

Abstract
Anthrolysin O (ALO) is a pore-forming, cholesterol-dependent cytolysin (CDC) secreted by Bacillus anthracis, the etiologic agent for anthrax. Growing evidence suggests the involvement of ALO in anthrax pathogenesis. Here, we show that the apical application of ALO decreases the barrier function of human polarized epithelial cells as well as increases intracellular calcium and the internalization of the tight junction protein occludin. Using pharmacological agents, we also found that barrier function disruption requires increased intracellular calcium and protein degradation. We also report a crystal structure of the soluble state of ALO. Based on our analytical ultracentrifugation and light scattering studies, ALO exists as a monomer. Our ALO structure provides the molecular basis as to how ALO is locked in a monomeric state, in contrast to other CDCs that undergo antiparallel dimerization or higher order oligomerization in solution. ALO has four domains and is globally similar to perfringolysin O (PFO) and intermedilysin (ILY), yet the highly conserved undecapeptide region in domain 4 (D4) adopts a completely different conformation in all three CDCs. Consistent with the differences within D4 and at the D2-D4 interface, we found that ALO D4 plays a key role in affecting the barrier function of C2BBE cells, whereas PFO domain 4 cannot substitute for this role. Novel structural elements and unique cellular functions of ALO revealed by our studies provide new insight into the molecular basis for the diverse nature of the CDC family.
Cholesterol-dependent cytolysins (CDCs)4 are a family of pore-forming toxins from many organisms, including but not limited to the genera Archanobacterium, Bacillus, Clostridium, Listeria, and Streptococcus. Recently, work in vertebrates has revealed that CDCs and membrane attack complex/perforin superfamily domain-containing proteins share a similar fold, suggesting that vertebrates use a similar mechanism for defense against infection (1, 2). A common feature of the CDC family is the requirement of cholesterol in the membrane to form pores (3). In addition to cholesterol, certain members of the family also require a cellular receptor, such as CD59 for the toxin ILY from Streptococcus intermedius (4). The specific mechanism by which CDCs form pores is not completely resolved; however, what is generally known is that ring-shaped oligomerization at the cellular membrane is followed by large conformational changes in each unit of the oligomer, resulting in the insertion of a β-barrel into the cellular membrane (5). Pore formation results in a variety of downstream signaling effects, including but not limited to the influx of Ca2+ into the cell (6).
A good deal is known about structures of the prepore conformation of CDCs. The crystal structures of prepore PFO, from Clostridium perfringens, and ILY have previously been elucidated (7, 8). Each structure shows a characteristic four-domain architecture, in which domain 4 (D4) is involved in membrane recognition, domain 3 (D3) is involved in β-sheet insertion, and domain 2 (D2) is the hinge region that undergoes a large conformational change (9-11). Nevertheless, despite the similarities, structural differences in D4 orientation and the conformation of a highly conserved segment named the undecapeptide region confer functional differences to PFO and ILY (8). Noting these differences, we decided to explore the structure and function of another member of the CDC family, anthrolysin O (ALO).
ALO is secreted by Bacillus anthracis, the etiologic agent for anthrax. ALO is chromosomally encoded by a gene whose regulation is poorly understood, and it is highly homologous to other members of the CDC family (12). ALO has been shown to have hemolytic and cytolytic activity (13, 14). Although clinical studies have shown that B. anthracis is weakly hemolytic (15), anthrax bacteria do produce biologically relevant amounts of hemolytic ALO, although the levels of expression are under complex regulation and are dependent on the culture media and growth conditions (12, 13, 16). At lower concentrations, ALO can disrupt cell signaling (13, 14). Search for a cellular receptor of ALO has lead to the conclusion that it is a TLR4 agonist (17). However, it is not known that ALO binds to TLR4 directly and, if so, whether ALO also binds other cellular receptors.
In addition to ALO, B. anthracis secrete ∼400 proteins, termed the anthrax secretome (18). Of those, two exotoxins, edema toxin (ET) and lethal toxin (LT) have been characterized in greatest detail. ET raises intracellular cAMP to pathologic levels, whereas LT impairs mitogenic and stress responses by inactivating mitogen-activating protein kinase kinase (19, 20). The complex interplay between these two toxins on various aspects of host cellular functions have been demonstrated (20-25). ALO could also work in conjunction with other anthrax virulence factors to modulate their cellular toxicity. For example, ALO and LF together induce macrophage apoptosis, whereas ALO and PLC play a redundant role in a murine inhalation anthrax model (17, 26). Interplay among anthrax secreted factors on cells relevant to anthrax infection is just beginning to be understood. This network of interactions is vital to the molecular basis of how anthrax bacteria interact with the hosts during anthrax infection.
Anthrax infection initiates when B. anthracis spores enter the host through one of three routes: cutaneous, inhalational, or gastrointestinal (GI) (27, 28). All three routes of infection can lead to systemic infection and are ultimately lethal. Different from inhalational anthrax, spores are ingested and germinate on or within the epithelium of the GI tract in GI anthrax (29). This is primarily based on pathological observations that primary lesions of the GI tract are found in GI anthrax, whereas no primary lesions of the lung are found in inhalational anthrax (29). Inhalational anthrax is a disease of choice for biological weapons because of its high infectivity and mortality (30). The initiation of GI anthrax requires much higher doses of spores than inhalational anthrax, and the molecular basis for the initiation of GI anthrax remains elusive (31).
Since the primary function of GI epithelia is to control the flux of material into the body, disruption of this barrier can lead to movement of bacteria into the surrounding tissue (32). The barrier is produced by a matrix of transmembrane and membrane-associated proteins. These cell to cell contacts, or tight junctions, are sometimes altered during bacterial infection to specifically disrupt the barrier function of epithelial cells. Using a functional model for the gut epithelium, human gut epithelial Caco-2 brush border expressor (C2BBE) cells, we report that ALO decreases the barrier function of C2BBE cells through disruption of tight junctions. We also show that ALO disruption of barrier function is dependent on epithelial cell polarity. We also present the crystal structure of the soluble state of ALO and compare it with the known structures of other CDCs. In addition, we show that ALO exists primarily as a monomer, in contrast to its closely related homologue PFO, which exists as a dimer. Finally, we used domain swapping to examine the structural components that confer specificity of ALO to gut epithelial cells.
EXPERIMENTAL PROCEDURES
Molecular Cloning of ALO and PFO—To construct the plasmid for recombinant ALO, the signal peptide-deficient ALO gene was isolated from the B. anthracis genome as described (13), and was cloned into the pProEx-1 expression vector, which contains an N-terminal hexahistidine tag. The expression plasmid for recombinant PFO was provided by Rodney Tweten (University of Oklahoma Health Sciences Center), and PFO was expressed and purified as described (10). To generate D4 swap mutants, an endogenous ScaI site within ALO was removed without changing the amino acid sequence by site-directed mutagenesis using the QuikChange kit (Stratagene). In addition, a single silent ScaI site was inserted into both ALO and PFO at the residues that code for the start of D4. The resulting ScaI mutations and the presence of a ScaI site in the Ampr gene allowed the excision of both D4s from ALO and PFO. The ALO D4 fragment was ligated into PFO and vice versa.
