

Abstract
The mechanism of the four-electron reduction of dioxygen by a multicopper oxidase, CueO, was studied based on reactions of single and double mutants with Cys500, a type I copper ligand, and the noncoordinating Asp112 and Glu506, which form hydrogen bonds with the trinuclear copper center directly and indirectly via a water molecule. The reaction of C500S containing a vacant type I copper center produced intermediate I in an EPR-silent peroxide-bound form. The formation of intermediate I from C500S/D112N was restricted due to a reduction in the affinity of the trinuclear copper center for dioxygen. The state of intermediate I was realized to be the resting form of C500S/E506Q and C500S of the truncated mutant Δα5–7CueO, in which the 50 amino acids covering the substrate-binding site were removed. Reactions of the recombinant CueO and E506Q afforded intermediate II, a fully oxidized form different from the resting one, with a very broad EPR signal, g < 2, detectable only at cryogenic temperatures and unsaturated with high power microwaves. The lifetime of intermediate II was prolonged by the mutation at Glu506 involved in the donation of protons. The structure of intermediates I and II and the mechanism of the four-electron reduction of dioxygen driven by Asp112 and Glu506 are discussed.
CueO is a multicopper oxidase involved in a copper efflux system of Escherichia coli (1–3). In contrast to other multicopper oxidases such as laccase and ascorbate oxidase (4), CueO exhibits strong activity toward cuprous ion but does not show activity toward most organic substrates such as 2,6-dimethoxyphenol, catechol, and guaiacol, except considerably low levels toward 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS)2 and p-phenylenediamine. This substrate specificity, unique to CueO, originates in the methionine-rich helical region covering the substrate-binding site (5–7). Nevertheless, CueO has the same catalytic copper centers as other multicopper oxidases: a type I copper that mediates electron transfer and a trinuclear copper center comprised of a type II copper and a pair of type III copper atoms, where dioxygen is reduced to two water molecules (5, 7). The type I copper is responsible for the intense charge transfer band at 610 nm due to Cys(S-)π → Cu2+ and the bands at 430, ∼500, and ∼750 nm due to the charge transfers His(N) → Cu2+ and Cys(S-)σ → Cu2+ and d-d transitions, respectively (4). The type III copper atoms bridged with a hydroxide ion afford an intense charge transfer band, OH- → Cu2+ at ∼330 nm, whereas the type II copper does not give a conspicuous band in the visible region. The type I and II coppers give rise to EPR signals with the hyperfine splitting of small (6.7 milliteslas (mT)) and normal (18.5 mT) magnitudes, respectively, whereas the type III copper atoms are EPR-silent because of the strong anti-ferromagnetic interaction (7–9).
Special attention has been paid to the four-electron reduction of dioxygen by multicopper and terminal oxidases because activated oxygen species such as superoxide, peroxide, etc. are not formed or, if formed, are effectively converted into water molecules without damage to protein molecules. Therefore, this four-electron reduction of dioxygen by multicopper oxidases has been expected to be applicable to biofuel cells (10–12). Two reaction intermediates have been detected during reactions of some multicopper oxidases. One of them, intermediate I, could be trapped by the following modified multicopper oxidases so as to interrupt the electron transfer from the type I copper: a plant laccase whose type I copper was substituted with mercury (13); a mixed valent laccase in which the type I copper was oxidized, but the trinuclear copper center was reduced (14); and a Cys → Ser mutant of bilirubin oxidase (15) and Fet3p (16) whose type I copper center became vacant. Although the trinuclear copper center must be fully reduced to produce intermediate I, it has been considered to be a two-electron reduced form and, therefore, also called the peroxide intermediate (13, 16). Another reaction intermediate, II, also called the native intermediate, has been detected at the final stage of a single turnover (15, 17–19). Four electrons have already been transferred to dioxygen in this intermediate, and accordingly, intermediate II is in a fully oxidized form to give the g < 2 EPR signal at cryogenic temperatures. Under catalytic conditions, intermediate II is not detected because of its prompt conversion to the fully reduced form for the next enzyme cycle without decaying to the resting form. Both intermediates have a half-life in the order of seconds to minutes, but information to directly show their structures has not been obtained yet. They afford analogous absorption bands at ∼330–350, 450–470, and 680 nm, of which the former two bands have been assigned to the charge transfer from a certain oxygen group to Cu2+ (σ and π transitions) and the latter to the d-d transitions of the trinuclear copper center in the cupric state. The d-d transitions of intermediate II are masked by strong absorption due to the oxidized type I copper (13–19).
In the present study, we succeeded in trapping intermediates I and II from reactions of a recombinant form of CueO (rCueO) and mutants altered at Cys500, a ligand to the type I copper, and at Asp112 and Glu506 located adjacent to the trinuclear copper center to modify the dioxygen reduction process. The Asp residue is conserved in every multicopper oxidase except for ceruloplasmin, which has Glu instead (Fig. 1). According to the x-ray crystal structures of rCueO (5) and the truncated mutant, Δα5–7CueO, missing the 50 amino acids covering the substrate-binding site (Fig. 2) (7, 20), Asp112 forms a hydrogen bond with His448, a ligand to a type III copper, and indirectly with the water molecule coordinating the type II copper through an ordered water molecule. In a preliminary study on the Asp112 mutants (21), we showed that this acidic amino acid functions in the binding of dioxygen at the trinuclear copper center and may also be involved in the donation of protons to the reaction intermediate(s). On the other hand, one to three acidic amino acids are present in the spacers to connect the copper ligands of multicopper oxidases, His-Cys-His-XXX-His-XXXX-Met-(Leu/Phe). Fig. 2 shows that Glu506 of CueO in this spacer is directly hydrogen-bonded with the His143 ligand to one of the type III copper atoms and indirectly with the hydroxide ion bridged between the type III copper atoms through an ordered water molecule. Therefore, Glu506 is also speculated to play a crucial role in the reduction of dioxygen. We singly and doubly mutated Cys500, Asp112, and Glu506 of CueO to trap intermediates I and II and to elucidate the mechanism behind the four-electron reduction of dioxygen.
