

Abstract
Proliferative retinal neovascularization occurring in response to ischemia is a common mechanism underlying many retinal diseases. In recent studies, retinal microglia have been shown to influence pathological neovascularization, likely through an exchange of cellular signals with associated vascular elements. CX3CR1 is a chemokine receptor located specifically on microglia; its ligand, CX3CL1 (also known as fractalkine or neurotactin) displays pro-angiogenic activity both in in vivo and in vitro. Discovering the regulatory role, if any, that CX3CR1 signaling may have in ischemic retinopathy will shed light on the molecular nature of microglial-vascular interactions and clarify potential targets for future therapy. In this study, we examined this question by inducing and comparing ischemic vascular changes in transgenic mice in which CX3CR1 signaling is either preserved or ablated. Using a well-known oxygen-induced retinopathy (OIR) model, we induced ischemic retinopathy in transgenic mice in which the gene for CX3CR1 has been replaced by green fluorescent protein (GFP) and their wild type controls. CX3CR1+/+, CX3CR1+/GFP, and CX3CR1GFP/GFP transgenic mice were exposed to 75% oxygen for 5 days starting from postnatal day (P) 7, and then transferred back to room air. At P12 and P17, the extents of vascular repair and neovascularization, and associated changes in retinal microglia distribution, were quantified and compared between mice of different genotypes. Neuronal loss in the retina following ischemia was also evaluated in paraffin sections. Our results show that: (1) CX3CR1 signaling is not required for normal vascular, microglial, and neuronal development in the retina in the first postnatal week, (2) the processes of retinal vascular repair and neovascularization following ischemia occur similarly with and without CX3CR1 signaling, (3) microglia redistribution in the retina and their association with vascular elements occurring concurrently is independent of CX3CR1, and (4) CX3CR1 does not influence the extent of neuronal cell loss in the retina following ischemia. Taken together, our findings indicate that the regulatory signals exchanged between microglia and vascular elements in the ischemic retinopathy animal model are unlikely to involve CX3CR1. These results have implications on therapeutic approaches to, pathological neovascularization involving the modulation of chemokine signaling in general, and the regulation of CX3CR1 signaling specifically.
Keywords: retina, microglia, ischemia, neovascularization, CX3CR1, vascular repair
INTRODUCTION
Retinal diseases resulting in vision loss, such as diabetic retinopathy, retinopathy of prematurity, and vein occlusions, involve the formation of abnormal blood vessels in a process called retinal neovascularization, developing as a pathological response to tissue ischemia. Recent studies have demonstrated that resident immune cells in the form of microglia or macrophages are involved in mediating these vascular changes in the retina; microglia are found intimately associated with retinal vessels during both vasculogenesis early in life during normal development.(Checchin et al., 2006) as well as during vascular repair and neovascularization under pathological conditions.(Checchin et al., 2006; Ritter et al., 2006; Yoshida et al., 2003) In models of ischemic retinopathy, retinal microglia cell density are dramatically increased (Davies et al., 2006), and when these cells are depleted artificially, increased vascular pathology ensues(Checchin et al., 2006; Ritter et al., 2006). Myeloid progenitors, when transplanted into ischemic retinas, were also found to differentiate into perivascular microglia and concurrently ameliorate vascular pathology.(Ritter et al., 2006) These findings strongly implicate retinal microglia as close associates in the pathogenesis of retinal neovascularization, and microglia-vascular interactions may represent a target for therapeutic approaches to neovascular retinal disease (Dorrell et al., 2007).
Discovery of the identity and nature of cellular signals exchanged between retinal microglia and vascular elements relevant to vascular repair and neovascularization is an ongoing area of investigation. In multiple organ systems, tissues stimulate vascular growth by producing a variety of factors that mobilize myeloid cells and encourage their extravasation into target tissues and their participation in neovascular processes (Avraamides et al., 2008; Sunderkotter et al., 1994). In particular, chemokines have been implicated in regulating retinal neovascularization. Levels of CCL2 (also termed MCP-1) and CCL4 (or MIP-1α), for example, are increased in the retina following ischemic retinal injury (Davies et al., 2006; Yoshida et al., 2003) and may play a role in regulating the regression of neovascular vessels (Davies et al., 2008). Although the recruitment of microglia into areas of retinal hypoxia and the interaction between microglia and vascular elements are likely critical in regulating vascular repair and neovascularization, knowledge on the repertoire of relevant chemokines that are involved in this process is still incomplete.
