

Abstract
Methylmalonic acidemia (MMA), an autosomal recessive metabolic disorder, is most often caused by mutations in methylmalonyl-CoA mutase (MUT). Severely affected patients typically present with metabolic crisis in the early neonatal period and can perish despite intervention. Survivors follow an unstable course and can require elective liver transplantation to prevent life-threatening metabolic decompensation. Therapeutic alternatives to liver transplantation such as hepatocyte-directed gene and cell therapies lack experimental validation. We have used a murine model of mut0 MMA to assess the efficacy of virus-mediated gene therapy to rescue the neonatal lethality seen in the Mut−/− mice. Affected pups and control littermates received either intramuscular or intrahepatic injections of adenovirus carrying the Mut gene expressed under the control of the cytomegalovirus promoter. All of the Mut−/− pups injected via the intramuscular route perished within the first 48 hr of birth. However, more than 50% of the Mut−/− pups that received intrahepatic injections survived beyond weaning (day 15). The treated mutants expressed methylmalonyl-CoA mutase mRNA and protein, and displayed decreased metabolite levels compared with uninjected Mut−/− mice. The results demonstrate that adenovirus-mediated, hepatic methylmalonyl-CoA mutase expression can rescue Mut−/− pups from neonatal mortality and provide proof-of-principle evidence for the efficacy of liver-directed gene delivery in methylmalonic acidemia.
INTRODUCTION
Methylmalonic acidemia(MMA) is a severe autosomal recessive inborn error of intermediary metabolism characterized by intermittent metabolic instability, multiorgan pathology, and poor prognosis for long-term survival (Fenton et al., 2001). The disorder exhibits genetic heterogeneity and can be caused by defective intracellular transport, processing, and metabolism of cobalamin or impaired activity of methylmalonyl-CoA mutase (MUT) (Fenton and Rosenblatt, 2001; Venditti, 2005). This mitochondrial enzyme uses 5′-de-oxyadenosylcobalamin, a vitamin B12 derivative, as a cofactor to catalyze the formation of succinyl-CoA from L-methylmalonyl-CoA at the terminal step of propionyl-CoA metabolism, as depicted in Fig. 1. Historically, patients with vitamin B12-unresponsive methylmalonic acidemia, typically associated with mutations in the MUT gene (mut0 class), have had severe clinical manifestations (Matsui et al., 1983) and high morbidity (van der Meer et al., 1994; de Baulny et al., 2005), with a median survival of 6 years in one retrospective study (Nicolaides et al., 1998). The use of expanded or supplemental screening to identify newborns with methylmalonic acidemia (Chace et al., 2002) underscores the need for improved therapies for these patients.
FIG. 1.
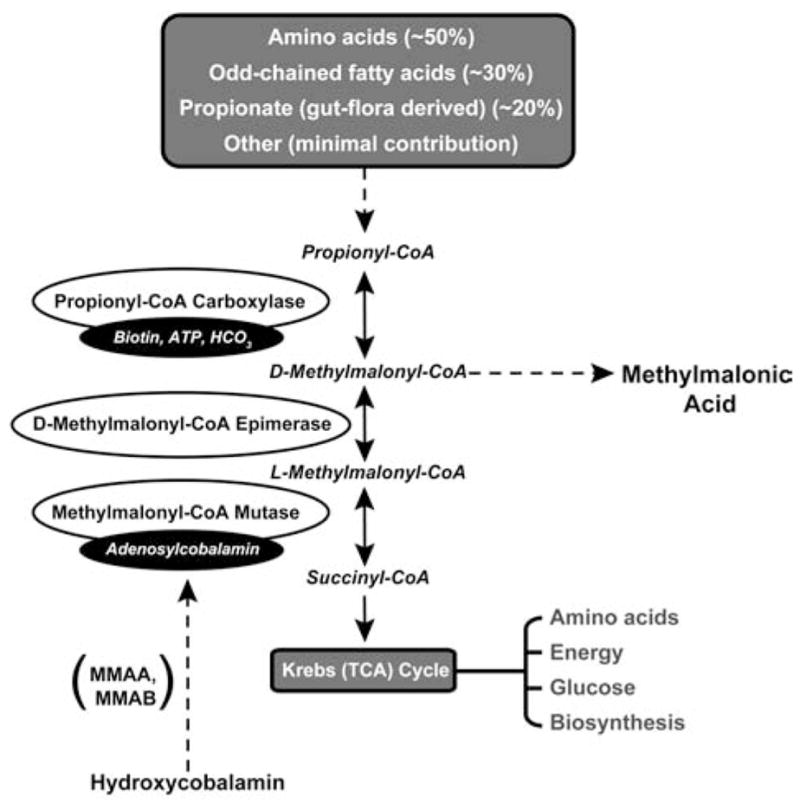
The metabolism of propionyl-CoA to succinyl-CoA, an important Krebs cycle intermediate, is displayed. Isolated methylmalonic acidemia results from deficiencies in the conversion of D-methylmalonyl-CoA to succinyl-CoA and can be caused by mutations in methylmalonyl-CoA mutase (MUT) as well as defects in the synthesis of adenosylcobalamin (MMAA, MMAB, cblD variant 2) and impaired activity of D-methylmalonyl-CoA epimerase (MCEE).