FIGURE 1.

Homology of amino acid sequence around the copper binding sites of multicopper oxidase. The numbers 1, 2, and 3 represent the type I, II, and III copper ligands, respectively. BO, Myrothecium verrucaria bilirubin oxidase; RvLc, Rhus vernicifera laccase; CpAO, Cucurbita pepo ascorbate oxidase; TvLc, Trametes versicolor laccase; CcLc, Coprinus cinereus laccase; Fet3p, multicopper oxidase from Saccharomyces cerevisiae; CumA, multicopper oxidase from Pseudomonas putida; CotA, multicopper oxidase from Bacillus subtilis; SLAC, small laccase from Streptomyces coelicolor; hCp, human ceruloplasmin. The single asterisk represents the conserved acidic amino acid residue in all multicopper oxidases, and the double asterisk represents Glu506 in CueO, which forms a hydrogen bond with a His residue coordinating a type III copper and the hydroxide ion bridged between type III coppers.
FIGURE 2.

Structure around the active site of the truncated mutant of CueO (7). Type I, II, and III coppers are represented as spheres. Small spheres, oxygen atoms. The two networks of hydrogen bonds lead to the exterior of the protein molecule, forming the pathway to let protons in and water molecules out. Mutated amino acid residues, Cys500, Glu506, and Asp112, and the networks of hydrogen bonds are indicated.
EXPERIMENTAL PROCEDURES
Mutagenesis, Expression, and Purification—The genes for C500S, D112N, E506Q, C500S/D112N, and C500S/E506Q were synthesized with a QuikChange kit (Stratagene) using the oligonucleotide primers listed below and the template plasmid pUCCueO′ as described (12). The gene fragment encoding Δα5–7CueO C500S was synthesized using the C500S primers and pUCCueOΔα5–7 (7) as a template: C500S(+), 5′-TATATGGCGCACTCCCATCTGCTG-3′; C500S(-), 5′-CAGCAGATGGGAGTGCGCCATATA-3′; D112N(+), 5′-CCGGGTGAAGTCAACGGCFGCC-3′; D112N(-), 5′-CTGCGGGCCGCCGTTGACTTCACC-3′; E506Q(+), 5′-CTGGAGCATCAAGATACGGGG-3′; and E506Q(-), 5′-CCCCGTATCTTGATGCTCCAG-3′.
E. coli Origami (DE3)/pLacI (Novagen) was transformed with the mutant plasmids. Cultivation of the transformants and purification of the mutant proteins were carried out as described previously (7, 20). Protein concentrations were determined using the BCA protein assay reagent (Pierce) and the absorption intensity at 280 nm (ε = 73,000 m-1 cm-1).
Enzyme Activities—Activities of the mutants for oxidizing ABTS, which functioned simply as an electron donor, were colorimetrically determined from the changes in absorption of the oxidized product of ABTS at 420 nm (ε = 36,000) in acetate buffer, pH 5.5 (3, 7, 20, 21) (see the legend to supplemental Fig. 2). One unit of activity was defined as the amount of enzyme that oxidized 1 μmol of ABTS/min.
Reactions—Reactions of rCueO and mutants with dioxygen were performed using a 5-mm path length quartz cell with a long neck capped with a rubber septum to introduce dithionite through a thin needle under argon. It took one night to fully reduce the single and double mutants with an altered Cys500 residue. After rCueO and mutants were fully reduced and no absorption at 314 nm due to the residual dithionite was observed, pure dioxygen gas was bubbled into the reaction mixture, and measurements of absorption spectra were started. In measurements of resonance Raman spectra, 18O2 (Isotec 99%) was also used. The sample for EPR measurements was withdrawn from the reaction mixture with a syringe and frozen with liquid nitrogen. The total amount of EPR-detectable Cu2+ was determined by the double integration method using Cu-EDTA as a standard. Signal intensities due to the differences in tuning conditions were calibrated using a Mn2+ marker (JEOL) as an external standard. The reactions were performed at least twice to ascertain reproducibility.
Measurements—The total copper content of a protein molecule was determined by atomic absorption spectroscopy on a Varian SpectrAA-50 spectrometer. Absorption spectra were measured on a JASCO V-560 spectrometer and a Shimadzu MultiSpec-1500 spectrometer with a diode allay detector for kinetic measurements. Circular dichroism (CD) spectra were measured on a JASCO J-500C spectropolarimeter. X-band EPR spectra were measured on a JEOL JES-RE1X spectrometer attached to an Oxford ESR900 cryostat between 77 and 3.5 K. Resonance Raman scattering was excited at 488 nm with an Ar+ laser (Spectra Physics 2017) and detected with a CCD (Princeton Instruments) attached to a triple monochrometer (JACSO NRS-1800). Fourier transform infrared spectra were measured using a 0.025-mm path length ZnSe cell on a JASCO FT/IR-4200 spectrometer.
