °C and was completed at about 62 °C with a calculated Tm of 54.1 ± 1.2 °C (Fig. 2B). The unfolding was reversible (Fig. 2A). Similar results were obtained by DSC (Tm = 55.4 °C) (Fig. 2C). Although we were not able to test the murine CH2 used in the previous study concurrently with human CH2, it appears that human γ1 CH2 is significantly more stable than its murine counterpart.
FIGURE 2.

Stability of human CH2 measured by CD and DSC. A, folding curve at 25 °C (—), unfolding at 90 °C (□□□), and refolding (– – –) at 25 °C measured by CD. B, fraction-folded of the protein (ff) was calculated as ff = ([θ] – [θM])/([θT] – [θM]). [θT] and [θM] were the mean residue ellipticities at 216 nm of folded state at 25 °C and unfolded state of 90 °C. The Tm value (54.1 ± 1.2 °C) from CD was determined by the first derivative [d(Fraction-folded)/dT] with respect to temperature (T). C, thermo-induced unfolding curve from DSC. Tm = 55.4 °C, which is similar to that from CD.
Design and Generation of Engineered Human γ1 CH2 Domains with an Additional Disulfide Bond—To further improve the stability of human CH2, we engineered an additional disulfide bond between the N-terminal strand A and the C-terminal one G. We reasoned that constraining the degrees of freedom of these two strands could lead to a decrease in the extent of unfolding. The mutants were initially designed based on the crystal structure of CH2 in an intact IgG1 (14), which is very similar to the crystal structure of isolated CH2, which we recently reported although there are certain differences in some loops and at both termini (15). Based on the distance between two α-carbons of two cysteines in proteins with known structure (24, 25) and the orientation of the bonds we selected five amino acid pairs, V10/E103, F11/K104, L12/T105, L12/K104, and V10/K104 (the numbering starts with 1:Ala (Fig. 1A) corresponding to number 231 in the γ1 heavy chain), which were substituted by Cys residues. Two mutants (L12/K104 to C12/C104, distance between the Cαs in L12 and K104 = 6.53 Å, and V10/K104 to C10/C104, distance between the Cαs in V10 and K104 = 7.25 Å) (Fig. 3), designated m01 and m02, respectively, were highly soluble and expressed at levels comparable or higher than that of CH2 (Fig. 4).
FIGURE 3.

Design of m01 and m02 based on the CH2 structure. The distance between two Cαs forming the native disulfide bond (indicated by black arrow) is 6.53 Å. An engineered disulfide bond was introduced between Leu-12 and Lys-104 (m01) or Val-10 and Lys-104 (m02), which were replaced by cysteines.
FIGURE 4.

High level of expression of m01 and m02. Soluble expression of m01 and m02 is compared with that of CH2 by SDS-PAGE. The expressed proteins are indicated by arrows.
The existence of an additional disulfide bond was confirmed by mass spectrometry. The number of disulfide bonds in CH2 was one, while the mutants m01 and m02 contained two, as expected (Table 1). These mutants were selected for further characterization.
TABLE 1.
Number of disulfide bonds determined by mass spectrometry
| Protein | Intact | Denatured (D) | Reduced (R) | Reduced/Alkylate (R/A) | Alkylated (A) | Ncysa | NSHb | Number of -S-S-c |
|---|---|---|---|---|---|---|---|---|
| Da | Da | Da | Da | Da | ||||
| CH2 | 14707.3607 | 14714.5977 | 14710.9160 | 14822.6719 | 14708.5791 | 2 | 0 | 1 |
| m01 | 14674.3447 | 14677.7539 | 14676.3398 | 14899.9238 | 14669.0400 | 4 | 0 | 2 |
| m02 | 14688.9561 | 14686.2461 | 14695.6543 | 14901.8076 | 14686.1230 | 4 | 0 | 2 |
Ncys = (MR/A-MR)/57.
NSH = (MA-MD)/57.
Number of disulfide bonds (-S-S-) = (Ncys-NSH)/2.
m01 and m02 Are Significantly More Stable Than CH2—The thermal stability of m01 and m02 was measured using CD and DSC, and their stability against chemical agents, by using urea and spectrofluorimetry. In all cases the two mutants were much more stable than CH2 (Fig. 5). The CD spectra of m01 and m02 showed that they had high β-sheet content at 25 °C (Fig. 5, A and B). The β-sheet structure was gradually disrupted as the temperature increased (Fig. 5C). At 90 °C, the structure was in an unfolded state (Fig. 5C). The sigmoidal curve was fitted by a two-state model, which was also previously used (11). Notably, 50% unfolding of m01 and m02 occurred at temperatures (Tm = 73.8 ± 1.7 °C and 65.3 ± 0.6 °C, respectively) that were significantly higher than that of native CH2 (54.1 ± 1.2 °C) (Fig. 5C). CH2 and m01 unfolding was reversible while that of m02 only partially reversible (Fig. 5, A and B).
FIGURE 5.