Gene- and cell-based therapies may hold promise as future treatments for organic acidemias such as MMA. The limited experience with liver (Chakrapani et al., 2002; Kayler et al., 2002; Nyhan et al., 2002; Hsui et al., 2003; Kaplan et al., 2006; Kasahara et al., 2006) and liver–kidney transplantation (van’t Hoff et al., 1998; Nagarajan et al., 2005) as a treatment for MMA patients indicates that organ-specific therapy can provide enough enzyme activity to prevent frequent decompensations. Although transplanted patients continue to exhibit substantial methylmalonic acidemia (van’t Hoff et al., 1998;Nagarajan et al., 2005; Kaplan et al., 2006), salutary effects have included increased protein tolerance and improved growth. Hence, liver transplantation has proven efficacious in restoring metabolic homeostasis in severely affected patients. Moreover, the liver is recognized to play a pivotal role in methylmalonyl-CoA metabolism (Frenkel and Kitchens, 1975). In addition, the demonstration that adenovirus-mediated gene transfer to primary human mut0 hepatocytes completely corrected the biochemical defect in vitro provides the foundation for a study of in vivo correction in an animal model (Chandler et al., 2007).
We have used a mouse model of methylmalonic acidemia that was generated by targeted deletion of a critical exon in the murine methylmalonyl-CoA mutase (Mut) gene (Chandler and Venditti, 2005; Venditti et al., 2005) to examine whether virus-mediated gene delivery can influence the fatal outcome seen in the mutant mice. The Mut−/− mice display early neonatal lethality and faithfully replicate the severe phenotype of affected humans and display early neonatal lethality, similar to what has been described in another murine model of Mut MMA (Peters et al., 2003). Because studies in the Mut−/− mice have demonstrated progressive hepatic pathology (Peters et al., 2003) and massive accumulation of methylmalonic acid in the liver near the time of death (Venditti et al., 2005), we hypothesized that reducing the metabolite load by restoring hepatic metabolism may rescue the uniform neonatal lethality observed in the homozygous mutants. To test this hypothesis, we used an E1,E3-deleted adenovirus to deliver the murine methylmalonyl-CoA mutase gene by direct injection into the liver of neonatal knockout mice and control littermates. Survival analysis, expression (mRNA and protein) studies, and metabolite levels in the treated mutants demonstrated the rescue of the Mut−/− pups from neonatal lethality and provide proof-of-principle evidence for hepatocyte-directed gene therapy approaches in methylmalonic acidemia.
MATERIALS AND METHODS
Murine model of methylmalonic acidemia
The targeted Mut allele harbors a deletion of exon 3 in the methylmalonyl-CoA mutase gene (Chandler and Venditti, 2005; Venditti et al., 2005). This exon encodes the putative substrate-binding pocket in the methylmalonyl-CoA mutase enzyme. The mutant allele does not produce detectable RNA, protein, or enzymatic activity (Chandler et al., 2007). Homozygous knockout animals on a mixed (C57BL/6 × 129Sv/Ev) background display a neonatal lethal phenotype and perish within the early neonatal period. Affected mice display massively elevated methylmalonic acid in the plasma that progressively rises to the 2 mM range until death occurs.
Adenoviral vector construction and production
Previous studies have demonstrated that the cytomegalovirus (CMV)-Mut bifunctional adenovirus (Ad-Mut-GFP) used in these experiments expressed enhanced green fluorescent protein (eGFP) in vivo and could correct the propionate metabolic defect in Mut−/− murine embryonic fibroblasts (MEFs) and primary hepatocytes derived from a mut0 patient (Chandler et al., 2007). Another adenovirus, carrying only the Rous sarcoma virus (RSV) eGFP expression cassette in the E3 region (Ad-GFP), was used as an injection control. Direct hepatic or intramuscular (hind leg) injections of 1.2 × 1010 adenoviral particles (3.3 × 108 plaque-forming units [PFU]) in a total volume of 10 μl were delivered to (C57BL/6 × 129Sv/Ev) neonatal mice, using a 32-gauge Hamilton syringe. Vector genomes and plaque-forming units were measured in the final adenoviral preparations. Animal studies were reviewed and approved by the National Human Genome Research Institute (Bethesda, MD) Animal User Committee.
Reverse transcription-polymerase chain reaction
One microgram or 500 ng of total RNA was used to amplify methylmalonyl-CoA mutase or glyceraldehyde-3-phosphate de-hydrogenase (GAPDH), respectively. The murine methylmalonyl-CoA mutase primers used were as follows: forward, 5′-CATGTTGAGAGCTAAGAATC-3′; and reverse, 5′-TAGAAGTTCATTCCAATCCC-3′, which yielded a band of 943 base pairs after amplification of the target. The murine GAPDH primers used were as follows: forward, 5′-TGAAGGTCGGTGTGAACGGATTTGG-3′; and reverse, 5′-CATGTAGGCCATGAGGTCCACCAC-3′, which yielded a band of 983 base pairs after amplification of the target.