Chemokine signaling mediated by CX3CL1 through its receptor CX3CR1 is one such candidate for microglial-vascular interactions in retinal neovascularization. CX3CR1, previously shown to regulate microglia migration in the CNS and mediate neurotoxicity of microglia (Cardona et al., 2006), also appears to promote inflammatory angiogenesis in a model of rheumatoid arthritis (Murphy et al., 2008; Volin et al., 2001). In ocular tissues, there have been mixed effects reported on the regulatory role of CX3CL1-CX3CR1 signaling in neovascularization; in one study, mice lacking CX3CR1 signaling showed decreased macrophage accumulation and less neovascularization in response to alkali-injury in the cornea (Lu et al., 2008), while in another study, the same CX3CR1 deficiency conversely resulted in greater subretinal microglial accumulation and more choroidal neovascularization in response to laser-induced injury (Combadiere et al., 2007).
In the present study, we investigated the role of CX3CL1-CX3CR1 signaling within the retina in regulating vascular repair and neovascularization, as well as microglia distribution in a well-described model of ischemic retinopathy, the oxygen-induced retinopathy (OIR) model (Smith et al., 1994). Using transgenic mice in which the gene for enhanced green fluorescent protein (eGFP) has replaced the coding sequence for one (CX3CR1+/GFP) or both (CX3CR1GFP/GFP) alleles for CX3CR1, we first examined the impact of CX3CR1-deficiency on retinal microglial and vascular development in the first postnatal week prior to entry into the OIR model. Subsequently, we subjected the transgenic animals to the OIR model and evaluated the effect of CX3CR1-deficiency on the processes of microglial redistribution, vascular repair, and retinal neovascularization in the aftermath of oxygen-induced ischemia. Our results indicate that the absence of CX3CR1 signaling did not affect either microglial or vascular development of the retina in the first postnatal week. Quantitative analysis of the vascular changes induced in the OIR model also showed no significant differences in the amount of vascular repair or retinal neovascularization between animals in which CX3CR1 signaling was either present or ablated. CX3CR1 deficiency also did not affect the changes in number or the distribution patterns of retinal microglia occurring concurrently with the vascular changes. Taken together, our results show that in the retina, regional recruitment of microglia in response to ischemia likely occurs independently of CX3CR1. In addition, the pro- and anti-angiogenic signals exchanged between microglia and vascular elements in the regulation of vascular repair and neovascularization in the present model are also not likely to involve CX3CR1. Our findings have significant implications on the design of chemokine-based therapies for neovascularization (Dorrell et al., 2007) and the applicability of CX3CR1-based therapies presently under development (Charo and Ransohoff, 2006).
MATERIALS AND METHODS
Experimental Animals
Homozygous CX3CR1GFP/GFP mice, which are CX3CR1 deficient, were generated on a C57BL/6 background as previously described (Jung et al., 2000) and were obtained from The Jackson Laboratory (Bar Harbor, Maine). These mice were bred to wild type C57BL/6 mice to generate heterozygous CX3CR1+/GFP mice. CX3CR1+/GFP mice were also crossed with each other in order to generate multiple genotypes in a single litter (CX3CR1GFP/GFP, CX3CR1+/GFP, and wild type CX3CR1+/+). As in these transgenic mice, the eGFP reporter gene replaces the CX3CR1 gene, CX3CR1GFP/GFP and CX3CR1+/GFP animals will be referred in subsequent sections as CX3CR1−/− and CX3CR1+/− animals respectively. Experiments were conducted according to protocols approved by a local Institutional Animal Care and Use committee.
Oxygen-induced retinopathy (OIR) model
Processes of retinal vessel degeneration, revascularization, and pathological neovascularization were induced in the OIR model as described previously (Smith et al., 1994). Briefly, P7 mice pups were transferred from room air to an environment of 75% oxygen until age P12. Retinal vessel degeneration was assessed at P12 after significant vascular obliteration has occurred. The remaining pups were transferred to room air at P12, whereupon the retinal areas lacking vascularization experience ischemia and induce processes of vascular repair and neovascularization. The extents of these processes were assessed in the transgenic animals at P17, when the neovascular response is most prominent.
Retinal Tissue Preparation
Eyecups were harvested from transgenic animals at different developmental ages (at P0 and P7, prior to the OIR model; and at P12 and P17, after induction in the OIR model), fixed in 4% paraformaldehyde for 1 hour, and transferred into buffered PBS containing 0.2% Tween 20 (Sigma),,0.5% bovine serum albumin, and sodium azide. Retinal vasculature was labeled by incubation with isolectin B4 conjugated with Alexa 568 (1:100; Invitrogen). For retinal flat-mount preparations, whole retinas were laid flat after radial relaxing incisions were made and then mounted on slides and cover-slipped. For vibratome sections, retinal tissue was embedded in 7% agarose and sectioned with a vibrating microtome (Leica, VT1000S). Retinal microglia were visualized in retinas of transgenic animals by the selective expression of green fluorescent protein in microglia.. In wild type animals, retinal microglia were visualized by immunohistochemical staining with Iba-1 (Wako Chemicals, Richmond, VA). 4′,6-diamidino-2-phenylindole (DAPI) (1:1000; Invitrogen) was used as a nuclear marker. For paraffin sections, fixed eyecups were embedded in paraffin, and 7 μm-thick sagittal sections were cut parallel to the optic nerve and stained with hematoxylin and eosin.