Increased stability of two mutants measured by CD (A–C), DSC (D), and spectrofluorimetry (E). Folding curve at 25 °C (—), unfolding at 90 °C (□□□), and refolding (– – –) at 25 °C of m01 (A) and m02 (B) are shown. C, fraction-folded and Tm of m01 and m02 were calculated by the same method as with CH2; Tm of m01 = 73.8 ± 1.7 °C, Tm of m02 = 65.3 ± 0.6 °C. D, thermo-induced unfolding curves of m01 (Tm = 73.4 °C) and m02 (Tm = 66.5 °C) were also recorded by DSC. E, urea-induced unfolding of CH2, m01, and m02 measure by spectrofluorimetry. Half-unfolding of CH2, m01, and m02 is at 4.2, 6.8, and 5.8 m, respectively.
Similar results were obtained by DSC. The Tm values of m01 (73.4 °C) and m02 (66.5 °C) were much higher (by about 20 and 10 °C, respectively) than that of native CH2 (Fig. 5D). Interestingly, the unfolding of m02 occurred over a much broader temperature range and with a lower maximum peak intensity than that observed for CH2 and m01. This phenomenon could be caused by the presence of dimers in m02 (see below).
The stability against chemically induced unfolding of m01 and m02 was also higher than that of CH2 (Fig. 5E). Urea was used as the chemical agent and intrinsic fluorescence was used to monitor protein unfolding. The unfolding dependence on the urea concentration can be also fitted using a two-state model. The 50% unfolding of m01 and m02 occurred at higher urea concentrations (6.8 and 5.8 m, respectively) than that of CH2 (4.2 m).
Only the monomeric state was observed for m01 while m02 contained small amounts of higher molecular species (mostly dimers) as determined by SEC (Fig. 6). Because of its superior physical properties m01 was selected for further characterization.
FIGURE 6.

Size exclusion chromatograms of m01 and m02. The same standard curve as in Fig. 1B is used. CH2 are m01 monomeric, while m02 includes a small amount of dimers.
Structural Conservation of m01—To examine structural perturbation caused by the cysteine mutations, solution NMR experiments were performed for the CH2 domain and the mutant m01. 1H-15N HSQC spectrum generally shows a correlation of nitrogen atoms and their directly bounded protons, and provides a “finger print” of the protein backbone. Each of the 1H-15N HSQC spectra of CH2 and m01 (recorded using identical experimental conditions) exhibited only one set of peaks, indicating that the protein was well-folded in solution (supplemental Fig. S1). Of the structure region of the proteins, the chemical shifts of backbone 15N, C′, and Cα were ∼75% assigned in both proteins. In m01, the measured chemical shifts for Cα and Cβ of residue Cys-12 were 57.6 ppm and 37.7 ppm, respectively, whereas the Cα and Cβ chemical shifts of Cys-104 were 54.5 and 34.2 ppm, respectively. These values fall within the expected range for oxidized cysteine residues (26), demonstrating that the additional disulfide bridge is formed in the mutant m01.
Comparison of the overall backbone chemical shits of N and Cα also showed the overall similarity of the protein structures between CH2 and m01 (Fig. 7). However, interestingly changes in chemical shifts were observed around residues Cys-31 and Cys-91 as well as around the newly introduced Cys residues 12 and 104. This is reasonable because the newly introduced disulfide bridge is proximal to the native Cys-31—Cys-91 by linking the adjacent β-strands in the same β-sheet with the Cys-31—Cys-91 bridge (Fig. 8A). The newly introduced disulfide bond in m01 most likely affected microscopic environments of the native disulfide bond between Cys-31 and Cys-91.
FIGURE 7.

Cα (A) and N (B) chemical shift differences between CH2 and m01. Asterisks at the bottom of graphs indicate the residues that were not assigned in one or both proteins. The p at the bottom of the graphs indicate proline residues.
FIGURE 8.