Quantitative real-time polymerase chain reaction
Quantitative polymerase chain reaction (PCR) was accomplished with TaqMan gene expression assays (mouse GAPD [4352932E] and murine methylmalonyl-CoA mutase [Mm00485312_m1]; Applied Biosystems, Foster City, CA), which were analyzed in an Applied Biosystems 7500 fast real-time PCR system, in accordance with the manufacturer’s protocols (1 μl of cDNA per reaction). All samples were assayed in triplicate and included RNA without reverse transcriptase added to the reaction as a negative control. Ten pooled mRNA liver samples from wild-type mice were used to determine the 100% wild-type methylmalonyl-CoA mutase mRNA expression level and 10 pooled mRNA Mut−/− liver samples were used to determine 0% wild-type methylmalonyl-CoA mutase mRNA expression. For each cDNA sample, total mRNA, which had not undergone reverse transcription, was used as a negative control for background noise and was subtracted from the resultant threshold cycle (CT) value of the cDNA sample.
Western blotting
Tissue samples were homogenized with a 2-ml Tenbroeck tissue grinder (Wheaton, Millville, NJ) in T-PER (Pierce Biotechnology, Rockford, IL) tissue protein extraction buffer in the presence of Halt (Pierce Biotechnology) protease inhibitor cocktail. Twenty micrograms of clarified extract was analyzed by immunoblotting and probed with affinity-purified, rabbit polyclonal antisera raised against the murine methylmalonyl-CoA mutase enzyme (Chandler et al., 2007). Albumin was used as a loading control and was also detected by immunoblotting [ALB (N-18): sc-46291; Santa Cruz Biotechnology, Santa Cruz, CA]. The anti-mutase antibody was used at a dilution of 1:750 and the anti-albumin antibody was used at a dilution of 1:2000. Horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (NA934; GE Healthcare Life Sciences, Piscat-away, NJ) or rabbit anti-goat IgG (sc-2768; Santa Cruz Biotechnology) was used as the secondary antibody and was visualized with chemiluminescence detection (Pierce Biotechnology).
Metabolic studies
Plasma was isolated from animals immediately after death. The samples were immediately centrifuged and the plasma was removed, diluted in water, and stored at −80°C in a screw-top tube for later analysis. Methylmalonic acid was analyzed by gas chromatography–mass spectrometry with stable isotopic internal calibration to measure MMA as previously described (Marcell et al., 1985; Allen et al., 1993).
Statistical analysis
Kaplan–Meier survival curves were used to compare groups defined on the basis of treatment with or without the Ad-Mut-GFP virus. The primary null hypothesis, that the two survival curves were identical, was tested with a log-rank statistic using SAS software version 9 (SAS Institute, Cary, NC). The end point for the survival analysis was defined as time at weaning (day 15). A paired Student t test was used to assess the significance of the difference between the plasma MMA concentrations between treated and untreated mutant mice. p Values less than 0.05 were considered significant.
RESULTS
Mut+/− mice, maintained on a mixed (C57BL/6 × 129Sv/Ev) background, were crossed to generate mutant animals. Newborn pups received either an intramuscular or intra-hepatic injection of 1.2 × 1010 adenoviral particles in a total volume of 10 μl immediately after birth. To reduce stress on the animals, they were not genotyped before injection. Intramuscular injections were performed on a total of 63 pups from 6 litters. All Mut−/− pups from this cohort perished in the first 48 hr of life. Only 39 animals (heterozygotes and wild type) were alive at weaning, indicating that our method of intramuscular adenoviral gene delivery was not effective at rescuing the neonatal lethal mutant phenotype and therefore it was not further analyzed.
Intrahepatic delivery was examined next. Seventy-seven pups from 8 litters were injected and 69 animals (13 Mut+/+, 42 Mut+/−, and 14 Mut−/−) were genotyped. Eight of the 14 treated Mut−/− pups, or 57% (Fig. 2), escaped the neonatal lethality uniformly associated with this genotype and lived 15 days or more. Estimations of the distribution of the genotypes of missing mice would not alter the conclusion that a significant fraction of the mutants were rescued from certain death after direct hepatic delivery of an adenoviral vector that expressed the Mut gene in the liver. A summary of the survival data is presented in Fig. 2. The log-rank test indicated that the observed difference in the Kaplan–Meier survival curves among the groups could not be explained by chance (χ2 = 91.48, df = 1, p < 0.0001), supporting the intuitive proof of principle for direct hepatic injection rescue.
FIG. 2.

Kaplan–Meier survival curves for untreated Mut−/− mice (n = 17), adenovirus-injected Mut−/− mice (n = 14), and control mice (n = 55). The number of mice in each group is indicated. Greater than 50% (8 of 14) of the Mut−/− mice hepatically injected with adenovirus (Ad) survived 15 days or more. The difference in survival between the mutant treated mice and untreated mice, calculated by log-rank statistics, was significant: p < 0.0001.
Carcasses removed at earlier times showed that Mut+/+ and Mut+/− animals had perished, suggesting morbidity was associated with the adenoviral delivery. To examine the extent of hepatic pathology, eight pups were treated by hepatic injection and sacrificed 48–72 hr later to assess microscopic changes in the liver. No evidence of necrosis or inflammation was detected (data not presented). Additional experiments using Ad-GFP delivered intrahepatically to 26 mice from three litters failed to produce surviving mutants. Table 1 summarizes the number of animals treated and survival data for the adenovirus-injected animals.
Table 1.