Reverse Transcription-PCR Amplification of CX3CR1
Cellular RNA was prepared from isolated retinal tissue using the RNeasy kit (Qiagen, Valencia, CA). Reverse transcription was performed using a cDNA synthesis kit (Ambion, Austin, TX), followed by amplification of cDNA using the following primer sequences for 38 cycles: 5′ttcacgttcggtctggtggg 3′ (forward) and 5′ ggttcctagtggagctaggg3′ (reverse). Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as an internal control to normalize the amount of mRNA between tissues of different genotypes.
Imaging of Vascular Structures and Microglia Distribution
High-resolution, 3-dimensional imaging of retinal vessels and microglia was performed using a confocal microscope (SP2; Leica) in retinal flatmounts and vibratome sections. Multiplane Z series image stacks were collected at a 1024×1024 pixel resolution, enabling the examination of vessel and microglia distributions at different levels of the retina. In addition, images for the quantitative analysis of vascular structures and microglia were obtained by capturing images using a Spot RT digital camera (Spot Diagnostic Instruments, Burroughs, MI). Whole retinal montages were generated from the individual images using automated alignment algorithms (Adobe Photoshop, San Jose, CA).
Quantification of Vascular Structures and Microglia Distribution
Quantifications of vasculature structures and microglia distribution were performed in a masked fashion without knowledge of the genotype of the animal analyzed. The extents of vascular obliteration at P12 and P17 were analyzed using the method described by Banin et al.(2006). Briefly, the areas of avascular zones in the central retina were outlined on retinal images using the freehand selection tool of the image analysis software (Adobe Photoshop) and the total area was quantified. The extent of retinal neovascularization was evaluated by measuring the area of neovascular tufts in retinal images; these were highlighted using the “magic wand” tool in the imaging software as a guide. As the neovascular tufts stain more strongly with conjugated lectin than the surrounding normal vasculature, this localized thresholding function was useful in a semi-automated delineation of the neovascular tufts.
Quantification of microglia cell density was performed in retinal flatmounts in 3 different vascular areas: avascular zones, areas of normal vascular structures, and areas of neovascularization. Images were taken at different retinal levels and all microglia in each of the 3 areas were counted and expressed in number of microglia per unit area of flatmounted retina.
Statistical analysis
Results in the figures were expressed as mean ± SEM. Statistical comparisons were performed using one-way ANOVAs and unpaired t-tests employing statistical software (GraphPad Prism, GraphPad Software, La Jolla, CA). Error bars in the graphs indicate standard error.
RESULTS
Early retinal development in the presence and absence of CX3CR1
In order to evaluate the contribution of CX3CR1 function to vascular changes in OIR model at P12 and P17, we first evaluated retinal development in neonatal CX3CR1+/− and CX3CR1−/− at P0 and P7 prior to the OIR intervention. At P0, vasculogenesis in both genotypes was limited to the inner central retina as a circular ring of peripapillary vessels, with retention of a prominent hyaloid vasculature (Fig. 1A and B). By P7, vascular development extended to the peripheral retina, with vessels extending into the deeper retinal layers (Fig. 1E–H). Early vascular patterning in the first postnatal week was not noticeably distinct between CX3CR1+/− and CX3CR1−/− retinas and occurred similarly as that occurring in wild type animals.(Connolly et al., 1988)
Figure 1.

Vascular development and microglia distribution occurs normally in CX3CR1+/− and CX3CR1−/− animals. At postnatal day (P) 0, the hyaloid vasculature is seen prominently and vasculogenesis in the retina extends only to the superficial central retina in both genotypes (A, B). Microglia are concentrated at the optic nerve and the retinal ganglion cell layer (GCL), with scattered microglia migrating into the neuroblastic layer (NBL) (C, D). At P7, vascular development progresses towards the peripheral and outer layers of the retina (E, F). Microglia are found in close association with retinal vessels and in a laminated distribution in the GCL, inner plexiform layer (IPL), inner nuclear layer (INL), and outer plexiform layer (OPL), similarly between both genotypes (G, H).
Early patterns of microglia distribution in the retina also appeared similar between CX3CR1+/− and CX3CR1−/− animals, akin to that seen in wild type animals.(Santos et al., 2008) At P0, retinae of both genotypes demonstrated a similar distribution of microglia in the hyaloid vasculature, optic nerve, ganglion cell layer, and neuroblastic layers (Fig. 1A–D). At this time point, microglia possessed a sparsely ramified morphology, and appeared to migrate from the ganglion cell layer where they are present in higher numbers to the deeper neuroblastic layer where they are more sparsely found. At P7, microglia acquired a more ramified morphology and were found in the deeper inner plexiform layer, inner nuclear layer, and outer plexiform layer in close proximity to the developing vasculature, but not in the outer nuclear layer or in the subretinal space. In CX3CR1+/− and CX3CR1−/− animals, the development of retinal plexiform and cell body lamina at P0 and P7 also occurred on a normal developmental time frame.