Purification of Recombinant Proteins—The hexahistidine-tagged ALO, PFO, ALO(D1-3)PFO(D4), and PFO(D1-3)-ALO(D4) constructs were transformed into Escherichia coli Bl21(DE3) cells harboring pUBS520. The cells were grown in T7 media with 50 μg/ml ampicillin and 30 μg/ml kanamycin to A600 = 0.5 and were then induced with 500 μm isopropyl-1-thiogalactopyranoside and harvested after 16 h at 30 °C. The cells were lysed in T20N100β5P0.1, pH 7.7, buffer (20 mm Tris-HCl, pH 7.7, 100 mm NaCl, 5 mm β-mercaptoethanol, and 0.1 mm phenylmethylsulfonyl fluoride) by 0.1 mg/ml lysozyme and sonication. The lysate was centrifuged at 35,000 rpm for 30 min at 4 °C, and the supernatant was loaded directly onto an Ni2+-nitrilotriacetic acid column that was equilibrated with T20 β5P0.1N100. The Ni 2+-nitrilotriacetic acid column was then washed with three column volumes of T20β5P0.1N500 and with T20β5P0.1N100 containing 10 mm imidazole. The proteins were then eluted with buffer T20β5P0.1N50 containing 150 mm imidazole. The peak fractions were then loaded onto a Source S column after a 10-fold dilution with C20E1P0.1 (20 mm sodium citrate, pH 5.5, 1 mm EDTA, 0.1 mm phenylmethylsulfonyl fluoride), and the proteins were eluted by an NaCl gradient. The peak fractions were then concentrated and loaded onto a Superdex 200 column and eluted using T20P0.1N100, pH 8.0, buffer. The purified proteins were concentrated to >10 mg/ml and stored at 80 °C.
Cell Culture—Caco-2 brush border expressor (C2BBE) cells, a subclone of human colonic adenocarcinoma Caco-2 cells, were used at passages 55-80 (33). The cells were cultured in high glucose Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 4 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10 μg/ml human transferrin (all from Invitrogen).
Cell Monolayer Permeability Quantification—C2BBE cells were seeded on 0.4-μm pore polyethylene terephthalate transwells (BD Biosciences) at 104/cm2, and resistance was monitored using a Millicell-ERS (Millipore, Medford, MA). Cells were used for experiments when the transepithelial resistance (TER) was between 200 and 300 ohms/cm2, which was ∼10-14 days after seeding. 2 h prior to treatment, the medium was changed to serum-free medium. A combination of ALO, ET, and/or LT was then added to either the apical or basolateral side, and TER was measured at various time points for 3 h. For the dextran flux assays, 10 μg/ml of a 3-kDa fluorescein-labeled dextran (Invitrogen) was also added to the apical side. Basolateral samples were taken at various time points, and fluorescence was quantified on a Safire2 fluorometer (Tecan, Research Triangle Park, NC).
Hemolytic Assay—Expired whole human blood was diluted 1:10 with sterile PBS. Various concentrations of ALO, PFO, ALO(D1-3)PFO(D4), and PFO(D1-3)ALO(D4) were added to the diluted blood and were allowed to incubate for 30 min at 37 °C. The samples were then centrifuged at 8000 × g for 3 min, and the A350 of the supernatant was measured. For hemolysis controls, 0.1% Triton X-100 was used as the positive control, and PBS only was used as the negative control.
Intracellular Ca2+ Imaging—Cells were cultured on 25-mm glass coverslips 7-10 days prior to experiments. Cells were washed with HBSS and then loaded for 30 min at 37 °C with 3 ml of HBSS containing 1 mg/ml bovine serum albumin (Sigma), 2.5 μl of a 25% (w/w in dimethyl sulfoxide) F127 detergent, and 5 μm fura2/AM (HBSS, F127, and fura2/AM; Invitrogen). After loading, the cells were washed with HBSS with or without 10 μm BAPTA-AM for 45 min 37 °C. This incubation allows time for full cleavage of fura2/AM, since uncleaved fura2/AM interferes with calcium measurements. The coverslip was mounted into a perfusion chamber system and was perfused with HBSS for up to 5 min prior to toxin treatment. During the experiment, the perfusion was stopped, and 1 ml of 2, 10, or 20 μg/ml ALO or 2 o r10 μm ionomycin was added to 1 ml of HBSS within the perfusion chamber. Intracellular fura2 fluorescence was measured every 10 s for up to 30 min, using the epifluorescent imaging system previously described (34). Determination of free intracellular Ca2+ in the samples was calculated by correlating the ratio of fura2 fluorescence at 340 and 380 nm to the 340/380 ratios obtained from a Ca2+ standard curve.
Immunofluorescence Experiments—C2BBE cells were grown within transwell cell culture inserts as previously stated. Briefly, after treating the C2BBE monolayers with ALO (0-10 μg/ml) for 2 h, the cells were fixed with 1% paraformaldehyde, followed by a wash of 50 mm NH4Cl to remove residual fixative. The cells were permeabilized and blocked with 0.05% saponin and 3% bovine serum albumin in PBS. The cells were then labeled with either mouse monoclonal anti-occludin or rabbit polyclonal antibodies against either ZO1 or E-cadherin (Invitrogen) and then stained with polyclonal Cy5-conguated anti-mouse IgG or Cy3-conjugated anti-rabbit IgG antibodies (Invitrogen). The transwell membranes were cut and mounted with SlowFade antifade reagent (Invitrogen) to be viewed on an Olympus Fluo-view 200 laser-scanning confocal microscope, as provided by the Microscopy Imaging Core at the University of Chicago. The images were processed using Image J and are average intensity projections of confocal Z-stacks, which covered the range of fluorescence within the treated and untreated C2BBE cells. For actin staining, C2BBE cells were grown and treated with ALO as described above. The cells were fixed with 3.75% formaldehyde followed by a wash with PBS. The cells were permeabilized with 0.1% Triton X-100 and blocked with 1% bovine serum albumin in PBS. The cells were stained with Alexa-Phalloidin (Invitrogen), and the transwell membranes were cut and mounted onto coverslides with Vectashield (Vector Laboratories) for fluorescence microscopy imaging.
Crystallization and Data Collection—Crystals of N-terminal hexahistidine-tagged ALO were obtained by hanging drop vapor diffusion at 18 °C. The drops contained 1 μl of 15 mg/ml protein plus 1 μl of precipitant solution, made of 3.5 m sodium formate, 100 mm HEPES, pH 7.0, and 10 mm taurine. The crystal used for data collection was directly loop-mounted at 100 K in nitrogen gas with 3.5 m sodium formate in the mother liquor providing sufficient cryoprotection. This procedure improved the diffraction properties of ALO crystals. X-ray diffraction data were collected at the beamline 14-BM-C (Advanced Photon Source, Argonne National Laboratories). The diffraction data were indexed, integrated, and scaled using HKL2000 software (35). Examination of the systematic absences and of the peaks of self-rotation functions is consistent with the orthorhombic space group C2221 with cell dimensions a = 141.74 Å, b = 141.75 Å, and c = 294.1 Å. Data collection statistics are shown in Table 1.
TABLE 1.
Summary of data collection and refinement statistics of ALO
| H6-ALO | |
|---|---|
| Data collection | |
| Beamline | APS 14BM-C |
| Wavelength (Å) | 0.90020 |
| Space group | C2221 |
| Cell dimensions (Å) | |
| a | 141.74 |
| b | 141.75 |
| c | 294.1 |
| Resolution (Å) | 50.0-3.1 |
| Rmerge (%)a,b | 6.3 (41.6) |
| I/σb | 30 (5.6) |
| Redunancyb,c | 7.0 (7.4) |
| Completeness (%)b | 99.0 (100.0) |
| Unique reflections | 53,120 |
| Refinement statistics | |
| Rwork (%)d | 26.9 |
| Rfree (%)e | 29.3 |
| No. of atoms | |
| Protein | 7,101 |
| Water | 27 |
| Average B-factors (Å2) | |
| Protein | 76 |
| Water | 65 |
| Rmsdf | |
| Bond lengths (Å) | 0.014 |
| Bond angles (degrees) | 1.64 |
| Ramachandran plot (%)g | |
| Favorable region | 73.8 |
| Allowed region | 23.1 |
| Generously allowed region | 2.6 |
| Disallowed region | 0.5 |
| Protein Data Bank accession code | 3CQF |
Rmerge = ∑ (I - <I>)/∑ <I>.
Values in parentheses indicate the highest resolution shell.
Nobs/Nunique.
Rwork = ∑hkl∥Fo| - k|Fc∥/∑hkl| Fo|.
Rfree is the Rwork value for 5% of the reflections excluded from the refinement.