Summary of Survival and Biochemical and Molecular Data for Groups of Animals Reported
| A. Survival Data | |||
|---|---|---|---|
| Group | Number of animals treated | Number alive at weaning (day 15) | Number of mutants Group alive (day 15) |
| Intramuscular Ad-Mut-GFP | 63 | 39 | 0 |
| Intrahepatic Ad-Mut-GFP | 77 | 54 | 8 |
| Intrahepatic Ad-GFP | 26 | 19 | 0 |
| Untreated | 69 | 52 | 0 |
| B. Biochemical and Molecular Data | ||||
|---|---|---|---|---|
| Group | Age (DOL) | Average plasma MMA concentration (μM) | Number of mutants studied | Comments |
| Intrahepatic Ad-Mut-GFP | 2–3 | 1549 | 10 | RNA and protein in some animals were present in quantities greater than in WT animals |
| Untreated | 2–3 | 2318 | 15 | No RNA or protein |
| Intrahepatic Ad-Mut-GFP | 19 | 1500 | 4 | RNA detected by RT-PCR and qPCR; no protein detected |
| Intrahepatic Ad-Mut-GFP | 25 | 1922 | 3 | RNA detected by RT-PCR and qPCR; no protein detected |
| Intrahepatic Ad-Mut-GFP | 78 | 2376 | 1 | NA |
Abbreviations: DOL, days of life; GFP, green fluorescent protein; MMA, methylmalonic acidemia; NA, not available; qPCR, quantitative polymerase chain reaction; RT-PCR, reverse transcription-polymerase chain reaction, WT, wild type.
On day 15, the 8 surviving Mut−/− mice appeared normal and could not be differentiated from their littermates. However, progressive weight loss starting at approximately 20 days of life occurred in the mutant survivor animals relative to their littermates. Eventually, the mutants became lethargic and distressed, at which time they were euthanized. Subcutaneous fluids and dextrose administration were able to temporarily ameliorate symptoms but could not prevent death. One treated Mut−/− mouse survived for 78 days and another for 8 months. Plasma methylmalonic acid levels ranged from 1 to 3 mM in the mice that were killed after day 15 (Table 1).
RT-PCR was first employed to detect the expression of the Mut gene in the treated mutant survivors. RNA obtained from the liver, the site of adenoviral injection, was extracted and used to determine whether Mut mRNA was present. A low level of Mut expression was observed in two mice sacrificed on days 19 and 25 (data not presented; and see Fig. 3A, columns 7 and 8). Both of these mice appeared distressed and experienced weight loss before sacrifice. However, earlier time points demonstrated robust expression in some treated animals. For example, one Mut−/− mouse, studied 48–72 hr after injection, expressed Mut at 384% of wild-type levels (Fig. 3A, column 3). This animal also had the lowest plasma methylmalonic acid level of the injected cohort at 529 μM as compared with 2332 μM for age-matched, uninjected Mut−/− mice. All of the other injected mice (Fig. 3A, columns 4 and 5) exhibited less Mut expression (from 0 to 13% of wild type) and had higher plasma methylmalonic acid levels (between 953 and 3876 μM) compared with this mouse yet exhibited significantly lower MMA levels than the untreated mutants (Fig. 3A, column 2). It should be noted that the 19-day-old animals (Fig. 3A, columns 6 and 7) were fed a diet supplemented with fruit and carbohydrates, which could have lowered the dietary precursor load and resulted in lower plasma methylmalonic acid levels.
FIG. 3.

(A) Quantitative RT-PCR of total liver RNA from a wild-type mouse and from Mut−/− mice and Mut−/− adenovirus-treated mice at various time points after injection. The adenovirus was designed to express methylmalonyl-CoA mutase from a CMV promoter. Methylmalonyl-CoA mutase expression is presented as a percentage of wild-type expression. GAPDH was used to normalize the samples. The plasma methylmalonic acid (MMA) levels for each mouse are shown below the graph. Mut−/− 1 Ad 48–72 hr had the highest expression at 384% of Mut wild-type RNA and the lowest plasma MMA level at 529 μM. Values for the other treated animals are similarly presented. (B) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis of liver protein extracts from the same mice depicted in (A), probed with antibodies against methylmalonyl-CoA mutase (mutase) or albumin. The Mut−/− 1 Ad 48–72 hr animal, which had the lowest plasma MMA level, exhibited the strongest methylmalonyl-CoA mutase signal (lane 3), which is in agreement with the quantitative RT-PCR results for this mouse, which showed 384% of wild-type expression.
Western blot analysis of protein extracts prepared from the livers of the mutant survivors was also performed (Fig. 3B). The Mut−/− mouse with the highest transcript level yielded the strongest methylmalonyl-CoA mutase band after probing with anti-mutase antibodies (Fig. 3B, lane 3), whereas no protein could be convincingly detected in the other injected Mut−/− mice. Consistent with the results of the initial RT-PCR studies, the mutase enzyme could not be detected by Western analysis in the livers of the 19- and 25-day-old mice. Because the CMV promoter functions ubiquitously, it was possible that some methylmalonyl-CoA mutase was being expressed in other tissues. Gross dissections of injected mice, using a microscope equipped to detect GFP, expressed from the cis minigene cassette in the E3 region of the viral genome, failed to show expression in other sites. In summary, the results indicate that high Mut RNA and protein expression in the liver correlate with lower methylmalonic plasma levels and represent a marker of functional enzymatic activity.