In young adult animals (aged 6–8 weeks) where vascular, microglial, and neuronal development are complete, CX3CR1+/− and CX3CR1−/− animals also displayed normal vascular structure, normal microglial morphology and distribution, as well normal retinal lamination and thickness which were similar to each other and to that in wild type animals (data not shown).
Vascular obliteration in the OIR model in the presence and absence of CX3CR1
After confirming that P7 CX3CR1+/− and CX3CR1−/− animals entering into the OIR model were similar in their retinal vascular and microglial anatomy, we evaluated the role of CX3CR1 on the subsequent processes of vascular change induced by the OIR model. We also confirmed using rt-PCR that there is an presence and absence of CX3CR1 mRNA transcripts in CX3CR1+/− and CX3CR1−/− retinas respectively (Fig. 2A). After 5 days in hyperoxic (75% oxygen) conditions, retinas were harvested from CX3CR1+/+, CX3CR1+/− and CX3CR1−/− animals (litter mates of a cross between CX3CR1+/− animals) at age P12, stained to reveal the retinal vasculature, and flatmounted. Central areas of vascular obliteration were observed in all genotypes (Fig. 2B–D), and the total quantified avascular areas were highly similar and not statistically distinct (p>0.05 for all comparisons) (Fig. 2E). These avascular areas, when expressed as a percentage of total retinal area, were also statistically similar between genotypes (data not shown). Statistical comparisons between animals of different genotypes originating from the same litter (i.e. offspring of a CX3CR1+/− x CX3CR1+/− cross) were also separately performed and did not reveal statistically significant differences were found between genotypic groups (p>0.05 for all comparisons, data not shown).
Figure 2.

Vascular obliteration in oxygen-induced retinopathy occurs to similar extents in CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− animals. (A) Reverse transcription-PCR analysis of CX3CR1 transcription in experimental animals, confirming the absence of CX3CR1 mRNA in CX3CR1−/− animals. GADPH amplification reflected equal retrotranscription of the RNA (B, C, D) Representative retinal flatmounts at P12 from the 3 genotypes, showing the central areas of vascular obliteration (outlined in yellow). (E) Quantifications of the central avascular areas CX3CR1+/+ (n = 41 eyes), CX3CR1+/−, (n = 42 eyes), and CX3CR1−/−, (n = 39 eyes) in P12 animals. All pair wise comparisons were not statistically distinct (p>0.05).
Vascular repair and neovascularization in the OIR model in the presence and absence of CX3CR1
At P17, 5 days after returning to conditions of room air, retinas from CX3CR1+/+, CX3CR1+/− and CX3CR1−/− animals were similarly harvested, stained, and flat-mounted. Compared to P12 animals, smaller avascular areas were observed centrally, indicating that the central retina had undergone interval vascular repair (Fig. 3A–C). Total central avascular areas, when quantified, were not statistically different between genotypes (p>0.05 for all comparisons) (Fig. 3D). At this age, all examined retinal flatmounts also exhibited prominent retinal neovascularization in the form of tufts of vessels rising above the inner limiting membrane (Fig. 3E–G). Quantification of the total areas occupied by the neovascular tufts also revealed similar extents of neovascular change between CX3CR1+/+, CX3CR1+/− and CX3CR1−/− animals (p>0.05 for all comparisons) (Fig. 3H).
Figure 3.

Vascular repair, and pathological neovascularization in oxygen-induced retinopathy occurs to similar extents in CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− animals. (A, B, C) Representative retinal flatmounts at P17 from 3 genotypes, showing the area of vascular obliteration (outlined in yellow). (D) Quantifications of the summed avascular areas in P17 CX3CR1+/+ (n = 32 eyes), CX3CR1+/− (n = 53 eyes), and CX3CR1−/− (n = 42 eyes) animals were not statistically distinct for all paired comparisons (p>0.05). (E, F, G) Representative retinal flatmounts at P17 from 3 genotypes, showing the distribution of neovascular vessel tufts (outlined in pink). (H) Quantifications of the summed areas of neovascular changes in P17 CX3CR1+/+ (n = 32 eyes), CX3CR1+/− (n = 53 eyes), and CX3CR1−/−, (n = 42 eyes) animals were not statistically distinct for all paired comparisons (p>0.05).\
In the OIR model, retinal hypoxia resulting from vascular obliteration has been demonstrated to cause retinal neuronal cell apoptosis and cell loss, as evidenced by a thinning of the inner nuclear layer at P17.(Sennlaub et al., 2001) We examined whether the presence or absence of CX3CR1 contributed to a difference in the degree of neuronal cell loss. Evaluation of retinal thickness and lamination in paraffin sections demonstrated that the interval thinning of the INL from P12 to P17 occurred to similar extents in CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− retinas, with approximately equal residual INL thickness in equivalent areas of the retina, with no other systematic differences in retinal lamination between the genotypes (Fig. 4).