Ribbon structure presentations of CH2 domain structure. In A, the regions that show largest (red), medium (orange), and small (yellow) chemical shift changes are shown by the colors in parentheses, the regions that show insignificant changes in chemical shifts are in blue. In B, the rigid regions (1H-15N NOE > 0.7) are shown in blue, and the mobile regions (1H-15N NOE < 0.7) are shown in red. See details of the criteria under “Experimental Procedures.” Native cysteines and engineered cysteines are also shown by cyan sticks and pink sticks. In both A and B, the regions that were not assigned are shown in white.
Relatively High Loop Flexibilities and Rigid Framework of CH2 and m01—To determine whether the loops are flexible in both CH2 and m01, 15N-1H NOE spectra were recorded. We found that the framework is rigid as indicated by the high NOE values (above 0.7); in contrast the loops were on average more flexible (Fig. 8B). Interestingly, the local dynamics of CH2 and m01 were comparable (Fig. 9), indicating that the conformational entropy of the m01 at the native states is likely very similar to that of CH2. Although the 15N-1H NOE is inherently not measurable for the proline residues and, therefore, the NOE reside-by-residue profile provides no information at the locations of the prolines it is most likely that the essential structure and dynamics of the CH2 domain is maintained while thermal stability is increased upon introduction of the cysteine mutation. Such a dynamics profile is also consistent with the b-factors observed for the CH2 crystal structure (PDB 3DJ9) (15). The increase in the flexibility of the loops also indicates that both CH2 and m01 could be used as scaffolds for grafting to or mutating residues in the loops.
FIGURE 9.