Methylmalonic acid plasma levels from treated versus untreated Mut−/− mice after hepatic injection with the Ad-Mut-GFP adenovirus at 48–72 hr of life were examined in two independent experiments to determine whether hepatic MUT expression could alter methylmalonic acid plasma levels. A total of 15 Mut−/− uninjected controls were sacrificed at 48–72 hr to establish a baseline uncorrected range. This control group had an average methylmalonic acid plasma concentration of 2318 μM, with the 95% confidence interval falling between 2010 and 2626 μM. Two groups of mutants totaling 10 animals, treated with the same preparation of Ad-Mut-GFP virus by the same operator, were also studied in the 48- to 72-hr period. The treated mutants displayed an average methylmalonic acid plasma concentration of 1550 μM, with the 95% confidence interval falling between 1008 and 2090 μM, which was significantly less than that of the untreated group (p = 0.015). Figure 4 depicts a scatter plot of the values with the 95% confidence intervals surrounding the average value for each group.
FIG. 4.
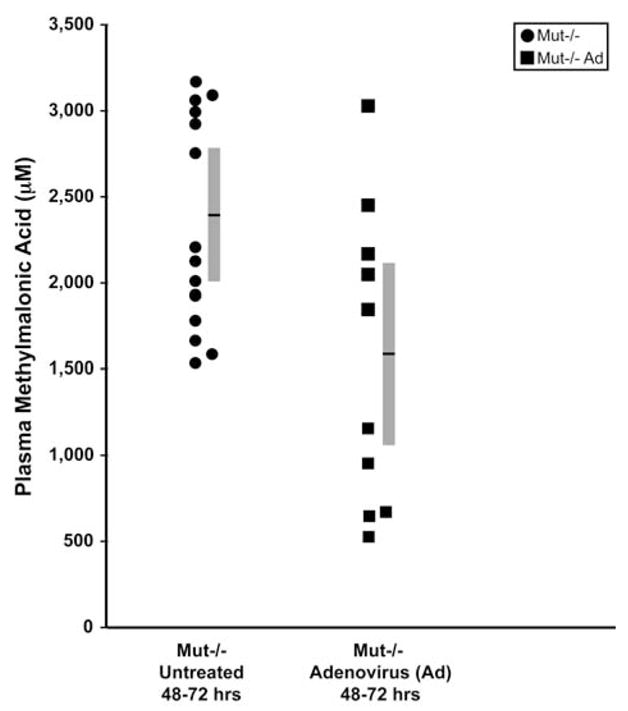
Methylmalonic acid levels at 48–72 hr of life from untreated Mut−/− mice (solid circles) and Mut−/− mice treated by hepatic injection of Ad-Mut-GFP adenovirus (solid squares). The 15 uninjected Mut−/− 48–72 hr animals had an average methylmalonic acid plasma concentration of 2318 μM (indicated by the horizontal black line within the gray bar, the ends of which indicate the 95% confidence intervals, falling between 2010 and 2626 μM), and served as an untreated control group. The 10 injected Mut−/− mice had an average methylmalonic acid plasma concentration of 1550 μM with 95% confidence intervals falling between 1008 and 2090 μM. The p value for comparison between the two groups, calculated by Student paired t test, was 0.015.
DISCUSSION
Murine models have been generated for two of the common human organic acidemias, propionic acidemia and methylmalonic acidemia, but have been difficult to manipulate. Both models feature almost immediate neonatal lethality in homozygote mutants, which has made basic investigations challenging (Miyazaki et al., 2001; Peters et al., 2003). Our initial experimental approach was to examine, in a proof-of-principle fashion, whether virus-mediated gene delivery could rescue the affected mice from uniform fatality in the neonatal period and extend the life span. We developed and tested an E1,E3-deleted adenovirus that expressed the murine methylmalonyl-CoA mutase gene in the E1 region under the control of the CMV promoter and, independently, eGFP from the E3 region under the control of the RSV promoter (Chandler et al., 2007). Intrahepatic delivery was rapid, taking only minutes to inject an average litter of mice, minimally stressful to the neonatal pups, and biologically effective. However, the nature of the intrahepatic injection procedure was variable and associated with general mortality because treated heterozygotes and wild-type animals also perished. Pathological investigations of the liver at a time when inflammation might be expected to be observed were unrevealing in the treated animals. The exact cause of mortality is unknown but was suspected to be related to the stress of the procedure, because survival on day 15 (Table 1) among the Ad-Mut-GFP-treated animals (54 of 77; 70.0%) was similar to that seen among the Ad-GFP-treated animals (19 of 26; 73%). Mechanistic explanations for the increased mortality will be important to examine in future studies.
A strong viral promoter driving the expression of the target gene was selected because we hypothesized that an immediate burst of methylmalonyl-CoA mutase expressed in the liver might provide enough enzyme to allow the neonatal pups to survive until weaning. The overall success of this approach was most obvious from the survival statistics, showing that >50% of the mutant mice that were treated by direct hepatic injection were alive at weaning, whereas uninjected controls (Fig. 2), mice that received the same virus injected in the skeletal muscle, and Ad-GFP-treated animals uniformly perished (Table 1).