Root mean square deviations calculated with REFMAC (37).
Values are from PROCHECK (71).
Structure Determination and Refinement—Examination of the self-rotation function suggested that two molecules occupy the asymmetric unit (Vm = 6 Å3/Da, 80% solvent content), arranged as a noncrystallographic dimer with a 2-fold axis. Initial phases were obtained by molecular replacement at 3.1 Å by the program PHASER (36), using as a search model coordinates of Perfringolysin O (Protein Data Bank code 1PFO) modified to polyalanine. The initial molecular replacement phases were refined by rigid body refinement in REFMAC 5.4 (37). Successive and iterative cycles of model rebuilding and refinement were performed in COOT (38) PHENIX (39), and REFMAC (37). Simulated annealing followed by isotropic individual and group B-factor refinement were performed in PHENIX (39). The last cycle of refinement was performed in REFMAC 5.4 (37), fixing the Babinet's bulk solvent B value. The final model yielded a crystallographic R-factor of 26.8% and a free R-factor of 29.4%. The MOLPROBITY server (40) was used to evaluate the Ramachandran plot that resulted in 81.6% of the residues in favored regions and 5.2% outliers. Refinement statistics are shown in Table 1. All figures were prepared using PyMOL (41).
Size Exclusion Chromatography Followed by Intrinsic Viscosity and Molecular Mass Measurements—A Superdex 200 column (GE Healthcare) was used for size exclusion chromatography (SEC). It was controlled by the GPCmax module and connected on-line to the triple detector array model 302 (Viscotek Ltd., Houston, Basingstoke, UK). The oven of the triple detector array contains (i) a static light scattering cell with two photodiode detectors, at 7° for low angle (LALS) and at 90° for right angle laser light scattering (RALS), (ii) a deflection refractometer, (iii) a photometer, and (iv) a differential viscometer. SEC experiments were performed at 20 °C, and the detections in the oven were done at 25 °C. The buffer was 20 mm Tris, 150 mm NaCl, pH 7.3. ALO concentration was 2 mg/ml (37 μm). Bovine serum albumin (2 mg/ml, various volumes) was used to calibrate internal instrument constants, using the same buffer at the same temperature. All data were acquired and processed using Omnisec software. Protein concentration was determined using a UV photometer. RALS and LALS data coupled to the concentration provide the molecular mass of the eluting protein using the Zimm equation. The differential viscometer is made of a balanced four-capillary bridge and provides intrinsic viscosity [η] by combining specific viscosity with concentration determined from the UV photometer.
Dynamic Light Scattering—Dynamic light scattering experiments were realized on a DynaPro MS800 (Wyatt) and acquired with Protein Solution Dynamics software version 6.2.05. The laser power was 100%, and its incident light wavelength was λqels: 824.7 nm. A microcuvette of dimensions 3 × 8.5 mm (105 251 QS) was loaded with 100 μl of ALO at 14.5 μm. Samples were allowed to thermally equilibrate for 10 min in the cell compartment at 25 °C. Acquisition time was 10 s, with an interval time of 1 s. At least 30 acquisitions were averaged to produce a data collection. A set of six independent data collections was produced and analyzed with SEDFIT 9.2 (42). The hydrodynamic radius, RH, is calculated from the Stokes-Einstein equation, RH = (kBT)/(6πηsDt), where kB is Boltzmann's constant, T is the temperature, Dt is the translational diffusion coefficient, and ηs is the viscosity of the solvent. Solvent viscosity is 0.009 poise at 25 °C in our experimental conditions.
Protein Shape, Hydration, and Volume Determination—The
intrinsic viscosity [η] of a protein in a defined solvent is related to
its shape, its molecular volume, and electroviscous effects. Its expression,
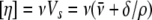 ,
is the product of a shape factor, the hydrodynamic shape function ν (called
the viscosity increment), and the swollen volume VS. The
viscosity increment is related to the semi-axes a/b of the
protein;
,
is the product of a shape factor, the hydrodynamic shape function ν (called
the viscosity increment), and the swollen volume VS. The
viscosity increment is related to the semi-axes a/b of the
protein; 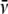 is the partial specific
volume of the protein (volume occupied by 1 g of protein), ρ is the
density of the solvent, and δ is the time-averaged apparent hydration of
the protein (g of water/g of protein). The hydration parameter δ of the
protein includes (i) the water molecules bound to the protein and (ii) the
water molecules entrained by the diffusion of the protein. The partial
specific volume of ALO is 0.7338 ml g-1, and solvent density is
1.0038 ml g-1. The parameters
is the partial specific
volume of the protein (volume occupied by 1 g of protein), ρ is the
density of the solvent, and δ is the time-averaged apparent hydration of
the protein (g of water/g of protein). The hydration parameter δ of the
protein includes (i) the water molecules bound to the protein and (ii) the
water molecules entrained by the diffusion of the protein. The partial
specific volume of ALO is 0.7338 ml g-1, and solvent density is
1.0038 ml g-1. The parameters
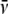 , ρ, and
ηs were computed using Sednterp.
, ρ, and
ηs were computed using Sednterp.
The Einstein viscosity relation, M[η] =
νVHNA, where VH is the
hydrodynamic volume defined by VH =
4πR3H/3, provides the viscosity
increment ν of ALO. In this relation, the molecular mass M came
from static light scattering and analytical ultracentrifugation, and the
hydrodynamic radius RH came from dynamic light scattering
(see above). In turn, knowing [η] and ν (ρ and
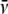 were computed using Sednterp), the
hydration δ was calculated from the relation
were computed using Sednterp), the
hydration δ was calculated from the relation
 .
The viscosity increment provides the axial ratio a/b. The
radius R0 of an anhydrous sphere of volume
V0 of equivalent mass of ALO was calculated by inverting
the relation
.
The viscosity increment provides the axial ratio a/b. The
radius R0 of an anhydrous sphere of volume
V0 of equivalent mass of ALO was calculated by inverting
the relation
 to
to
 ,
with
,
with  .
.
Analytical Ultracentrifugation—Purified H6-ALO was run through a S200 size exclusion column in either 20 mm sodium citrate, pH 5.5, plus 100 mm NaCl, 20 mm HEPES, pH 7.0, plus 100 mm NaCl or 20 mm Tris, pH 8.0, plus 100 mm NaCl. The protein was concentrated to 0.8 mg/ml and was run on a Beckman Optima XLA for sedimentation velocity data collection. The data were processed using Sedfit (42).
RESULTS
ALO Decreases the Barrier Function of Human Gut Epithelial Cells—One of the signs of active anthrax infection within the gastrointestinal tract is the presence of primary lesions within the intestinal tissue (29). Potentially, these lesions could be caused by secretion of anthrax toxins by vegetative bacteria. The presence of anthrax toxins (such as ET or LT) could then work to weaken the gut epithelial cell lining, allowing for paracellular movement of B. anthracis across the important epithelial cell barrier. Therefore, we first examined whether ET or LT could alter movement across the intestinal epithelial barrier. Since the interactions between secreted virulence factors and the host can be very complex, we utilized a simplified and well studied cell culture model of the gastrointestinal epithelium. C2BBE cells were chosen to represent the GI epithelium due to their ability to form polarized monolayers with tight junctions and apical brush borders (43, 44). To quantify barrier integrity of the C2BBE monolayer, we measured two parameters characteristic of the level of barrier function: translocation of fluorescein labeled dextran across the monolayer and TER.