RESULTS
C500S—CueO lost exactly one copper ion with the mutation of Cys500 to Ser and did not show catalytic activity. C500S was colorless because the strong charge transfer band, Cys(S-) → Cu2+ at 610 nm, was absent due to the loss of the type I copper (absorption spectra of C500S (blue line) and rCueO (black line) in Fig. 3A). On the other hand, a strong band derived from the OH- → Cu2+ charge transfer was observed at 320 nm (ε = 5,200), a 10-nm shorter wavelength from that of rCueO, indicating that the electronic state of the trinuclear copper center was slightly affected due to the absence of the type I copper at the remote site. The d-d transition band due to the type II and III copper atoms, which had been masked by the intense d-d band of the type I copper at ∼750 nm (ε = 2,000) in rCueO, became observable at ∼710 nm (ε =∼500). The CD spectrum of C500S (blue line in Fig. 3B) afforded bands at 323 (Δε = -2.6), 374 (Δε =+0.3), 504 (Δε =+0.8), 602 (Δε =-0.5), 698 (Δε =+0.5), and ∼890 nm (Δε =-1.0). (The spectrum at 800–1,000 nm is not shown.) The CD bands were similar in number and intensity to those of the corresponding Cys → Ser mutant of bilirubin oxidase (15) and Fet3p (16), although some of them were inverted in sign. The EPR spectrum afforded only the signal due to the type II copper (g∥ = 2.24, g⊥ = 2.04, and A∥ = 17.8 mT) with the five superhyperfine splittings originating from the coordination of the two His residues (AN = 1.7 mT) at ∼318 mT (blue line in Fig. 3C). The total EPR-detectable type II Cu2+ was 1.0/protein molecule. The type III copper atoms were EPR-silent as in rCueO.
FIGURE 3.

Absorption (A), CD (B), and EPR (C) spectra of rCueO (black) and mutants, C500S (blue), C500S/D112N (red), E506Q (purple), and C500S/E506Q (green). Absorption and CD spectra of ∼100 μm proteins in 0.1 m phosphate buffer, pH 7, were measured at room temperature using a 1-cm path length quartz cell. The units of the ordinate are based on the protein molecule. EPR spectra were measured at 77 K, with a frequency of 9.2 GHz, microwave power of 4 milliwatts, modulation of 1 mT at 100 kHz, filter of 0.3 s, sweep time of 4 or 8 min, and amplitude of 200–400. The EPR spectra are normalized to give analogous signal intensities except for C500S/E506Q. The total numbers of the EPR-detectable Cu2+ in rCueO, C500S, C500S/D112N, E506Q, and C500S/E506Q are 2.0, 1.0, 1.0, 1.0, and 0.36/protein molecule, respectively.
It took almost one day to reduce C500S with a slight excess of dithionite under anaerobic conditions. This unusually slow reduction of the trinuclear copper center, as was also observed in the case of the bilirubin oxidase mutant (15), originated from the absence of the type I copper to mediate the electron transfer between the substrate and the trinuclear copper center. Soon after the start of the reaction of the reduced C500S with dioxygen, the transient spectrum coming from a reaction intermediate was obtained. Subtraction of the absorption spectrum of the reduced C500S afforded bands at 340 (ε = 9,000), 470, and 680 nm, which were assigned to the O-σ → Cu2+ charge transfer, O-π → Cu2+ charge transfer, and d-d transitions, respectively (blue line in Fig. 4A), based on similarities to those of intermediate I derived from plant laccase, bilirubin oxidase, and Fet3p derivatives (14–16). The lifetime of the trapped intermediate I was very long even at room temperature (blue squares in the inset in Fig. 4A for the initial 8% decay showing a biphasic process with the half-life, t½ = 320 min, in the second phase starting at ∼10 min), and the absorption spectrum finally returned to that of the form isolated after 1 day (data not shown). Because of the long lifetime, we could observe the CD spectrum of intermediate I (Fig. 4B), which is very similar to that of C500S/E506Q as isolated (green line in Fig. 3B; see below). EPR spectra measured at between 77 and 3.5 K indicated that intermediate I was EPR-silent (blue line in Fig. 4C at 3.5 K). The signal intensity of the type II copper was ∼0.1/protein molecule at the start of the reaction (1.0 Cu2+ before the reaction, dotted blue line in Fig. 4C) and increased concomitantly with the decay of intermediate I. A clear dependence on pH was not observed in the decay of intermediate I, differing from the derivatives of laccase, bilirubin oxidase, and Fet3p to suggest the participation of an acidic amino acid residue as a proton donor (14–16). Intermediate I could be obtained again using the recovered C500S, indicating that the trinuclear copper center was not fatally damaged, even after a prolonged period in the state of intermediate I.
FIGURE 4.

Absorption (A), CD (B), and EPR (C) spectra of intermediate I derived from C500S (blue) and C500S/D112N (red). A, inset, the absorbance decay of intermediate I at 340 and 330 nm formed from C500S and C500S/D112N, respectively. Sample conditions for measurements of absorption and CD spectra are the same as those in Fig. 3 except that a 0.5-cm path length cell was used for ∼100–200 μm proteins. EPR spectra were measured at 3.5 K, with a frequency of 9.0 GHz, microwave power of 1 milliwatt, modulation of 1 mT at 100 kHz, filter of 0.3 s, sweep time of 4 or 8 min, and amplitude of 400. The spectrum of C500S before the reaction (dotted blue line) is also shown for comparison.