During the period between injection and weaning, hepatically treated mutants were indistinguishable from heterozygote and wild-type littermates and had normal body weights, habitus, and behaviors. However, when the surviving Mut−/− mice were weaned and placed on regular chow, they slowly stopped growing, failed to gain weight, and eventually perished. One animal survived 8 months after a single viral treatment but most died shortly after weaning. The reason for the death is uncertain but was likely related to the expected diminution of enzymatic activity from dilution effects, immune responses, or silencing effects on viral gene expression (Coude et al., 1990; Guo et al., 1996; Connelly, 1999). Because adenovirus is a nonintegrating vector and the liver rapidly expands between the neonatal period and the time at weaning, dilution effects may be the most significant factor contributing to the diminution of MUT expression in the treated mice. Other variables, such as the change in diet when the animals were transitioned from milk to chow, were also likely contributory to the deaths seen after weaning.
To assess the efficacy of virus-mediated delivery, time course studies were conducted, using a validated qPCR assay to examine RNA expression; and Western analysis, using highly specific anti-mutase antibodies, to demonstrate virus-directed protein production. As might be expected for a low-volume, direct injection method, some variability in the degree of expression at the RNA and protein levels was observed (Fig. 3A and B). However, mRNA and protein levels exceeding those seen in wild-type animals were present in some treated mice (Fig. 3A, column A; and Fig. 3B, lane 3), whereas other injected mutants showed lower level expression but definite evidence of transcripts. As final support for the restoration of enzymatic activity in the livers of treated mice, plasma MMA levels were measured and showed a statistically significant decrease between treated and untreated mutants (Fig. 4). The lower methylmalonic acid levels seen in these mice indicate that significant hepatic methylmalonyl-CoA mutase activity was restored and was responsible for the lowered metabolite levels. It further suggests that the survival observed in the treated Mut−/− cohort likely correlates with more efficient adenoviral transduction and lower MMA levels, because roughly the same proportion of treated mutant mice in this study had lower metabolite levels when compared with survival at weaning. The fact that other regimens, including intramuscular injections, failed to produce survivors indicates that successful rescue gene delivery should be targeted to the liver in neonatal animals.
The demonstration of survival-based, adenovirus-mediated gene delivery to treat neonatal Mut−/− mice has implications for future treatment strategies. The studies described here prove that virus-mediated rescue of neonatal lethality in Mut−/− mice, and by extension, other lethal organic acidemia murine models, can be successfully accomplished. These results are surprising when the pleiotropic nature of the enzyme defect in MMA is considered (Wilkemeyer et al., 1993) and suggest that metabolic cooperativity of propionate metabolism (Wilkemeyer et al., 1992; Stankovics and Ledley, 1993) may contribute to the biological effects observed by transient restoration of metabolism in the hepatocyte. The rapidity and severity of symptom onset in this model differ when compared with other inborn error of metabolism models that have been treated successfully with integration viruses and transposons as gene delivery agents to achieve permanent correction, such as lysosomal storage disorders (Ponder et al., 2002), aminoacidopathies (Chen and Woo, 2005), and bilirubin metabolic defects (Nguyen et al., 2005). Such considerations led to the selection of an adenovirus in our initial proof-of-principle correction experiments because these vectors can be highly concentrated, have been demonstrated to have biological effects in other intermediary metabolic mouse models, such as ornithine transcarbamylase (OTC) deficiency (Stratford-Perricaudet et al., 1990; Ye et al., 1996), citrullinemia (Patejunas et al., 1998; Ye et al., 2000), and glycogen storage disorder type I (Nguyen et al., 2005), and can be easily engineered with strong heterologous expression cassettes. Our results encourage the application of other vector systems, if efficient in early transduction of hepatocytes, to treat Mut−/− mice. Other promoters and viral delivery systems, in particular adeno-associated viral serotype 8 vectors, might be especially effective in this application and will be the subject of future gene delivery studies in this murine model.
Adenovirus-mediated correction to extend the life span of Mut−/− mice has additional utility to generate older mice with severe methylmalonic acidemia and diminished or absent viral gene expression. Such mice have been used in pilot experiments to study the effects of dietary manipulations and provide an animal model of childhood methylmalonic acidemia that is analogous to the state of severely affected patients who have survived a neonatal crisis. Furthermore, rescued Mut−/− mice may also be useful to study the unusual pathology that is seen in MMA patients, such as pancreatitis, renal disease, and stroke syndrome, which have not been detected in neonatal animals.
Acknowledgments
R.J.C. and C.P.V. were supported, in part, by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Footnotes
AUTHOR DISCLOSURE STATEMENT: No competing financial interests exist.