Figure 4.

Inner retinal cell loss following oxygen-induced retinopathy in CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− animals. Retinal degeneration and thinning of the inner nuclear layer (INL) occurring between P12 and P17 (top versus bottom panels) following ischemic retinopathy is comparable between the genotypes.
Microglia number and distribution in the presence and absence of CX3CR1
As CX3CR1 signaling has previously been implicated in microglia migration and distribution (Cardona et al., 2006; Combadiere et al., 2007), and changes in retinal microglial cell density are induced by ischemic changes (Davies et al., 2006; Yoshida et al., 2003), we examined microglia number and distribution in CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− retinas following ischemia induction in the OIR model. At P12, when animals emerge from hyperoxia into normal room air, retinal microglia are found widely and evenly distributed across the retina in both vascularized and avascular zones (Fig. 5A–F). This uniform distribution is similarly observed in the retinas of all genotypes; quantification of microglial cell density separately in avascular areas and areas of preserved vascularization did not reveal any statistical differences between retinal zones or between genotypes (Fig. 6A, F).
Figure 5.

Distribution of retinal microglia in oxygen-induced retinopathy at P12 and P17. At P12, microglia are distributed uniformly across the retina from center to periphery (A, C, E) in avascular and vascular areas of the retina (B, D, F) in CX3CR1+/+, CX3CR1+/−, and CX3CR1+/− animals. At P17, retinal microglia become densely aggregated (H,K,N) in areas of neovascularization (G, J, M) on the surface of the tufts themselves as shown in the yellow areas in composite panels (I, L, O). At both P12 and P17, patterns of microglia distribution and the nature of microglia-vessel associations were similar between animals of all 3 genotypes.
Figure 6.
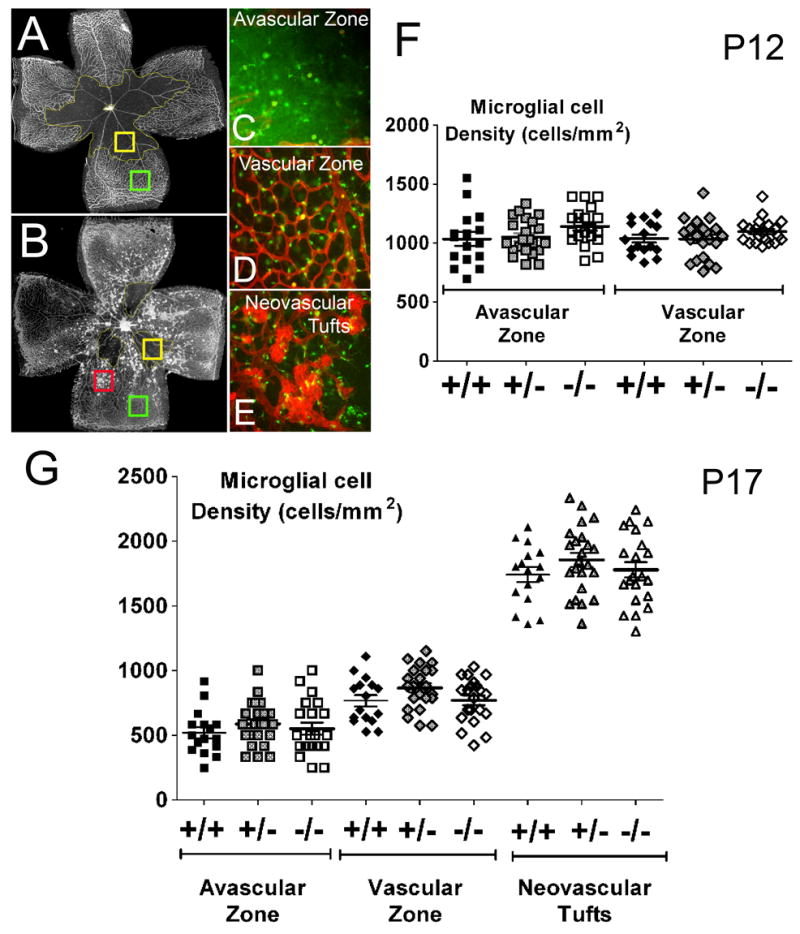
Quantitation of microglia distribution in oxygen-induced retinopathy. At P12 and P17, microglial cell density was quantified separately in avascular areas (A and B, yellow box; inset shown in C), areas with normal vasculature (A and B, green box; inset shown in D), and in areas of neovascularization (B, red box; inset shown in E). (F) At P12, overall microglia cell density was uniform between avascular and vascular zones, and similar between CX3CR1+/+ (n = 16 eyes, black symbols), CX3CR1+/− (n = 20 eyes, gray symbols), and CX3CR1−/− (n = 22 eyes, white symbols) animals (p >0.05). (G) At P17, microglia cell densities were decreased in avascular zones but markedly elevated in areas of neovascularization, indicating a redistribution of microglia to sites of vascular repair and pathological neovascularization. Statistical comparisons between microglia density in avascular zones, vascular zones, and neovascular zones showed significant (p<0.05) differences. The extent of microglia redistribution was similar between CX3CR1+/+ (n = 16 eyes, black symbols), CX3CR1+/− (n = 23 eyes, gray symbols), and CX3CR1−/− (n = 21 eyes, white symbols) animals (p >0.05).