C500S/D112N—C500S/D112N also contained 3 copper ions/protein molecule within an experimental error of 10% and did not show any catalytic activity, similar to C500S. C500S/D112N also lacked absorption bands derived from the type I copper (red line in Fig. 3A). A strong band due to the OH- → Cu2+ charge transfer characteristic of the resting trinuclear copper center was observed at 315 nm (ε = 4,800), 15 and 5 nm shorter than that of rCueO and C500S, respectively. The d-d transition band was observed at ∼720 nm (ε =∼500). The CD spectrum of C500S/D112N (red line in Fig. 3B) afforded bands at 317 (Δε =-2.2), ∼380 (Δε =-0.5), 507 (Δε =+0.1), 600 (Δε =-0.7), 690 (Δε =+1.2), and ∼880 nm (Δε =-1.2), (The spectrum at 800–1,000 nm is not shown.) The EPR spectrum (red line in Fig. 3C) showed only the signal due to the type II copper (g∥ = 2.24, g⊥ = 2.04, and A∥ = 17.8 mT) with five superhyperfine splittings (AN = 1.7 mT) at ∼318 mT as in C500S. The total number of EPR-detectable type II copper atoms was 1.0/protein molecule, ensuring that the type III copper atoms were EPR-silent.
The anaerobic reduction of C500S/D112N with a slight excess of dithionite was also very slow, similar to that of C500S. The formation of intermediate I was, unlike in the case of C500S, very slow, despite the continuous bubbling of pure O2 into the reduced C500S/D112N solution. It took ∼3 min for maximum formation of intermediate I, which was only ∼20% compared with C500S (red line in Fig. 4A). The bands derived from intermediate I began to decrease with a half-life of t½ = 99 min (red squares in the inset in Fig. 4A), despite expectations that intermediate I would be stabilized by introducing the mutation at Asp112. EPR spectra observed between 77 and 3.5 K also indicated that intermediate I was EPR-silent (red line in Fig. 4C). The absorptions in the near UV regions began to increase very slowly after ∼3 h and finally gave an absorption spectrum similar to that of the form isolated after one day (data not shown), suggesting that auto-oxidation of the reduced C500S/D112N took place.
rCueO—The reaction of rCueO was performed to detect intermediate II. In contrast to C500S and C500S/D112N, rCueO was immediately reduced with the four-electron equivalent of dithionite. The absorption spectrum obtained soon after starting the reaction of the reduced rCueO with dioxygen was already analogous with that of the resting rCueO except for slightly higher absorption intensities at 300–500 nm. The lifetime of intermediate II was too short to obtain an absorption spectrum, although the difference spectrum between the transient spectra (data not shown) was similar to that obtained from the reaction of E506Q (Fig. 5A; see below) with much lower intensities. The EPR spectrum of the sample frozen within 5 s after starting the reaction (black line in Fig. 5C) gave a very broad signal with a trough, g = 1.85, at <40 K, in addition to the type I and II copper signals. The intensity of this novel EPR signal increased as the temperature dropped, indicating a rapid relaxation. Furthermore, the increase in signal intensity was proportional to the square root of microwave power at 3.5 K (black squares in supplemental Fig. 1 in the range of 1–100 milliwatts), in contrast to the type I and II copper signals, which began to saturate at <1 milliwatt. Therefore, it is apparent that the novel EPR signal originated not from an isolated species but from a coupled species magnetically. This g = 1.85 EPR signal disappeared after the sample was thawed and frozen again, indicating that it was derived from intermediate II.
FIGURE 5.

Absorption (A), CD (B), and EPR (C) spectra of intermediate II derived from rCueO (black) and E506Q (purple). The absorption spectrum of intermediate II formed from E506Q (A) was obtained from supplemental Fig. 1B. Sample conditions are the same as those for Fig. 3. A, inset, the absorbance decay of intermediate II at 350 nm. The measurement of the CD spectrum at <500 nm (B) was accomplished within 5 min after starting the reaction to avoid the masking effect due to the oxidized type I copper. C, the EPR spectrum of rCueO (dotted black line) and the spectra obtained soon after the reactions of rCueO (black) and E506Q (purple) at 3.5 K, with a frequency of 9.0 GHz, microwave power of 1–100 or 200 milliwatts, modulation of 1 mT at 100 kHz, filter of 0.3 s, sweep time of 4 or 8 min, and amplitude of 400–2,000. C, inset, the overall EPR signal of intermediate II, which was obtained by subtracting the type I copper signal.
E506Q—E506Q contained 4 copper ions/protein molecule, as determined by atomic absorption spectroscopy, and afforded absorption, CD, and EPR spectra (type I copper: g∥ = 2.24, g⊥ = 2.04, and A∥ = 6.3 mT; type II copper: g∥ = 2.24, g⊥ = 2.04, and A∥ = 17.3 mT) similar to those of rCueO, except for a blue shift by 5 nm and a slight decrease in the intensity of the charge transfer band, OH- → Cu2+ (purple line in Fig. 3). However, the catalytic activity shown by E506Q was extremely low (Vmax = 0.0041 units/mg) (supplemental Fig. 2) compared with those of rCueO (Vmax = 1.1 units/mg for rCueO) (7, 21). E506Q was reduced with a slight excess of dithionite as rapidly as rCueO and gave intermediate II soon after the start of the reaction with dioxygen (supplemental Fig. 1B). Subtraction of the reduced E506Q spectrum gave the absorption spectrum of intermediate II with absorption maxima at 310 and 350 nm (ε = 4,200) and a shoulder at ∼410 nm (purple line in Fig. 5A). The electron transfer from the type I copper to dioxygen has already finished, as is evident from the full recovery of absorption at 610 nm. The absorptions at 350 nm exponentially decayed with t½ = 64 min (inset in Fig. 5A), and the spectrum returned to that of the original E506Q after 1 day. The CD spectrum between 300 and 500 nm, measured within 5 min after the start of the reaction (purple line in Fig. 5B), gave a negative band at 332 nm, which was red-shifted by 12 nm from that of E506Q (dotted purple line). The EPR spectrum at 3.5 K (purple line in Fig. 5C) gave a clear trough at g = 1.94, which was not saturated with increasing microwave power (purple squares in supplemental Fig. 1A in the range of 1–100 milliwatts). On subtraction of the type I copper signal, the presence of the g = 2.12 component became evident (inset in Fig. 4C), as has been reported in a laccase study (27). Except for this novel signal observable at below 40 K, only the type I copper EPR signal was observed. The type II copper signal became observable with the disappearance of the g = 1.94 signal (spectra not shown).