References
- ALLEN RH, STABLER SP, SAVAGE DG, LINDENBAUM J. Elevation of 2-methylcitric acid I and II levels in serum, urine, and cerebrospinal fluid of patients with cobalamin deficiency. Metabolism. 1993;42:978–988. doi: 10.1016/0026-0495(93)90010-l. [DOI] [PubMed] [Google Scholar]
- CHACE DH, KALAS TA, NAYLOR EW. The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu Rev Genomics Hum Genet. 2002;3:17–45. doi: 10.1146/annurev.genom.3.022502.103213. [DOI] [PubMed] [Google Scholar]
- CHAKRAPANI A, SIVAKUMAR P, McKIERNAN PJ, LEONARD JV. Metabolic stroke in methylmalonic acidemia five years after liver transplantation. J Pediatr. 2002;140:261–263. doi: 10.1067/mpd.2002.121698. [DOI] [PubMed] [Google Scholar]
- CHANDLER RJ, VENDITTI CP. Genetic and genomic systems to study methylmalonic acidemia. Mol Genet Metab. 2005;86:34–43. doi: 10.1016/j.ymgme.2005.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDLER RJ, TSAI MS, DORKO K, SLOAN J, KORSON M, FREEMAN R, STROM S, VENDITTI CP. Adenoviral-mediated correction of methylmalonyl-CoA mutase deficiency in murine fibroblasts and human hepatocytes. BMC Med Genet. 2007;8:24. doi: 10.1186/1471-2350-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN L, WOO SL. Complete and persistent phenotypic correction of phenylketonuria in mice by site-specific genome integration of murine phenylalanine hydroxylase cDNA. Proc Natl Acad Sci USA. 2005;102:15581–15586. doi: 10.1073/pnas.0503877102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- CONNELLY S. Adenoviral vectors for liver-directed gene therapy. Curr Opin Mol Ther. 1999;1:565–572. [PubMed] [Google Scholar]
- COUDE M, CHADEFAUX B, RABIER D, KAMOUN P. Early amniocentesis and amniotic fluid organic acid levels in the prenatal diagnosis of organic acidemias. Clin Chim Acta. 1990;187:329–332. doi: 10.1016/0009-8981(90)90118-c. [DOI] [PubMed] [Google Scholar]
- DE BAULNY HO, BENOIST JF, RIGAL O, TOUATI G, RABIER D, SAUDUBRAY JM. Methylmalonic and propionic acidaemias: Management and outcome. J Inherit Metab Dis. 2005;28:415–423. doi: 10.1007/s10545-005-7056-1. [DOI] [PubMed] [Google Scholar]
- FENTON WA, ROSENBLATT DS. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver CR, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3897–3933. [Google Scholar]
- FENTON WA, GRAVEL RA, ROSENBLATT DS. Disorders of propionate and methylmalonate metabolism. In: Scriver CR, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2165–2192. [Google Scholar]
- FRENKEL EP, KITCHENS RL. Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats. Br J Haematol. 1975;31:501–513. doi: 10.1111/j.1365-2141.1975.tb00885.x. [DOI] [PubMed] [Google Scholar]
- GUO ZS, WANG LH, EISENSMITH RC, WOO SL. Evaluation of promoter strength for hepatic gene expression in vivo following adenovirus-mediated gene transfer. Gene Ther. 1996;3:802–810. [PubMed] [Google Scholar]
- HSUI JY, CHIEN YH, CHU SY, LU FL, CHEN HL, HO MJ, LEE PH, HWU WL. Living-related liver transplantation for methylmalonic acidemia: Report of one case. Acta Paediatr Taiwan. 2003;44:171–173. [PubMed] [Google Scholar]
- KAPLAN P, FICICIOGLU C, MAZUR AT, PALMIERI MJ, BERRY GT. Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl-CoA mutase deficiency. Mol Genet Metab. 2006;88:322–326. doi: 10.1016/j.ymgme.2006.04.003. [DOI] [PubMed] [Google Scholar]
- KASAHARA M, HORIKAWA R, TAGAWA M, UEMOTO S, YOKOYAMA S, SHIBATA Y, KAWANO T, KURODA T, HONNA T, TANAKA K, SAEKI M. Current role of liver transplantation for methylmalonic acidemia: A review of the literature. Pediatr Transplant. 2006;10:943–947. doi: 10.1111/j.1399-3046.2006.00585.x. [DOI] [PubMed] [Google Scholar]
- KAYLER LK, MERION RM, LEE S, SUNG RS, PUNCH JD, RUDICH SM, TURCOTTE JG, CAMPBELL DA, Jr, HOLMES R, MAGEE JC. Long-term survival after liver transplantation in children with metabolic disorders. Pediatr Transplant. 2002;6:295–300. doi: 10.1034/j.1399-3046.2002.02009.x. [DOI] [PubMed] [Google Scholar]
- MARCELL PD, STABLER SP, PODELL ER, ALLEN RH. Quantitation of methylmalonic acid and other dicarboxylic acids in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal Biochem. 1985;150:58–66. doi: 10.1016/0003-2697(85)90440-3. [DOI] [PubMed] [Google Scholar]
- MATSUI SM, MAHONEY MJ, ROSENBERG LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857–861. doi: 10.1056/NEJM198304143081501. [DOI] [PubMed] [Google Scholar]