C2BBE monolayers were grown to confluence in transwell cell culture inserts for 10-14 days after seeding. The barrier function of these monolayers was measured using fluorescein labeled dextran or TER. Incubation of the C2BBE monolayers with either ET or LT or together resulted in no significant change to barrier function within 2 h when compared with the untreated cells (data not shown). We then expanded our search for potential secreted virulence factors, which could affect the barrier function of epithelial cells. Since other CDCs have been reported to act on epithelial cells, we proposed that ALO could potentially function to alter barrier integrity of C2BBE monolayers (45). Therefore, we examined whether ALO could alter movement across the intestinal epithelial barrier. ALO was added to the apical side of polarized C2BBE cells, and barrier function was measured as a function of time. ALO incubation resulted in a dose-dependent increase in the flux of the dextran across the monolayer, and such an increase could be observed within 20 min. Neither untreated cells (data not shown) nor heat-inactivated ALO (Fig. 1A) caused noticeable changes. Interestingly, cells that were basolaterally treated with ALO showed no change in dextran flux. Since the movement of fluorescein-dextran can be either via a transcellular or paracellular route, we utilized TER to distinguish whether ALO altered the junctions in between the cells, as is indicative of paracellular translocation (46). Within 20 min of ALO treatment, the TER of the C2BBE monolayers dropped ∼80%, indicating that the movement of dextran occurred via a paracellular route, as opposed to a transcellular route (Fig. 1B).
FIGURE 1.

ALO induces C2BBE monolayer permeability. A, 3-kDa dextran-fluorescein flux in C2BBE monolayers treated apically (AP) or basolaterally (BL) with ALO or heat-inactivated ALO (ALO-Φ) at 1 μg/ml or 10 μg/ml. Flux is quantified by basolateral relative fluorescence units (RFU). B, transepithelial resistance of C2BBE monolayers treated with ALO at 1 or 10 μg/ml. Symbols represent the same conditions as seen in A. C, intracellular Ca2+ levels in C2BBE monolayers during ALO (thick lines) or ionomycin (thin lines) treatment. The monolayers were perfused with HBSS (which contains 1.26 mm Ca2+) and treated with either ALO or ionomycin at the indicated concentrations at time = 0 s (vertical line). D, dextran-fluorescein flux across C2BBE monolayers treated apically with ALO (0, 5, or 10 μg/ml) or ionomycin (1 or 5 μm) for 2 h. E, ALO (1 μg/ml) induced dextran-fluorescein flux across the C2BBE monolayer after pretreatment of C2BBE monolayers with either the proteasome inhibitor, MG132 (10 μm), for 2 h or BAPTA-AM (10 μm) for a 30-min pretreatment and 2-h treatment. Error bars, S.E. CHX, cycloheximide.
Intracellular Calcium Increases Caused by ALO or Ionomycin Increase Monolayer Permeability—Previous studies have shown that one modulator of Caco-2 and T84 gut epithelial cell monolayer permeability is calcium influx caused by calcium-specific ionophores (48, 49). In addition, Listeriolysin O, another member of the CDC family, has been shown to cause an increase in intracellular calcium levels (50). Therefore, we decided to probe the role of calcium in ALO-dependent C2BBE monolayer disruption. By monitoring the level of intracellular calcium within fura2-loaded C2BBE monolayers, we were able to observe a dose-dependent increase of intracellular calcium within seconds of ALO treatment (Fig. 1C). In addition, we tested the effect of the calcium elevator ionomycin on intracellular calcium in C2BBE cells. Although both ALO and ionomycin induced potent increases in intracellular calcium, ionomycin caused a transient increase, whereas ALO caused a sustained increase (Fig. 1C). We then compared monolayer dextran flux after either ALO or ionomycin treatment of C2BBE monolayers (Fig. 1D). Both ALO and ionomycin caused a dose-dependent increase in dextran flux. Interestingly, although 1 and 5 μg/ml ALO caused a much larger and more prolonged calcium increase than 1 or 5 μm ionomycin, respectively, monolayer dextran flux was favored in the ionomycin treated cells by as much as 2-6-fold. However, when comparing the early stages (0-200 s) of ALO and ionomycin incubation, the levels of intracellular calcium correlated well with the amount of monolayer disruption (5 μm ionomycin > 5 μg/ml ALO > 1 μm ionomycin > 1 μg/ml ALO). Also, the duration of the intracellular calcium flux did not correlate with the amount of monolayer disruption.
Since early increases in intracellular calcium can affect the permeability of epithelial monolayers, we investigated whether ALO-dependent intracellular calcium increases were required for C2BBE monolayer permeability. When cells were loaded with the calcium chelator BAPTA-AM to block ALO-induced calcium increase (supplemental Fig. 1), ALO-dependent fluorescein-dextran flux across C2BBE monolayers was inhibited by ∼48% (Fig. 1E). This confirms that intracellular calcium increase plays a role in increasing monolayer permeability, although this finding does not preclude the involvement of other cellular signaling event(s).
We then investigated whether de novo protein synthesis and/or degradation was required for the ALO-mediated increase of monolayer permeability. By incubating the C2BBE monolayers with a potent transition state analogue of the 26 S proteosome (MG132; 10 μm) (51), we showed a decreased ALO-mediated fluorescein-dextran flux of ∼72% (Fig. 1E). We also examined the effect of the protein synthesis inhibitor, cycloheximide, and showed that the presence of 10 μg/ml cycloheximide did not alter ALO-induced fluorescein-dextran flux across the epithelial monolayer of C2BBE cells (Fig. 1E).
ALO Treatment Causes Specific Rearrangement of the Tight Junction Protein, Occludin—Another marker of epithelial monolayer permeability is disruption of the tight junction architecture through translocation of tight junction proteins. The intracellular signaling network involved in tight junction regulation is quite complex (for a review, see Ref. 47). Calcium is known to be involved in both tight junction formation and disruption (47-49). Knowing that ALO can increase dextran flux and that calcium is implicated in this process, we then examined the effects of ALO on certain tight junction proteins. Using immunofluorescence microscopy, we observed that ALO treatment (10 μg/ml; 120 min) of C2BBE monolayers changed the cellular localization of the tight junction protein occludin. Occludin staining within untreated C2BBE monolayers showed well defined, continuous peripheral localization of occludin in the typical cobblestone pattern (Fig. 2, top left). ALO treatment resulted in major reorganization of occludin staining, including a large increase in intracellular punctate staining as well as loss of definition around the cell periphery (Fig. 2, top right). Occludin is an integral membrane protein, and its level is regulated by endocytosis and proteasome degradation. The increase in intracellular punctate staining of occludin is indicative of endocytosis, which could potentially lead to degradation via the proteasome. We then examined the specificity of ALO-mediated disruption of other cellular junction proteins. ALO-dependent occludin rearrangement was compared with another tight junction protein, ZO-1. We found that ALO had little or no effect on the distribution of ZO-1 at 10 μg/ml (Fig. 2, middle).
FIGURE 2.

ALO induces alteration to occludin localization. As visualized by confocal immunofluorescence microscopy, the location of three junctional proteins was observed both prior to (left column) and after ALO (10 μg/ml) treatment for 2 h (right column). Occludin labeling (top panel) of untreated C2BBE monolayers revealed continuous staining around the cell periphery. When C2BBE monolayers are treated with ALO (10 μg/ml), occludin becomes internalized as granular intracellular puncta, and gaps within the continuous cell periphery localization become apparent. Another tight junction protein, ZO-1 (middle), shows similar peripheral and intracellular localization both prior to and after ALO treatment. E-cadherin (bottom), an adherens junction protein not associated with occludin or ZO-1, remains on the cell periphery both prior to and after ALO treatment.
Additionally, we examined whether ALO caused global disruption of actin, a cytoskeletal component linked to the tight junction complex, as well as E-cadherin, a component of the adherens junction. Phalloidin labeling of polymerized actin showed that ALO treatment did not alter actin localization within C2BBE monolayers (supplemental Fig. 2). We also found that E-cadherin localization was not affected when C2BBE monolayers were treated with ALO at 10 μg/ml (Fig. 2, bottom). Other CDCs are known to form pores upon insertion into the plasma membrane of eukaryotic cells, which might cause cell lysis. However, we found that there was no observable cell lysis within 1 h of ALO treatment (data not shown). To monitor the alteration of cell permeability by ALO, we also treated ALO-treated C2BBE cells with the cell-impermeable dye, Hoechst 33258. We found that only a small fraction of C2BBE cells had nuclear DNA staining, and such staining was increased with higher concentrations of ALO (data not shown). This is consistent with the notion that ALO treatment leads to increased membrane permeability without causing cell death. Together, these observations suggest that ALO specifically targets certain components of the tight junction, such as occludin, without affecting other tight junction proteins and cytoskeleton architecture or causing cell lysis.