At P17, when vascular repair and neovascularization are underway, the uniform distribution of retinal microglia seen earlier was replaced by a markedly non-uniform pattern, with large numbers of microglia aggregating in and around the neovascular tufts on the vitreal surface of the retina (Fig. 5G–O), and more sparsely in the residual avascular areas. In the areas of neovascular tufts, three-dimensional analyses of confocal images confirm the close association of retinal microglia to the surface of neovascular vessels in a perivascular location. Microglia were also present in the outer retinal layers but were at a lower density compared to that in the inner retina (data not shown). Similar association between microglia and vascular elements were observed in retinas of all 3 genotypes. Quantification of microglial cell densities by region according to avascular areas, areas with normal vascularization, and areas with neovascular tufts confirmed that microglial cell densities were lowest in the avascular zone, significantly higher in the normally vascularized zones, and markedly elevated in the neovascular zones (Fig. 6G) (p<0.05 for all comparisons). No statistical differences however were found in microglial cell densities between equivalent areas between CX3CR1+/+, CX3CR1+/−, and CX3CR1−/− retinas (p>0.05 for all comparisons). Overall microglia cell density, averaged across all retina areas, was also found to be statistically similar between the 3 genotypes (p>0.05, data not shown).
DISCUSSION
In this study, we addressed the role of the chemokine receptor CX3CR1 in vascular repair and neovascularization in ischemic retinopathy. Its only known ligand, CX3CL1, is highly expressed in vascular endothelial cells but also found at lower and more diffuse levels throughout the retina.(Silverman et al., 2003) CX3CR1 is found expressed specifically in microglia (Combadiere et al., 2007), but not in CNS neurons, macroglia, or astrocytes (Cardona et al., 2006). Given the above expression pattern, as well as the close association between microglia and vascular elements during normal vascular development (Checchin et al., 2006) and pathological neovascularization (Davies et al., 2006; Ritter et al., 2006), CX3CR1 appeared as a candidate chemokine signal in the microglial-mediated regulation of pathological vascular disease. In models of rheumatoid arthritis, CX3CL1 has been demonstrated to act as a potent pro-angiogenic agent both in vitro and in vivo.(Volin et al., 2001) In ocular tissues, the pro-angiogenic nature of CX3CL1-CX3CR1 signaling has also been reported; in a recent study, You et al., (2007) described that administration of CX3CL1 in a corneal pocket assay increased corneal neovascularization while administration of anti-CX3CL1 antibody in an OIR model decreased retinal neovascularization. Conversely, another report(Lu et al., 2008) described that topical application of CX3CL1 inhibited corneal neovascularization and that in the absence of CX3CR1 signaling, CX3CR1-deficient mice instead displayed enhanced corneal neovascularization. Also, CX3CR1 deficiency was reported to result in a greater degree of choroidal neovascularization in a laser-induced model (Combadiere et al., 2007) and its absence over time may facilitate the accumulation of microglia in the subretinal space and encourage the formation of age-related choroidal neovascularization Chan et al., 2008; Tuo et al., 2007). As such, the contribution of CX3CR1 signaling to ocular neovascularization remains uncertain, differing between various tissue contexts.
In this report, we evaluated the contribution of CX3CR1-mediated signaling to the regulation of pathological retinal neovascularization in the OIR model by careful quantitation of the vascular phenotype in CX3CR1-deficient mice. We first verified that CX3CR1-deficiency did not result in any developmental vascular or microglial changes in the first postnatal week prior to entry into the OIR model. Next, we performed masked quantitation of different aspects of vascular change using previously verified methodology (Banin et al., 2006) in multiple experimental animals, taking into account variations that can occur within, as well as between, mouse litters. In addition, we examined, in a quantitative and area-specific manner, whether changes in microglial cell distribution occurring between P12 and P17 were influenced by the presence or absence of CX3CR1 signaling. The aggregation and depletion of microglia have each been associated with pro-angiogenic (Combadiere et al., 2007; Espinosa-Heidmann et al., 2003; Sakurai et al., 2003) as well as anti-angiogenic effects (Apte et al., 2006; Lu et al., 2008; Ritter et al., 2006). In addition, CX3CR1-deficiency in CNS microglia has been associated with abnormal microglial homing, migration, and distribution.(Cardona et al., 2006; Chinnery et al., 2007; Raoul et al., 2008) For these reasons, the nature of microglia redistribution produced by the OIR model in the presence and absence of CX3CR1 signaling was also of interest and relevant to the understanding of pathological neovascularization.