- MIYAZAKI T, OHURA T, KOBAYASHI M, SHIGEMATSU Y, YAMAGUCHI S, SUZUKI Y, HATA I, AOKI Y, YANG X, MINJARES C, HARUTA I, UTO H, ITO Y, MULLER U. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- NAGARAJAN S, ENNS GM, MILLAN MT, WINTER S, SARWAL MM. Management of methylmalonic acidaemia by combined liver–kidney transplantation. J Inherit Metab Dis. 2005;28:517–524. doi: 10.1007/s10545-005-0517-8. [DOI] [PubMed] [Google Scholar]
- NGUYEN TH, BELLODI-PRIVATO M, AUBERT D, PICHARD V, MYARA A, TRONO D, FERRY N. Therapeutic lentivirus-mediated neonatal in vivo gene therapy in hyperbilirubinemic Gunn rats. Mol Ther. 2005;12:852–859. doi: 10.1016/j.ymthe.2005.06.482. [DOI] [PubMed] [Google Scholar]
- NICOLAIDES P, LEONARD J, SURTEES R. Neurological outcome of methylmalonic acidaemia. Arch Dis Child. 1998;78:508–512. doi: 10.1136/adc.78.6.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYHAN WL, GARGUS JJ, BOYLE K, SELBY R, KOCH R. Progressive neurologic disability in methylmalonic acidemia despite transplantation of the liver. Eur J Pediatr. 2002;161:377–379. doi: 10.1007/s00431-002-0970-4. [DOI] [PubMed] [Google Scholar]
- PATEJUNAS G, LEE B, DENNIS JA, HEALY PJ, REEDS PJ, YU H, FRAZER M, MULL B, WARMAN AW, BEAUDET AL, O’BRIEN WE. Evaluation of gene therapy for citrullinaemia using murine and bovine models. J Inherit Metab Dis. 1998;21(Suppl 1):138–150. doi: 10.1023/a:1005322010854. [DOI] [PubMed] [Google Scholar]
- PETERS H, NEFEDOV M, SARSERO J, PITT J, FOWLER KJ, GAZEAS S, KAHLER SG, IOANNOU PA. A knock-out mouse model for methylmalonic aciduria resulting in neonatal lethality. J Biol Chem. 2003;278:52909–52913. doi: 10.1074/jbc.M310533200. [DOI] [PubMed] [Google Scholar]
- PONDER KP, MELNICZEK JR, XU L, WEIL MA, O’MALLEY TM, O’DONNELL PA, KNOX VW, AGUIRRE GD, MAZRIER H, ELLINWOOD NM, SLEEPER M, MAGUIRE AM, VOLK SW, MANGO RL, ZWEIGLE J, WOLFE JH, HASKINS ME. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc Natl Acad Sci USA. 2002;99:13102–13107. doi: 10.1073/pnas.192353499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANKOVICS J, LEDLEY FD. Cloning of functional alpha propionyl CoA carboxylase and correction of enzyme deficiency in pccA fibroblasts. Am J Hum Genet. 1993;52:144–151. [PMC free article] [PubMed] [Google Scholar]
- STRATFORD-PERRICAUDET LD, LEVRERO M, CHASSE JF, PERRICAUDET M, BRIAND P. Evaluation of the transfer and expression in mice of an enzyme-encoding gene using a human adenovirus vector. Hum Gene Ther. 1990;1:241–256. doi: 10.1089/hum.1990.1.3-241. [DOI] [PubMed] [Google Scholar]
- VAN DER MEER SB, POGGI F, SPADA M, BONNEFONT JP, OGIER H, HUBERT P, DEPONDT E, RAPOPORT D, RABIER D, CHARPENTIER C, PARVY P, BARDET J, KAMOUN P, SAUDUBRAY JM. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr. 1994;125:903–908. doi: 10.1016/s0022-3476(05)82005-0. [DOI] [PubMed] [Google Scholar]
- VAN’T HOFF WG, DIXON M, TAYLOR J, MISTRY P, ROLLES K, REES L, LEONARD JV. Combined liver–kidney transplantation in methylmalonic acidemia. J Pediatr. 1998;132:1043–1044. doi: 10.1016/s0022-3476(98)70407-x. [DOI] [PubMed] [Google Scholar]
- VENDITTI CP. Methylmalonic acidemia. [(accessed November 2007)];GeneReviews at GeneTests (medical genetics information resource on the Internet) 2005 Available at www.genetests.org.
- VENDITTI CP, CHANDLER R, TSAI MS, WEHRLI N, DEERING R, KAESTNER K, GAIVER G, KAZAZIAN HH. Genetic and genomic systems to study methylmalonic acidemia (MMA) Mol Genet Metab. 2005;84:207–208. doi: 10.1016/j.ymgme.2005.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKEMEYER M, STANKOVICS J, FOY T, LEDLEY FD. Propionate metabolism in cultured human cells after over-expression of recombinant methylmalonyl CoA mutase: Implications for somatic gene therapy. Somat Cell Mol Genet. 1992;18:493–505. doi: 10.1007/BF01232646. [DOI] [PubMed] [Google Scholar]
- WILKEMEYER MF, ANDREWS ER, LEDLEY FD. Genomic structure of murine methylmalonyl-CoA mutase: Evidence for genetic and epigenetic mechanisms determining enzyme activity. Biochem J. 1993;296:663–670. doi: 10.1042/bj2960663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE X, ROBINSON MB, BATSHAW ML, FURTH EE, SMITH I, WILSON JM. Prolonged metabolic correction in adult ornithine transcarbamylase-deficient mice with adenoviral vectors. J Biol Chem. 1996;271:3639–3646. doi: 10.1074/jbc.271.7.3639. [DOI] [PubMed] [Google Scholar]
- YE X, WHITEMAN B, JEREBTSOVA M, BATSHAW ML. Correction of argininosuccinate synthetase (AS) deficiency in a murine model of citrullinemia with recombinant adenovirus carrying human AS cDNA. Gene Ther. 2000;7:1777–1782. doi: 10.1038/sj.gt.3301303. [DOI] [PubMed] [Google Scholar]