C500S/E506Q—C500S/E506Q contained 3 copper ions/protein molecule, as determined by atomic absorption spectroscopy, and did not show enzyme activities. The absorption spectrum of C500S/E506Q (green line in Fig. 3A) was quite different from that of C500S or C500S/D112N, showing bands at 330 (ε = 7,000), 480, and 680 nm, which are characteristic of intermediate I (Fig. 4A). The CD spectral features (Fig. 3B) were also practically the same as those of intermediate I (Fig. 4B). Therefore, it is apparent that the state of the reaction intermediate I is realized in the resting C500S/E506Q. Although the type II copper signal was observed in the EPR spectrum (Fig. 4C), its intensity was 0.37 Cu2+ ions/protein molecule. Therefore, the residual ∼2/3 type II copper ions were in the EPR-undetectable Cu+ state. Analogous spectral features were obtained in the C500S mutant of the Δα5–7CueO, in which the 50 amino acids covering the substrate-binding site were removed from the rCueO molecule, showing only the 0.1 type II copper EPR signal (g∥ = 2.24, g⊥ = 2.04, and A∥ = 18.3 mT) (supplemental Fig. 3).
DISCUSSION
Effects of Mutations at Asp112, Cys500, and Glu506 on Electronic State of Trinuclear Copper Center—The electronic state of the trinuclear copper center was slightly modified by the mutations at Cys500, Asp112, and Glu506 as reflected in the blue shift of the OH- → Cu2+ charge transfer band: ∼10 nm in C500S, ∼15 nm in C500S/D112N, and ∼5 nm in E506Q. These shifts in the 330 nm band presumably became observable because of the much lower absorption intensity of the 280 nm band in CueO (ε = 73,000) compared with other multicopper oxidases, for example, ε = 120,000 in bilirubin oxidase (15). The modifications induced by the absence of a type I copper were due to the type I copper center and the trinuclear copper center being connected with the His-Cys-His sequence and a peptide backbone, despite their separation by ∼13 Å (Figs. 1 and 2) (5–7). On the other hand, modifications induced by the mutations at Asp112 and Glu506 were derived from the breaking down of hydrogen bonds with a His ligand to a type III copper and the coordinated water molecule to the type II copper or the coordinated hydroxide ion between type III copper atoms through a water molecule. These modifications at the remote type I copper site and the noncoordinating Asp112 and Glu506 were mild as reflected in the absorption, CD, and EPR spectra (Fig. 3) but prominent enough to allow us to trap intermediates I and II by shutting down the steps to supply electrons or protons and by affecting the affinity of the trinuclear copper center for dioxygen (see below).
In contrast to the above mutants, C500S/E506Q (Fig. 3) and the C500S mutant of Δα5–7CueO as isolated (supplemental Fig. 3) gave absorption, CD, and EPR spectra typical of intermediate I (Fig. 4). The classical definition of a resting trinuclear copper center is that the type III copper atoms are antiferromagnetically coupled with a bridged hydroxide ion, and the type II copper is magnetically isolated. However, trinuclear copper centers in an exceptional state have been discovered by analyzing x-ray crystal structures of fungal laccases (22, 23) and CotA (24) and with improvements in resolution of the x-ray crystallography of human ceruloplasmin (25). Dioxygen or a reaction intermediate is located between type III copper atoms in a side-on fashion, or an oxygen atom is asymmetrically positioned near one of the type III copper atoms. These modes of binding of dioxygen might involve those in intermediate I, and it may not necessarily be unusual that intermediate I is the dominant resting form in C500S/E506Q and the C500S mutant of Δα5–7CueO (see below), although further studies on these mutants are required.
Intermediate I—Intermediate I trapped by the reaction of C500S unequivocally showed absorption bands at 340, 470, and 680 nm. These spectral features are very similar to those produced by a mercury-substituted or mixed valent laccase (13, 14) and Cys-to-Ser mutants of bilirubin oxidase (15) and Fet3p (16), indicating that multicopper oxidases commonly pass through intermediate I. The intensity of intermediate I band was much higher at 340 nm (ε = 9,000) than at 315–330 nm due to the resting trinuclear copper center (ε = 4,500–5,300) (Figs. 3A and 4A). This fact and small molecule studies (26) suggest that the origin of the oxygen atom concerned in the charge transfer (O → Cu2+) of intermediate I is not hydroxide but peroxide. The CD spectrum indicated that the absorption band at 340 nm is composed of two bands at 315 and 365 nm, and accordingly, the two oxygen atoms are not equivalent (26). Thus, the structure shown in Fig. 6 is depicted by taking the fact that intermediate I is EPR-silent into consideration. Dioxygen is bound as peroxide between type III copper atoms in the μ-η2:η2 fashion as in oxyhemocyanin (27), with an additional interaction with the reduced type I copper. Other possible structures, in which an oxygen atom of hydroperoxide ion is bridged between type III copper atoms in an end-on fashion (28) and the five-centered structure composed of 2Cu2+, 1Cu3+, and 2O2- (8), may be excluded, given that the two oxygen atoms are not equivalent. A three-electron reduced form might also be excluded because a radical species derived from a break of the O–O bond was not detected by cryogenic EPR measurements. We excited the Raman scattering of intermediate I at 488 nm, which was close to the maximum absorption wavelength of a new absorption band of intermediate I. However, we observed no oxygen isotope-sensitive band in the region at 500–1,200 cm-1, although fluorescence was not as strong as in bilirubin oxidase (spectra not shown) (15). Intermediate I was very sensitive to the laser light due to photoreduction of the metal site or photodissociation of the oxygenated species and, accordingly, we could not accumulate the Raman spectrum for a long time. In addition, Fourier transform infrared measurements did not give any band derived from intermediate I.