Our results indicate that within the confines of the present model, CX3CR1 signaling plays no detectable role in influencing microglial redistribution within the retina following ischemia or changing the physical association that microglia have with vascular elements during pathological neovascularization. The extent of vascular repair and neovascular changes induced by the OIR model also did not vary with the presence or absence of CX3CR1. Although CX3CR1-deficiency had been associated with increased neurotoxicity and degeneration in other experimental systems (Cardona et al., 2006; Combadiere et al., 2007), we also did not find any differences in ischemia-related inner nuclear cell loss between CX3CR1+/− and CX3CR1−/− animals. Although there are likely multiple cross interactions, it is possible that the regulation of neovascular changes induced primarily in response to retinal ischemia may differ in key ways to changes induced in response to inflammation. In retinal ischemia, oxygen-regulated growth factors, such as erythropoietin and vascular endothelial growth factor, may have a primary influence on endothelial cell proliferation, cytoprotection of neurons, and microglial regulation.(Chong et al., 2002; Li et al., 2006; Noguchi et al., 2007) CX3CL1-CX3CR1 signaling, which is upregulated in chronic inflammation (Fang et al., 2005; Hughes et al., 2002), may have a minimal effect in situations where vascular changes are not directly prompted by inflammation. In support of this, vascular maturation and microglial migration and distribution in the retinal and the uvea that occur during normal development in the absence of inflammation are uninfluenced by the presence or absence of CX3CR1.(Kezic et al., 2008) With the advent of potential novel therapies involving the manipulation of chemokine signaling, knowledge about the effect, or lack of effect, of specific chemokines in each clinical situation is important for both efficacy and safety concerns. Additional studies examining the relative contributions of different chemokines in mediating vascular versus microglial changes in retinal vascular diseases will be critical to a basic understanding of microglial-vascular interactions and in their translation into novel therapeutic approaches.
Acknowledgments
The authors would like to acknowledge Drs. Maria Campos and Jen-yue Tsai in the NEI Biological Imaging Core and the staff in the NEI Histology Core for technical assistance, and Katharine Liang and Yunqing Wang for critical comments on the manuscript. This work is supported by the National Eye Institute, Intramural Division.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apte RS, Richter J, Herndon J, Ferguson TA. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Med. 2006;3:e310. doi: 10.1371/journal.pmed.0030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008 doi: 10.1038/nrc2353. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin E, Dorrell MI, Aguilar E, Ritter MR, Aderman CM, Smith AC, Friedlander J, Friedlander M. T2-TrpRS inhibits preretinal neovascularization and enhances physiological vascular regrowth in OIR as assessed by a new method of quantification. Invest Ophthalmol Vis Sci. 2006;47:2125–2134. doi: 10.1167/iovs.05-1096. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Huang D, Sasse ME, Ransohoff RM. Isolation of murine microglial cells for RNA analysis or flow cytometry. Nat Protoc. 2006;1:1947–1951. doi: 10.1038/nprot.2006.327. [DOI] [PubMed] [Google Scholar]
- Chan CC, Ross RJ, Shen D, Ding X, Majumdar Z, Bojanowski CM, Zhou M, Salem N, Jr, Bonner R, Tuo J. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S. Potential role of microglia in retinal blood vessel formation. Invest Ophthalmol Vis Sci. 2006;47:3595–3602. doi: 10.1167/iovs.05-1522. [DOI] [PubMed] [Google Scholar]
- Chinnery HR, Ruitenberg MJ, Plant GW, Pearlman E, Jung S, McMenamin PG. The chemokine receptor CX3CR1 mediates homing of MHC class II-positive cells to the normal mouse corneal epithelium. Invest Ophthalmol Vis Sci. 2007;48:1568–1574. doi: 10.1167/iovs.06-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Angiogenesis and plasticity: role of erythropoietin in vascular systems. J Hematother Stem Cell Res. 2002;11:863–871. doi: 10.1089/152581602321080529. [DOI] [PubMed] [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SE, Hores TA, Smith LE, D’Amore PA. Characterization of vascular development in the mouse retina. Microvasc Res. 1988;36:275–290. doi: 10.1016/0026-2862(88)90028-3. [DOI] [PubMed] [Google Scholar]