Structural Analysis of the Soluble State of ALO—To begin our understanding of the structural basis for the action of ALO, we solved the structure of the soluble form of ALO. ALO was initially crystallized using an extensive screen of crystallization conditions. Large crystals of ALO formed within 2 days, but they were highly unstable and shattered within a few days from seeding. Probably, high solvent content (80%) and weak lattice contacts contributed to the inherent instability of these crystals. After sequential optimization of temperature, pH, precipitant, and additives, diffracting quality crystals of ALO were obtained using 3.5 m sodium formate as a precipitant. The high salt content of the mother liquor provided sufficient cryoprotection when the crystals were directly loop-mounted under the nitrogen gas stream for data collections. Screening of common cryoprotective compounds as well as freezing of crystals in liquid nitrogen resulted in lower resolution diffracting crystals. The structure of ALO was solved in a prepore conformation at 3.1 Å resolution (Table 1).
Structural analysis shows that the secondary structure content is in agreement with that observed from circular dichroism (data not shown). Similar to other CDCs, including PFO and ILY, ALO is composed of four distinct domains (named here D1, D2, D3, and D4), each of which is hypothesized to play a different role in pore formation (Fig. 3A). D1 consists of a network of loops, α-helices, and a single β-sheet. D2 consists of an extended amino acid chain, which extends from D1 and runs along the axial backbone of the protein to form a three-strand antiparallel β-sheet. This sheet has extensive hydrophobic interactions with both D3 and D4. D3 consists of a single antiparallel β-sheet that is sequestered by two α-helical regions termed transmembrane hairpins 1 and 2. It is postulated that these helices reorient into β-hairpins during insertion into the host cell membrane (11). D4 is connected to D2 by a Ser-Ser-Ala flexible linker. D4 is composed of two sandwiched antiparallel β-sheets. The ends of these β-sheets form a series of loops, one of which is the highly conserved CDC undecapeptide region. The global arrangement of ALO is similar to that of CDCs, whose structures are known (Fig. 3B and supplemental Table 1).
FIGURE 3.

Structural comparison of ALO with PFO (crystal form I), PFO (crystal form III), and ILY. A, side view of ALO colored by domain. D1 is colored magenta, D2 is colored cyan, D3 is colored orange, and D4 is colored green. B, stereo view of ALO aligned with PFO (I), PFO (III), and ILY using LSQ superimposition in COOT on all Cα′ atoms in D1 and D3 (70). ALO is colored red, PFO (I) is colored orange, PFO (III) is colored blue, and ILY is colored green. C, front view of ALO, PFO (I), PFO (III), and ILY aligned by D2. D4 has been omitted for clarity. Lower panels highlight the interactions or lack there of between the β-turn of D2 and transmembrane hairpin 2 of D3. D, side view of ALO and PFO (I) aligned by D2. The center panels show a close-up of the hydrophobic networks at the D2-D4 interface. The right panels show the interactions on the flip side of the D2-D4 interface.
We used dynamic light scattering and SEC coupled on-line to differential pressure transducers to measure the hydrodynamic radius and intrinsic viscosity of ALO, respectively (Fig. 4, A and B, and supplemental Table 2). From these data, we have calculated the viscosity increment and the hydration of ALO using the Einstein viscosity relation. Hydration is the sum of water bound to and entrained by the protein. The hydration of ALO is close to values obtained for native proteins (0.2-0.4 g of water/g of protein), indicating that ALO adopts a stable and folded state in solution. The viscosity increment is related to the axial ratio of a/b (with the semiaxis a > b). A viscosity increment of 4.5 corresponds to an axial ratio a/b of 3.8, which indicates that ALO adopts an elongated shape in solution, as in the crystal.
FIGURE 4.

Oligomerization analysis and hypothetical dimer configurations. A, chromatograms of ALO from S200 size exclusion chromatography coupled to differential pressure transducers and SLS and UV detectors. Data lines show UV (solid), RALS (dot-dash), LALS (dot-dot-dash), and DP (dotted). B, molecular weight (MW) and intrinsic viscosity (IV) patterns of ALO. Data lines show molecular weight (solid), LALS (dot-dot-dash), and IV (dotted). The retention volume of ALO (22.2 ml) is probably due to interactions with the column media. Using standard proteins for calibration, such a retention volume corresponds to a 15-20-kDa protein (not shown). Hence, measurement of molecular mass by SEC-SLS, which is independent from the retention volume on SEC, is appropriate for ALO. C, sedimentation velocity analysis of ALO from analytical ultracentrifugation at three different pH values: pH 5.5 (dashed), pH 7.0 (solid), and pH 8.0 (dotted). D, potential dimer interactions. Crystallographic dimers are found in ALO and PFO (I) crystals, and noncrystallographic dimers are seen in PFO (III) and ILY crystals. Domains 1-3 are colored blue, and D4 is colored orange.
Structural Comparison of the Interactions of D2 with D3 and D4 in ALO, PFO, and ILY—Although the prepore structures of ALO, PFO, and ILY are globally similar, there are noticeable differences. The orientation of the four domains within the prepore structures of ALO, PFO, and ILY is notably different. ALO and the other CDCs undergo large conformational changes during pore formation, so the prepore orientation of the domains plays an important role. The residues of D2 form interactions with all three of the other domains (Fig. 3, C and D). Together with their location, they most likely constitute a hinge region. Particularly, D2 has extensive side chain interactions with D4, which could play a role in orienting D4 (Fig. 3D). The nature of these interactions varies between ALO, PFO, and ILY. The relative orientation of D2 with D4 is different in ALO compared with PFO. One side of the D2-D4 interface of ALO is characterized by the presence of a strong hydrophobic network of residues that stabilize this conformation (Fig. 3D). This network is less profound in PFO. On the opposite side, the D2-D4 interface of ALO shows no interactions between residue side chains. PFO, in contrast, has a salt bridge in this region. These differences in the D2-D4 interface may play a role in constraining the orientation of D4 relative to D2.
Oligomerization of Soluble ALO—Among CDCs, oligomerization of the soluble state varies. Pneumolysin can form a soluble oligomer that resembles the membrane-bound prepore oligomer both in size and shape (52); however, the monomeric form is predominant in solution (53). In contrast, PFO may exist in equilibrium between the monomer and dimer form (54) without the ability to form higher order oligomers (53). To accurately determine the molecular mass of ALO in solution, we then performed static light scattering (SLS) coupled on-line to SEC (SEC-SLS). The measurement provides a molecular mass of 53.6 kDa (Fig. 4, A and B, and supplemental Table 2). This indicates that ALO exists as a monomer in solution. We also preformed sedimentation velocity analysis via analytical ultracentrifugation, which confirmed ALO as a monomer in solution (Fig. 4C and supplemental Table 3). Together, our results show unambiguously that ALO is a monomer in solution under native conditions.
Consistent with this notion, while there are two ALO molecules in the asymmetric unit of the soluble ALO structure, there is only limited contact between two ALO monomers (Fig. 4D). On the contrary, two PFO molecules in the dimeric soluble PFO structure form extensive contacts, which is consistent with the notion that PFO exists as a dimer in solution (Fig. 4D). Interestingly, the ILY crystal structure also reveals extensive contacts between two ILY molecules (Fig. 4D). The oligomeric state of ILY in solution remains elusive.