FIGURE 6.

The four-electron reduction of dioxygen by CueO. The structure of
the resting CueO is based on the x-ray crystal structure of rCueO
(5) and the truncated mutants
(7). It is not known whether
water molecules are present near the active site in the reduced CueO. The most
probable peroxide-bound structure for intermediate I is figured to account for
the production of the charge transfer bands due to
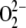 → Cu2+ and to be
EPR-silent. Intermediate I is converted into intermediate II with the supply
of electrons from type I and II coppers and of a proton with the assistance of
Glu506. In intermediate II, four electrons are transferred to
dioxygen, and the O–O bond is cleaved. Type II and III coppers are
magnetically interacted to give the g < 2 EPR signal. In the decay
of intermediate II, another proton is donated with the assistance of
Glu506.
→ Cu2+ and to be
EPR-silent. Intermediate I is converted into intermediate II with the supply
of electrons from type I and II coppers and of a proton with the assistance of
Glu506. In intermediate II, four electrons are transferred to
dioxygen, and the O–O bond is cleaved. Type II and III coppers are
magnetically interacted to give the g < 2 EPR signal. In the decay
of intermediate II, another proton is donated with the assistance of
Glu506.
In the reaction of the single mutant, D112N, with dioxygen, a very slow recovery of the blue color was observed (data not shown). This is consistent with our previous finding that the Km value for the binding of O2 changed from 0.019 to 0.035 mm, and enzyme activities were reduced to ∼2–10% by the mutation (21). Therefore, it is apparent that Asp112 is involved not only in the binding of dioxygen by forming a hydrogen bond with His448 but also in the later steps of a catalytic cycle influencing in the overall activities of the enzyme. An analogous study on the D94N mutant of Fet3p (19, 29) proposed that deprotonation from the water molecule coordinated to the cuprous type II copper was assisted by the hydrogen bond involving Asp94, leading to the strong binding of the peroxide ion to the trinuclear copper center, although the coordination of the hydroxide to the cuprous type II copper has not been proved. The critical role of Asp112 is also reflected in the remarkably slow and partial formation of intermediate I from the double mutant C500S/D112N (Fig. 4). Considering these essential roles of Asp112 in the formation of intermediate I, the possibility that Asp112 directly interacts with the peroxide ion cannot be excluded. One Gly residue is followed by this Asp residue according to the amino acid sequence of multicopper oxidases (Fig. 1), affording a flexibility or space in this region. Furthermore, an additional Gly residue is followed by this Asp-Gly sequence in CueO and a structural homologue of multicopper oxidase, SufI (FtsP), involved in cell division (30).
Glu506 might also contribute to the binding of peroxide at the trinuclear copper center by forming hydrogen bonds with His143 coordinated to a type III copper and with the peroxide ion directly or indirectly through a water molecule (Fig. 6). The carboxyl group in the side chain of Glu506 and one of the oxygen atoms in the peroxide ion are close enough to directly form a hydrogen bond, if the peroxide ion is bound between the type III copper atoms in the μ-η2:η2 fashion (Fig. 2). However, the first step, in which the reduced E506Q reacts with O2, was not inhibited (see below) in contrast to D112N, indicating that the major role of Glu506 is to donate a proton to intermediate I and perhaps also to intermediate II for their prompt decay (see below). The discovery of the very stable intermediate I state in the resting C500S/E506Q (Fig. 3) indirectly supports this, whereas supplies of electrons from the type I and II copper atoms are also required for the decay of intermediate I.
The decay of intermediate I did not show a clear dependence on pH, presumably because it occurred very slowly without a supply of protons directly or indirectly from Glu506 and electrons from the type I copper. In the cases of laccase and Fet3p, intermediate I decayed faster with decreasing pH, suggesting the involvement of an acidic amino acid with a pKa value of 5.0–5.6 (14, 16, 28). In addition, the decay of intermediate I did not involve the formation of intermediate II. We added a small amount of dithionite to intermediate I under argon, expecting to observe its conversion to intermediate II. However, there was no conversion because of the absence of the type I copper to mediate the prompt transfer of electrons. Nevertheless, intermediate I reached the same state as C500S isolated after one day, presumably due to an unknown autooxidation process. In the catalytic cycle of CueO, supplies of electrons from the type I and II coppers and protons from Glu506 will realize the prompt conversion of intermediate I to II, and this is why intermediate I has not been detected during the reaction of CueO.