- Davies MH, Eubanks JP, Powers MR. Microglia and macrophages are increased in response to ischemia-induced retinopathy in the mouse retina. Mol Vis. 2006;12:467–477. [PubMed] [Google Scholar]
- Davies MH, Stempel AJ, Powers M. MCP-1 Deficiency Delays Regression of Pathologic Retinal Neovascularization in a Model of Ischemic Retinopathy. Invest Ophthalmol Vis Sci. 2008 doi: 10.1167/iovs.07-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell M, Uusitalo-Jarvinen H, Aguilar E, Friedlander M. Ocular neovascularization: basic mechanisms and therapeutic advances. Surv Ophthalmol. 2007;52(Suppl 1):S3–19. doi: 10.1016/j.survophthal.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3586–3592. doi: 10.1167/iovs.03-0038. [DOI] [PubMed] [Google Scholar]
- Fang IM, Lin CP, Yang CM, Chen MS, Yang CH. Expression of CX3C chemokine, fractalkine, and its receptor CX3CR1 in experimental autoimmune anterior uveitis. Mol Vis. 2005;11:443–451. [PubMed] [Google Scholar]
- Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia. 2002;37:314–327. [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kezic J, Xu H, Chinnery HR, Murphy CC, McMenamin PG. Retinal microglia and uveal tract dendritic cells and macrophages are not CX3CR1 dependent in their recruitment and distribution in the young mouse eye. Invest Ophthalmol Vis Sci. 2008;49:1599–1608. doi: 10.1167/iovs.07-0953. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Microglial integrity is maintained by erythropoietin through integration of Akt and its substrates of glycogen synthase kinase-3beta, beta-catenin, and nuclear factor-kappaB. Curr Neurovasc Res. 2006;3:187–201. doi: 10.2174/156720206778018758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Li L, Kuno K, Wu Y, Baba T, Li YY, Zhang X, Mukaida N. Protective roles of the fractalkine/CX3CL1-CX3CR1 interactions in alkali-induced corneal neovascularization through enhanced antiangiogenic factor expression. J Immunol. 2008;180:4283–4291. doi: 10.4049/jimmunol.180.6.4283. [DOI] [PubMed] [Google Scholar]
- Murphy G, Caplice N, Molloy M. Fractalkine in rheumatoid arthritis: a review to date. Rheumatology (Oxford) 2008 doi: 10.1093/rheumatology/ken197. [DOI] [PubMed] [Google Scholar]
- Noguchi CT, Asavaritikrai P, Teng R, Jia Y. Role of erythropoietin in the brain. Crit Rev Oncol Hematol. 2007;64:159–171. doi: 10.1016/j.critrevonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul W, Keller N, Rodero M, Behar-Cohen F, Sennlaub F, Combadiere C. Role of the chemokine receptor CX3CR1 in the mobilization of phagocytic retinal microglial cells. J Neuroimmunol. 2008 doi: 10.1016/j.jneuroim.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Ritter MR, Banin E, Moreno SK, Aguilar E, Dorrell MI, Friedlander M. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J Clin Invest. 2006;116:3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44:3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- Santos AM, Calvente R, Tassi M, Carrasco MC, Martin-Oliva D, Marin-Teva JL, Navascues J, Cuadros MA. Embryonic and postnatal development of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- Sennlaub F, Courtois Y, Goureau O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest. 2001;107:717–725. doi: 10.1172/JCI10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman MD, Zamora DO, Pan Y, Texeira PV, Baek SH, Planck SR, Rosenbaum JT. Constitutive and inflammatory mediator-regulated fractalkine expression in human ocular tissues and cultured cells. Invest Ophthalmol Vis Sci. 2003;44:1608–1615. doi: 10.1167/iovs.02-0233. [DOI] [PubMed] [Google Scholar]
- Smith LE, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J Leukoc Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan CC. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volin MV, Woods JM, Amin MA, Connors MA, Harlow LA, Koch AE. Fractalkine: a novel angiogenic chemokine in rheumatoid arthritis. Am J Pathol. 2001;159:1521–1530. doi: 10.1016/S0002-9440(10)62537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Yoshida A, Ishibashi T, Elner SG, Elner VM. Role of MCP-1 and MIP-1alpha in retinal neovascularization during postischemic inflammation in a mouse model of retinal neovascularization. J Leukoc Biol. 2003;73:137–144. doi: 10.1189/jlb.0302117. [DOI] [PubMed] [Google Scholar]
- You JJ, Yang CH, Huang JS, Chen MS, Yang CM. Fractalkine, a CX3C chemokine, as a mediator of ocular angiogenesis. Invest Ophthalmol Vis Sci. 2007;48:5290–5298. doi: 10.1167/iovs.07-0187. [DOI] [PubMed] [Google Scholar]