ALO and PFO Domain 4 Structural and Functional Analysis—ALO, PFO, and ILY have differences both in primary sequence and structure in D4 and the D2-D4 interface (Fig. 5, A and B). As shown above, ALO decreases epithelial cell barrier integrity. We then examined whether PFO has a similar effect using the dextran flux assay. As expected, ALO showed a characteristic increase in flux. However, PFO showed an ∼15-fold decrease in activity relative to ALO (Fig. 5C). This experiment was repeated with the addition of toxins to the basolateral side, and neither toxin had an effect, as expected from previous results (data not shown). Both ALO and PFO have similar potency in their hemolytic activities, confirming the integrity of these purified toxins in our assays (Fig. 5D).
FIGURE 5.

Structural comparison of domain 4 and functional results from domain swap experiments. A, on the top are surface representations of D4. The red ribbon represents the main chain of the highly conserved undecapeptide region. The Trp side chains of the undecapeptide region are shown in yellow. The side chains under the translucent surface are thought to play a role in the orientation of the Trp side chains. Hydrophobic surfaces are colored coral. On the bottom are representations of D4 aligned with each other. The undecapeptide region is again highlighted in red. B, sequence alignment of D4s using ClustalW. Plus signs, residues not in the undecapeptide that are displayed in A. Asterisks, identical residues. Colons, conserved substitutions. Periods, semiconserved substitutions. C, 3-kDa dextran-fluorescein flux in C2BBE monolayers treated apically at 10 μg/ml. D, hemolysis of human erythrocytes after a 30-min treatment. RFU, relative fluorescence units.
To explore this functional difference between ALO and PFO, we returned to their structural differences. We hypothesize that ALO has a structural property that allows it to interact with C2BBE cells in such a way that triggers a decrease in barrier function, whereas PFO lacks this property. D4 is critical in binding of CDCs to the host cell membrane (7, 55, 56). The completely conserved undecapeptide region contained in D4 adopts a different conformation, both in main and side chain location (Fig. 5A). In the PFO undecapeptide, the Trp464 residue is buried in a hydrophobic pocket, whereas in ALO, this hydrophobic pocket does not exist. Therefore, the lack of a hydrophobic pocket for Trp favors the reorientation of the undecapeptide in ALO.
To test the involvement of D4 in the ability of ALO to affect the barrier function of C2BBE cells, we generated D4 swap mutants. ALO(D1-3)PFO(D4) is composed of ALO domains 1-3 and PFO domain 4, whereas PFO(D1-3)ALO(D4) is made of PFO domains 1-3 and ALO domain 4. Both mutants exhibited a potency of hemolytic activity similar to that of ALO and PFO (Fig. 5D). We found that ALO(D1-3)PFO(D4), the ALO mutant with the swapped PFO D4, had a 6-fold reduced effect compared with wild type ALO (Fig. 5C). Consistent with these data, PFO(D1-3)ALO(D4), the PFO mutant with the swapped ALO D4, had a 15-fold enhanced effect compared with PFO and had virtually the same effect on barrier function as wild type ALO (Fig. 5C). This observation indicates that the D4 domain of ALO plays a key role in altering the barrier function of C2BBE cells. The role of the D4 domain to modulate C2BBE barrier function could stem from a site internal to the D4 structure or from the interface between D2 and D4.
DISCUSSION
Our cellular studies have extended the potential roles of ALO in anthrax infection. We show that ALO can impair the epithelial cell barrier, one of the major obstructions to infection in the GI tract. Additionally, we have identified the specificity of ALO to the apical membrane of human gut epithelial cells, the primary contact with secreted virulence factors during the initiation of infection. Since there is no evidence of massive cell death at the ALO concentrations we tested and given that the two well known anthrax toxins, LT and ET, have no effect on epithelial barrier function (data not shown), we propose that ALO may be the primary secreted virulence factor that affects bacterial movement through the GI epithelium. Additional research would be required to evaluate the validity of this hypothesis and whether other secreted factors of anthrax bacteria modulate this process.
One mechanism by which ALO could affect epithelial layer permeability is through a regulated increase in intracellular calcium. The increase in intracellular calcium concentrations occurs within seconds after ALO treatment, and, as the ionomycin-ALO comparison indicates, it is the early influx of calcium that correlates to monolayer disruption. This time frame is so short that the elevation of intracellular calcium by ALO is probably due to pore formation within the plasma membrane, as opposed to pores formed within endocytic vesicles. Although the expected pore size of cholesterol-dependent cytolysins is relatively large (150-400 Å in diameter) (7, 57), the increase in intracellular calcium is only modest when compared with extracellular concentrations and is tightly regulated, as indicated by the prolonged plateau of the intracellular calcium concentration after ALO treatment (6). This regulated process is probably mediated by complex cellular machinery to regulate intracellular concentrations (6, 58). The endocytosis of ALO pores or the interaction of ALO with cellular proteins to plug or disrupt the pore could also contribute to such regulation or inactivation.
Using pharmacological agents, we show that increased intracellular calcium is necessary for epithelial barrier dysfunction. By loading C2BBE cells with BAPTA-AM to buffer intracellular calcium levels, the paracellular movement of fluorescein-dextran was partially blocked. We also show that ALO induces monolayer disruption by specifically targeting the tight junction protein occludin. Since occludin degradation is known to be altered by calcium- and ubiquitin-linked targeting to the proteasome, our experiments show a possible mechanistic link between ALO, calcium, and occludin degradation.
The epithelial lining of mucosal barriers forms an important permeability barrier separating luminal contents from the underlying tissue compartment (59, 60). This permeability barrier is controlled by blockage of paracellular movement through regulation of fluid and electrolyte diffusion across tight junctions. The tight junction barrier complex is a dynamic barrier regulated by a multitude of intracellular signaling mechanisms. How certain second messengers such as intracellular calcium levels induce tight junction dysfunction is currently the subject of much research (61, 62). In fact, intracellular calcium changes regulate the function of occludin through regulating occludin dephosphorylation and ubiquitination (63). ALO-induced increase of intracellular calcium to alter tight junction architecture could benefit movement of the vegetative anthrax bacteria or other bacterial toxins through this barrier for systemic infection. Future work will be required to elucidate the complex events involving ALO intoxication, intracellular increase of calcium, and differential rearrangement within junctional and cytoskeletal complexes of epithelial cells.
Our biophysical characterization reveals that ALO exists as a monomer in solution. Interestingly, previous studies show that PFO dimerizes in solution (53). It is likely that this state of PFO is the antiparallel dimer, which is seen in its crystal structure (64). The most obvious explanation for this difference is the overall shape of these two cytolysins. The angle between D1-3 and D4 in PFO is quite straight, whereas ALO is somewhat kinked at the D2-D4 interface. The nature of this kink is due to the difference in the network of interactions between the two domains. Analysis of the dimer interface of symmetry-related molecules of PFO and ALO reveals how the kink observed in ALO could affect head-to-tail dimerization. The interactions between β-strands of D2 and D4 observed in PFO are not present in ALO.
The fact that ALO does not form dimers in solution, whereas PFO does, suggests that dimerization of CDCs in solution could serve functions not required by ALO. First, it may increase the solubility of PFO by masking nonpolar surfaces. Second, it may also regulate the rate of formation of membrane-bound monomer. It seems likely that once one of the monomers in the dimer pair binds to the membrane, the structural transitions that are initiated in D3 by binding (65-67) cause the dissociation of the other monomer, which would then be free to also bind the membrane.
Our ALO structural analyses also suggest that disruption of the D2-D3 interface of ALO may proceed more slowly than for PFO and so affect the rate at which the prepore is formed. This transition is regulated by conformational changes in D4, which in turn is transmitted to D3 (9, 66-68). This allosteric trigger results in the loss of many contacts in D3, which exposes the transmembrane regions for insertion into the membrane (10, 69). Our crystallographic studies have shown that ALO contains additional intramolecular contacts between D2 and the transmembrane region in D3. This suggests that the free energy required for dissociation of D2 and D3 may be higher in ALO than PFO and may affect the rate at which the monomer is converted to the prepore state.