Intermediate II—In contrast to intermediate I, all four copper centers should be reduced to form intermediate II. The absorption and EPR spectra clearly showed that the electron transfer from the type I copper had finished in intermediate II (Fig. 5). However, intermediate II formed from E506Q did not show the type II copper EPR signal (Fig. 5C, bottom panel), whereas the signal was observed in the reaction of rCueO (middle panel) probably because most of rCueO had already reached the resting state due to the rapid reaction of the unmodified enzyme. However, intermediate II formed from E506Q afforded a unique EPR signal (g = 2.12 and 1.94) detectable at <40 K, which increased in intensity as temperature decreased, and was not saturated with high power microwaves (supplemental Fig. 1A). Therefore, all copper atoms in the trinuclear copper center are magnetically coupled (3Cu2+ system) and in a low-lying excited state (27). The difference in the parameters derived from laccase (g = 1.6 or 1.8) (18, 28), rCueO (g = 1.85), and E506Q (g = 1.94) suggests that the difference in the magnitude of the magnetic interaction is in the order laccase > rCueO > E506Q.
Intermediate II formed from E506Q gave an absorption maximum at 315 nm (noticeable by subtracting the spectrum of the reduced E506Q) and 350 nm (ε = 4,200) and a shoulder at ∼410 nm, whereas absorption between 500 and 800 nm was masked by the strong absorption derived from the oxidized type I copper (Fig. 5A). The absorption band at 350 nm was not as intense as those of intermediate I. This strongly suggests that the O–O bond is cleaved, as shown by the study of small binuclear copper molecules (26). In harmony with the shift in the absorption band from 325 to 350 nm, the corresponding CD band shifted from 318 (Δε =-2.0) to 332 nm (Δε =-2.1). Intermediate II disappeared after one day, and the absorption spectrum returned to that of E506Q as isolated. The prominent slowing down in the decay of intermediate II by the mutation at Glu506 (t½ = 64 min) indicates that this amino acid plays a critical role in the supply of a proton to intermediate II.
The structure shown in Fig. 6 satisfies the properties of intermediate II: the presence of the charge transfer band O2- → Cu2+, which was red-shifted and reduced in intensity compared with that from intermediate I, the presence of the re-oxidized type I copper, the absence of the type II copper EPR signal, and production of the g < 2 EPR signal. Another isoelectronic form might be that an oxygen-centered radical is bound to the cuprous type II copper (18) or the radical center is delocalized between an oxygen atom and the type II copper. These structures have been excluded in laccase and Fet3p studies (19, 28, 29), whereas no direct evidence is available yet.
The present detection of intermediate II and characterizations are due to the mutation at Glu506, indicating that the role of this amino acid is to donate a proton to intermediate II for its prompt decay. We did not study the temperature dependence of the decay process due to the very long lifetime of intermediate II, and accordingly, we could not ascertain whether a large change takes place in the framework of the trinuclear copper center during the conversion of intermediate II to the resting form (18, 31). The location of the amino acid corresponding to Glu506 in CueO, Glu510 in ascorbate oxidase (32), Glu499 or Glu504 in plant laccase (33), Glu487 in Fet3p (19), and Glu498 in CotA (24), is not rigorously conserved in the amino acid sequence of multicopper oxidase (Fig. 1). However, all these Glu residues form a hydrogen bond with a His ligand to a type III copper, as indicated in the three-dimensional structures (32, 34, 35) with the exception of Asp1025 in ceruloplasmin (25).
Conclusions—In summary, the four-electron reduction of dioxygen by CueO is accomplished in two two-electron steps through intermediates I and II. The binding of dioxygen to the trinuclear copper is independent of the presence of the type I copper and reaches intermediate I with the assistance of the reduced type II copper and Asp112, although the involvement of this intermediate in the catalytic process has not been proved. In the second two-electron transfer step, the type I and II coppers function as electron donors and Glu506 assists the transfer of a proton to intermediate I. Conversion of intermediate II to the resting form is markedly slowed by the mutation at Glu506, indicating the direct or indirect assistance of Glu506 to donate a proton to intermediate II. In the catalytic cycle, intermediate II may be promptly reduced for the next cycle without reaching the resting form. Thus, the present study on intermediates I and II of CueO unequivocally shows that the assistance of Asp112 and Glu506 located at the “outersphere” of the trinuclear copper center is indispensable in the four-electron reduction of dioxygen by CueO. A different mechanism, whereby dioxygen is captured between type III copper atoms in the resting form, has been proposed for CotA (35), although two intermediates corresponding to I and II are also suggested.
Supplementary Material
Acknowledgments
We thank Kyoto-Advanced Nanotechnology Network for the resonance Raman measurements.
This work was supported by Grant-in-aid for Scientific Research 19350081 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T. S.), NEDO, Mandom Corp., and Toyota Motor Corp.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
Footnotes
The abbreviations used are: ABTS, 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid); mT, millitesla(s); rCueO, recombinant CueO.