1H-15N NOE values of CH2 (closed triangles) and m01 (open squares). Asterisks and at the bottom of graphs indicate the residues that were not assigned in one or both proteins. The p at the bottom of the graphs also indicate proline residues and cannot measure the 1H-15N NOE values. The large errors indicated for the 1H-15N NOE values of a few residues result from the weak peak intensity in the spectra, leading to large uncertainty.
DISCUSSION
The major findings of this study are that isolated unglycosylated human γ1 CH2 domain is relatively stable and that its stability can be significantly increased by engineering an additional disulfide bond between the N- and C-terminal strands. Human CH2 is significantly more stable (Tm = 54.1 °C) than previously thought based on the results for a murine CH2 (Tm = 41 °C) as measured by CD (11). We have not investigated possible molecular mechanisms that determine the higher stability of the human CH2, but in general such differences can be attributed to the significant differences in their amino acid sequences and composition (Fig. 1A).
It has been recently shown that an additional disulfide bond naturally occurring or engineered in llama heavy chain variable domains (VHH) can significantly increase their stability (27, 28). Interestingly, in this case disulfide bonds between strands C′ and D, and C and E resulted in significant stabilization. Previously, shark new antigen receptor (IgNAR) domain exhibiting an Ig fold was also found to have some noncanonical Cys residues that form disulfide bonds linking frameworks regions 2 and 4 to CDR3 (Type I) and CDRs 1 to 3 (Type II) (29). For the human constant CH2 domain, we found that an additional disulfide bond between strands A and G leads to significant stabilization. There is a similarity between the CH2 domain and the IgNAR V domain in that the C strand directly connects to the D strand without the C′ and C″ strands as seen within human variable domains. Further studies are needed to determine whether a combination of these disulfide bonds as observed in VHH and IgNAR variable domains could lead to additional gain in stability and whether there are differences between variable and constant domains in terms of best localization of stabilizing disulfide bonds.
We also found that the introduction of an additional disulfide bond between strands A and G does not perturb to any significant degree the structural integrity and in general the structure of the CH2. In addition, in both CH2 and the mutant m01 the frameworks are relatively rigid and the loops more flexible. These data indicate that grafting of loops (e.g. from antibody variable domains) may not significantly affect the overall structure of the CH2 and m01 (i.e. they are both promising as scaffolds for generations of binders). In addition, both are highly expressible, soluble, and monomeric, which makes them appropriate for further development with potential implications as scaffolds for libraries of binders.
These findings could have implications for exploration of the unfolding mechanisms of antibody domains and for the development of candidate therapeutic antibodies with increased stability. Whether the observed increase in stability to temperature and chemical agents in vitro will result in increased stability in vivo remains to be seen.
Supplementary Material
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Dept. of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
This work was supported, in whole or in part, by National Institutes of Health Grants from the Intramural AIDS Targeted Antiviral Program (IATAP), the NIAID Intramural Biodefense Program, the NCI, Center for Cancer Research Intramural Research Program (to D. S. D.) and Contract N01-CO-12400 from the NCI. This work was also supported by the Gates Foundation.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: mAb, monoclonal antibody; dAbs, domain antibodies; SEC, size exclusion chromatography; DSC, differential scanning calorimetry; PBS, phosphate-buffered saline.
References
- 1.Kolmar, H., and Skerra, A. (2008) FEBS J. 275 2667. [DOI] [PubMed] [Google Scholar]
- 2.Skerra, A. (2007) Curr. Opin. Biotechnol. 18 295–304 [DOI] [PubMed] [Google Scholar]
- 3.Nygren, P. A., and Skerra, A. (2004) J. Immunol. Methods 290 3–28 [DOI] [PubMed] [Google Scholar]
- 4.Binz, H. K., Amstutz, P., and Pluckthun, A. (2005) Nat. Biotechnol. 23 1257–1268 [DOI] [PubMed] [Google Scholar]
- 5.Hey, T., Fiedler, E., Rudolph, R., and Fiedler, M. (2005) Trends Biotechnol. 23 514–522 [DOI] [PubMed] [Google Scholar]
- 6.Holliger, P., and Hudson, P. J. (2005) Nat. Biotechnol. 23 1126–1136 [DOI] [PubMed] [Google Scholar]
- 7.Holt, L. J., Herring, C., Jespers, L. S., Woolven, B. P., and Tomlinson, I. M. (2003) Trends Biotechnol. 21 484–490 [DOI] [PubMed] [Google Scholar]
- 8.Saerens, D., Ghassabeh, G. H., and Muyldermans, S. (2008) Curr. Opin. Pharmacol. 8 600–608 [DOI] [PubMed] [Google Scholar]
- 9.Ward, E. S., Gussow, D., Griffiths, A. D., Jones, P. T., and Winter, G. (1989) Nature 341 544–546 [DOI] [PubMed] [Google Scholar]
- 10.Muyldermans, S., Cambillau, C., and Wyns, L. (2001) Trends Biochem. Sci. 26 230–235 [DOI] [PubMed] [Google Scholar]
- 11.Feige, M. J., Walter, S., and Buchner, J. (2004) J. Mol. Biol. 344 107–118 [DOI] [PubMed] [Google Scholar]
- 12.Dimitrov, D. S. (2009) mAbs 1 26–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995) Protein Sci. 4 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saphire, E. O., Parren, P. W., Pantophlet, R., Zwick, M. B., Morris, G. M., Rudd, P. M., Dwek, R. A., Stanfield, R. L., Burton, D. R., and Wilson, I. A. (2001) Science 293 1155–1159 [DOI] [PubMed] [Google Scholar]
- 15.Prabakaran, P., Vu, B. K., Gan, J., Feng, Y., Dimitrov, D. S., and Ji, X. (2008) Acta Crystallogr. B. 64 1062–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphrey, W., Dalke, A., and Schulten, K. (1996) J Mol. Graph. 14 33–38 [DOI] [PubMed] [Google Scholar]
- 17.Koenig, B. W., Rogowski, M., and Louis, J. M. (2003) J. Biomol. NMR 26 193–202 [DOI] [PubMed] [Google Scholar]
- 18.Grzesiek, S., and Bax, A. (1993) J. Am. Chem. Soc. 115 12593 [Google Scholar]
- 19.Gong, Q., and Ishima, R. (2007) J. Biomol. NMR 37 147–157 [DOI] [PubMed] [Google Scholar]
- 20.Kay, L. E., Ikura, M., Tschudin, R., and Bax, A. (1990) J. Magn. Reson. 89 496–514 [DOI] [PubMed] [Google Scholar]
- 21.Muhandiram, D. R., and Kay, L. E. (1994) J. Magn. Res. Series B. 103 203–216 [Google Scholar]
- 22.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277–293 [DOI] [PubMed] [Google Scholar]
- 23.Masse, J. E., and Keller, R. (2005) J. Magn. Reson. 174 133–151 [DOI] [PubMed] [Google Scholar]
- 24.Dani, V. S., Ramakrishnan, C., and Varadarajan, R. (2003) Protein Eng. 16 187–193 [DOI] [PubMed] [Google Scholar]
- 25.Pellequer, J. L., and Chen, S. W. (2006) Proteins 65 192–202 [DOI] [PubMed] [Google Scholar]
- 26.Sharma, D., and Rajarathnam, K. (2000) J. Biomol. NMR 18 165–171 [DOI] [PubMed] [Google Scholar]
- 27.Saerens, D., Conrath, K., Govaert, J., and Muyldermans, S. (2008) J. Mol. Biol. 377 478–488 [DOI] [PubMed] [Google Scholar]
- 28.Hagihara, Y., Mine, S., and Uegaki, K. (2007) J. Biol. Chem. 282 36489–36495 [DOI] [PubMed] [Google Scholar]
- 29.Stanfield, R. L., Dooley, H., Flajnik, M. F., and Wilson, I. A. (2004) Science 305 1770–1773 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.