Our studies also reveal novel differences between ALO and PFO. Our functional studies have shown that ALO can effectively disrupt the barrier function of C2BBE cells, whereas PFO cannot. We also show that the D4 region plays a key role in the ability of ALO to weaken the barrier function of C2BBE cells, whereas it does not change the hemolytic activities of these two cytolysins. One of the distinguishing features of the CDC family is the highly conserved undecapeptide region. This region is required for binding to the cellular membrane and plays an integral role in triggering pore formation. Interestingly, we observe a striking structural difference in the undecapeptide between ALO and PFO. Although the functional implication of this conformational difference between ALO and PFO is yet to be determined, it is tempting to speculate that the structure of this undecapeptide may play an important role in determining the potency of ALO on C2BBE cells. In addition to the difference in the undecapeptide conformation, it is also possible that the difference in the relative orientation of the D2-D4 interface may confer the functional difference between ALO and PFO.
Our ALO structure provides the molecular basis as to how other regions in D4 could affect the conformation of the undecapeptide, which is identical in ALO and PFO. Based on our structure, the difference in the orientation of the undecapeptide region is due to the existence of a hydrophobic pocket in PFO that is absent in ALO. This hydrophobic pocket sequesters a tryptophan residue, which causes a change in the orientation of the other tryptophans in the undecapeptide region. The lack of this pocket in ALO allows increased accessibility of these tryptophans to the solvent.
The strong preference for ALO to target the apical side of C2BBE cells leads us to hypothesize that the presence of cholesterol alone is not enough to trigger certain functions of ALO. The effect of ALO on barrier function may be mediated by a receptor specific to the apical side of these gut epithelial cells. The need for a receptor for the function of a CDC has been established by the studies of ILY (4, 8). Further work is needed to elucidate this receptor and determine the molecular basis behind the apical selectivity for ALO.
Supplementary Material
Acknowledgments
We thank the University of Chicago Biophysics Core Facility and Elena Solomaha for assistance with various biophysical characterizations; the University of Chicago BSD Microscopy Core Facility and Christine Labno for the confocal microscopy; Marika Manolopoulou for cell culture; Raymond Hulse for imaging processing of confocal microscopy; Dr. David Boone, Jeanette Messer, and James Lodolce for helpful discussion on epithelial tight junction regulation and related experimentation; and Dr. Rodney Tweten (Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center) for helpful discussion of the manuscript.
The atomic coordinates and structure factors (code 3CQF) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant DK38510 (to E. B. C.), National Institutes of Health Grants GM62548 and AI66503, and a pilot grant from Digestive Disease Research Core Center of the University of Chicago (National Institutes of Health Grant DK42086). This work was also supported by the American Health Assistance Foundation (to E. M. M.), a pilot grant from the Great Lakes Regional Center of Excellence, and a University of Chicago FACCTS grant (to W. J. T.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and Tables 1-3.
Footnotes
The abbreviations used are: CDC, cholesterol-dependent cytolysin; ALO, anthrolysin O; PFO, perfringolysin O; ILY, intermedilysin; C2BBE, Caco-2 brush border expressor; TER, transepithelial resistance; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester; ET, edema toxin; LT, lethal toxin; GI, gastrointestinal; PBS, phosphate-buffered saline; HBSS, Hanks' balanced salt solution; SEC, size exclusion chromatography; LALS, low angle laser light scattering; RALS, right angle laser light scattering; SLS, static light scattering.
References
- 1.Rosado, C. J., Buckle, A. M., Law, R. H., Butcher, R. E., Kan, W. T., Bird, C. H., Ung, K., Browne, K. A., Baran, K., Bashtannyk-Puhalovich, T. A., Faux, N. G., Wong, W., Porter, C. J., Pike, R. N., Ellisdon, A. M., Pearce, M. C., Bottomley, S. P., Emsley, J., Smith, A. I., Rossjohn, J., Hartland, E. L., Voskoboinik, I., Trapani, J. A., Bird, P. I., Dunstone, M. A., and Whisstock, J. C. (2007) Science 317 1548-1551 [DOI] [PubMed] [Google Scholar]
- 2.Rosado, C. J., Kondos, S., Bull, T. E., Kuiper, M. J., Law, R. H., Buckle, A. M., Voskoboinik, I., Bird, P. I., Trapani, J. A., Whisstock, J. C., and Dunstone, M. A. (2008) Cell. Microbiol. 10 1765-1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giddings, K. S., Johnson, A. E., and Tweten, R. K. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 11315-11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giddings, K. S., Zhao, J., Sims, P. J., and Tweten, R. K. (2004) Nat. Struct. Mol. Biol. 11 1173-1178 [DOI] [PubMed] [Google Scholar]
- 5.Tweten, R. K. (2005) Infect. Immun. 73 6199-6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gekara, N. O., Westphal, K., Ma, B., Rohde, M., Groebe, L., and Weiss, S. (2007) Cell. Microbiol. 9 2008-2021 [DOI] [PubMed] [Google Scholar]
- 7.Rossjohn, J., Feil, S. C., McKinstry, W. J., Tweten, R. K., and Parker, M. W. (1997) Cell 89 685-692 [DOI] [PubMed] [Google Scholar]
- 8.Polekhina, G., Giddings, K. S., Tweten, R. K., and Parker, M. W. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 600-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran, R., Heuck, A. P., Tweten, R. K., and Johnson, A. E. (2002) Nat. Struct. Biol. 9 823-827 [DOI] [PubMed] [Google Scholar]
- 10.Shepard, L. A., Heuck, A. P., Hamman, B. D., Rossjohn, J., Parker, M. W., Ryan, K. R., Johnson, A. E., and Tweten, R. K. (1998) Biochemistry 37 14563-14574 [DOI] [PubMed] [Google Scholar]
- 11.Czajkowsky, D. M., Hotze, E. M., Shao, Z., and Tweten, R. K. (2004) EMBO J. 23 3206-3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross, C. L., and Koehler, T. M. (2006) J. Bacteriol. 188 7823-7829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shannon, J. G., Ross, C. L., Koehler, T. M., and Rest, R. F. (2003) Infect. Immun. 71 3183-3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosser, E. M., and Rest, R. F. (2006) BMC Microbiol. 6 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drobniewski, F. A. (1993) Clin. Microbiol. Rev. 6 324-338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mock, M., and Mignot, T. (2003) Cell. Microbiol. 5 15-23 [DOI] [PubMed] [Google Scholar]
- 17.Park, J. M., Ng, V. H., Maeda, S., Rest, R. F., and Karin, M. (2004) J. Exp. Med. 200 1647-1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chitlaru, T., Gat, O., Gozlan, Y., Ariel, N., and Shafferman, A. (2006) J. Bacteriol. 188 3551-3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duesbery, N. S., Webb, C. P., Leppla, S. H., Gordon, V. M., Klimpel, K. R., Copeland, T. D., Ahn, N. G., Oskarsson, M. K., Fukasawa, K., Paull, K. D., and Vande Woude, G. F. (1998) Science 280 734-737 [DOI] [PubMed] [Google Scholar]