References
- 1.Outten, F. W., Huffman, D. L., Halem, J. A., and O'Halloran, T. V. (2001) J. Biol. Chem. 276 30670-30677 [DOI] [PubMed] [Google Scholar]
- 2.Rensing, C., and Grass, G. (2003) FEMS Microbiol. Rev. 27 197-213 [DOI] [PubMed] [Google Scholar]
- 3.Grass, G., Thakali, K., Klebba, P. E., Thieme, D., Muller, A., Wilder, G. F., and Rensing, C. (2004) J. Bacteriol. 186 5826-5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messerschmidt, A. (ed) (1997) Multi-copper Oxidases, World Scientific, Singapore
- 5.Roberts, S. A., Weichsel, A., Grass, G., Thakail, K., Hazzard, J. T., Tollin, G., Rensing, C., and Montfort, W. R. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 2766-2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh, S., Grass, G., Rensing, C., and Montfort, W. R. (2004) J. Bacteriol. 186 7815-7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kataoka, K., Komori, H., Ueki, Y., Konno, Y., Kamitaka, Y., Kurose, S., Tsujimura, S., Higuchi, Y., Kano, K., and Sakurai, T. (2007) J. Mol. Biol. 373 141-152 [DOI] [PubMed] [Google Scholar]
- 8.Sakurai, T., and Kataoka, K. (2007) Chem. Rec. 7 220-229 [DOI] [PubMed] [Google Scholar]
- 9.Sakurai, T., and Kataoka, K. (2007) CMLS Cell. Mol. Life Sci. 64 2642-2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miura, Y., Tsujimura, S., Kamitaka, Y., Kurose, S., Kataoka, K., Sakurai, T., and Kano, K. (2007) Chem. Lett. 36 132-133 [Google Scholar]
- 11.Tsujimura, S., Miura, Y., and Kano, K. (2008) Electrochim. Acta 53 5716-5720 [Google Scholar]
- 12.Miura, Y., Tsujimura, S., Kurose, S., Kataoka, K., Sakurai, T., and Kano, K. (2009) Fuel Cells 9 70-78 [Google Scholar]
- 13.Shin, W., Sundaram, U. M., Cole, J. L., Zhang, H. H., Hedman, B., Hodgson, K. O., and Solomon, E. I. (1996) J. Am. Chem. Soc. 118 3202-3215 [Google Scholar]
- 14.Zoppellaro, G., Sakurai, T., and Huang, H.-W. (2001) J. Biochem. 129 949-953 [DOI] [PubMed] [Google Scholar]
- 15.Kataoka, K., Kitagawa, R., Inoue, M., Naruse, D., Sakurai, T., and Huang, H.-W. (2005) Biochemistry 44 7004-7012 [DOI] [PubMed] [Google Scholar]
- 16.Palmar, A. E., Quintanar, L., Severance, S., Wang, T., Kosman, D. J., and Solomon, E. I. (2002) Biochemistry 41 6438-6448 [DOI] [PubMed] [Google Scholar]
- 17.Lee, S., George, S. D., Antholine, W. E., Hedman, B., Hodgson, K. O., and Solomon, E. I. (2002) J. Am. Chem. Soc. 124 6180-6193 [DOI] [PubMed] [Google Scholar]
- 18.Huang, H.-W., Zoppellaro, G., and Sakurai, T. (1999) J. Biol. Chem. 274 32718-32724 [DOI] [PubMed] [Google Scholar]
- 19.Augustine, A., Quintanar, L., Stoj, C. S., Kosman, D. J., and Solomon, E. I. (2007) J. Am. Chem. Soc. 129 13118-13126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurose, S., Kataoka, K., Otsuka, K., Tsujino, Y., and Sakurai, T. (2007) Chem. Lett. 36 232-233 [Google Scholar]
- 21.Ueki, Y., Inoue, M., Kurose, S., Kataoka, K., and Sakurai, T. (2006) FEBS Lett. 580 4069-4072 [DOI] [PubMed] [Google Scholar]
- 22.Hakulinen, N., Kruus, K., Koivula, A., and Rouvinen, J. (2006) Biochim. Biophys. Res. Commun. 350 929-934 [DOI] [PubMed] [Google Scholar]
- 23.Garvaglia, S., Cambria, M. T., Miglio, M., Ragusa, S., Lacobazzi, V., Palmieri, F., D'Ambrosio, C., Scaloni, A., and Rizzi, M. (2004) J. Mol. Biol. 342 1519-1531 [DOI] [PubMed] [Google Scholar]
- 24.Durao, P., Bento, I., Fernandes, T., Melo, E. P., Lindley, P. F., and Martins, L. O. (2006) J. Biol. Inorg. Chem. 11 514-526 [DOI] [PubMed] [Google Scholar]
- 25.Bento, I., Zaitzev, V. N., and Lindley, P. F. (2007) Acta Cryst. D63 240-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis, E. A., and Tolman, W. B. (2004) Chem. Rev. 104 1047-1076 [DOI] [PubMed] [Google Scholar]
- 27.Magnus, K. A., Hazes, B., Ton-That, H., Bonaventura, C., Bonaventura, J., and Hol, W. G. J. (1994) Proteins 19 302-309 [DOI] [PubMed] [Google Scholar]
- 28.Solomon, E. I., Chen, P., Metz, M., Lee, S., and Palmer, A. E. (2001) Angew. Chem. 40 4570-4590 [DOI] [PubMed] [Google Scholar]
- 29.Quintanar, L., Stoj, C., Wang, T., Kosman, D. J., and Solomon, E. I. (2005) Biochemistry 44 6081-6091 [DOI] [PubMed] [Google Scholar]
- 30.Kato, J., Nishimura, Y., Yamada, M., Suzuki, H., and Hirota, Y. (1988) J. Bacteriol. 170 3967-3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon, J., and Solomon, E. I. (2007) J. Am. Chem. Soc. 129 13127-13136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Messerschmidt, A., Ladenstein, R., Huber, R., Bolognesi, M., Avigliano, L., Petrusselli, R., Posi, A., and Finazzi-Agro, A. (1992) J. Mol. Biol. 224 179-205 [DOI] [PubMed] [Google Scholar]
- 33.Nitta, K., Kataoka, K., and Sakurai, T. (2002) J. Inorg. Biochem. 91 125-131 [DOI] [PubMed] [Google Scholar]
- 34.Taylor, A. B., Stoj, C. S., Ziegler, L., Kosman, D. J., and Hart, P. J. (2005) Proc. Natl. Acad. Sci. 102 15459-15464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bento, I., Martins, L. O., Lopes, G. G., Carrondo, M. A., and Lindley, P. F. (2005) J. Chem. Soc. Dalton Trans. 3507-3513 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.