- 20.Leppla, S. H. (1982) Proc. Natl. Acad. Sci. U. S. A. 79 3162-3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beauregard, K. E., Wimer-Mackin, S., Collier, R. J., and Lencer, W. I. (1999) Infect. Immun. 67 3026-3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paccani, S. R., Tonello, F., Ghittoni, R., Natale, M., Muraro, L., D'Elios, M. M., Tang, W. J., Montecucco, C., and Baldari, C. T. (2005) J. Exp. Med. 201 325-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar, P., Ahuja, N., and Bhatnagar, R. (2002) Infect. Immun. 70 4997-5007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tournier, J. N., Quesnel-Hellmann, A., Mathieu, J., Montecucco, C., Tang, W. J., Mock, M., Vidal, D. R., and Goossens, P. L. (2005) J. Immunol. 174 4934-4941 [DOI] [PubMed] [Google Scholar]
- 25.Raymond, B., Leduc, D., Ravaux, L., Goffic, R. L., Candela, T., Raymondjean, M., Goossens, P. L., and Touqui, L. (2007) PLoS Pathog. 3 e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heffernan, B. J., Thomason, B., Herring-Palmer, A., and Hanna, P. (2007) FEMS Microbiol. Lett. 271 98-105 [DOI] [PubMed] [Google Scholar]
- 27.Dixon, T. C., Meselson, M., Guillemin, J., and Hanna, P. C. (1999) N. Engl. J. Med. 341 815-826 [DOI] [PubMed] [Google Scholar]
- 28.Inglesby, T. V., O'Toole, T., Henderson, D. A., Bartlett, J. G., Ascher, M. S., Eitzen, E., Friedlander, A. M., Gerberding, J., Hauer, J., Hughes, J., Mc-Dade, J., Osterholm, M. T., Parker, G., Perl, T. M., Russell, P. K., and Tonat, K. (2002) J. Am. Med. Assoc. 287 2236-2252 [DOI] [PubMed] [Google Scholar]
- 29.Beatty, M. E., Ashford, D. A., Griffin, P. M., Tauxe, R. V., and Sobel, J. (2003) Arch. Intern. Med. 163 2527-2531 [DOI] [PubMed] [Google Scholar]
- 30.Inglesby, T. V., Henderson, D. A., Bartlett, J. G., Ascher, M. S., Eitzen, E., Friedlander, A. M., Hauer, J., McDade, J., Osterholm, M. T., O'Toole, T., Parker, G., Perl, T. M., Russell, P. K., and Tonat, K. (1999) J. Am. Med. Assoc. 281 1735-1745 [DOI] [PubMed] [Google Scholar]
- 31.Glomski, I. J., Piris-Gimenez, A., Huerre, M., Mock, M., and Goossens, P. L. (2007) PLoS Pathog. 3 e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg, R. D. (1999) Adv. Exp. Med. Biol. 473 11-30 [DOI] [PubMed] [Google Scholar]
- 33.Peterson, M. D., and Mooseker, M. S. (1992) J. Cell Sci. 102 581-600 [DOI] [PubMed] [Google Scholar]
- 34.Wu, X., Zagranichnaya, T. K., Gurda, G. T., Eves, E. M., and Villereal, M. L. (2004) J. Biol. Chem. 279 43392-43402 [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 36.Qian, B., Raman, S., Das, R., Bradley, P., McCoy, A. J., Read, R. J., and Baker, D. (2007) Nature 450 259-264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 38.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 39.Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., Mc-Coy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., and Terwilliger, T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58 1948-1954 [DOI] [PubMed] [Google Scholar]
- 40.Lovell, S. C., Davis, I. W., Arendall, W. B., III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S., and Richardson, D. C. (2003) Proteins 50 437-450 [DOI] [PubMed] [Google Scholar]
- 41.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 42.Schuck, P. (2000) Biophys. J. 78 1606-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peterson, M. D., and Mooseker, M. S. (1993) J. Cell Sci. 105 445-460 [DOI] [PubMed] [Google Scholar]
- 44.Peterson, M. D., Bement, W. M., and Mooseker, M. S. (1993) J. Cell Sci. 105 461-472 [DOI] [PubMed] [Google Scholar]
- 45.Tsuchiya, K., Kawamura, I., Takahashi, A., Nomura, T., Kohda, C., and Mitsuyama, M. (2005) Infect. Immun. 73 3869-3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madara, J. L. (1990) Am. J. Pathol. 137 1273-1281 [PMC free article] [PubMed] [Google Scholar]
- 47.Gonzalez-Mariscal, L., Tapia, R., and Chamorro, D. (2008) Biochim. Biophys. Acta 1778 729-756 [DOI] [PubMed] [Google Scholar]
- 48.Tai, Y. H., Flick, J., Levine, S. A., Madara, J. L., Sharp, G. W., and Donowitz, M. (1996) J. Membr. Biol. 149 71-79 [DOI] [PubMed] [Google Scholar]
- 49.Rutten, M. J., Cogburn, J. N., Schasteen, C. S., and Solomon, T. (1991) Pharmacology 42 156-168 [DOI] [PubMed] [Google Scholar]
- 50.Dramsi, S., and Cossart, P. (2003) Infect. Immun. 71 3614-3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee, D. H., and Goldberg, A. L. (1998) Trends Cell Biol. 8 397-403 [DOI] [PubMed] [Google Scholar]
- 52.Gilbert, R. J., Rossjohn, J., Parker, M. W., Tweten, R. K., Morgan, P. J., Mitchell, T. J., Errington, N., Rowe, A. J., Andrew, P. W., and Byron, O. (1998) J. Mol. Biol. 284 1223-1237 [DOI] [PubMed] [Google Scholar]
- 53.Solovyova, A. S., Nöllmann, M., Mitchell, T. J., and Byron, O. (2004) Biophys. J. 87 540-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert, R. J. (2005) Structure 13 1097-1106 [DOI] [PubMed] [Google Scholar]
- 55.Tweten, R. K., Harris, R. W., and Sims, P. J. (1991) J. Biol. Chem. 266 12449-12454 [PubMed] [Google Scholar]
- 56.Iwamoto, M., Ohno-Iwashita, Y., and Ando, S. (1990) Eur. J. Biochem. 194 25-31 [DOI] [PubMed] [Google Scholar]
- 57.Tilley, S. J., Orlova, E. V., Gilbert, R. J., Andrew, P. W., and Saibil, H. R. (2005) Cell 121 247-256 [DOI] [PubMed] [Google Scholar]
- 58.Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003) Nat. Rev. Mol. Cell Biol. 4 517-529 [DOI] [PubMed] [Google Scholar]
- 59.Yu, D., and Turner, J. R. (2008) Biochim. Biophys. Acta 1778 709-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mitic, L. L., and Anderson, J. M. (1998) Annu. Rev. Physiol. 60 121-142 [DOI] [PubMed] [Google Scholar]
- 61.Somosy, Z., Bognar, G., Horvath, G., and Koteles, G. J. (2003) Cell Mol. Biol. (Noisy-Le-Grand) 49 59-63 [PubMed] [Google Scholar]
- 62.Park, J. H., Okayama, N., Gute, D., Krsmanovic, A., Battarbee, H., and Alexander, J. S. (1999) Am. J. Physiol. 277 C1066-C1074 [DOI] [PubMed] [Google Scholar]
- 63.Ma, T. Y., Tran, D., Hoa, N., Nguyen, D., Merryfield, M., and Tarnawski, A. (2000) Microsc. Res. Tech. 51 156-168 [DOI] [PubMed] [Google Scholar]
- 64.Rossjohn, J., Polekhina, G., Feil, S. C., Morton, C. J., Tweten, R. K., and Parker, M. W. (2007) J. Mol. Biol. 367 1227-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soltani, C. E., Hotze, E. M., Johnson, A. E., and Tweten, R. K. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 20226-20231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soltani, C. E., Hotze, E. M., Johnson, A. E., and Tweten, R. K. (2007) J. Biol. Chem. 282 15709-15716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramachandran, R., Tweten, R. K., and Johnson, A. E. (2004) Nat. Struct. Mol. Biol. 11 697-705 [DOI] [PubMed] [Google Scholar]
- 68.Heuck, A. P., Hotze, E. M., Tweten, R. K., and Johnson, A. E. (2000) Mol. Cell 6 1233-1242 [DOI] [PubMed] [Google Scholar]
- 69.Shatursky, O., Heuck, A. P., Shepard, L. A., Rossjohn, J., Parker, M. W., Johnson, A. E., and Tweten, R. K. (1999) Cell 99 293-299 [DOI] [PubMed] [Google Scholar]
- 70.Guttmann, D. M., and Ellar, D. J. (2000) FEMS Microbiol. Lett. 188 7-13 [DOI] [PubMed] [Google Scholar]
- 71.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.