

Abstract
Uncontrolled cell death is a fundamental cause of organ disease in humans. However, despite the need for us to delineate the molecular machinery that underlies cardiomyocyte death, our knowledge of these lethal cellular processes is still limited. The discovery that mitochondrial dysfunction, and in particular the mitochondrial permeability transition (MPT) pore, is often a common cause of the cardiac cell mortality that underlies numerous cardiac diseases has been a first crucial step. This purpose of this review is to outline our current understanding of the molecular identity of the MPT pore and the many questions that still need to be answered.
Keywords: Mitochondrial permeability transition, voltage-dependent anion channel, adenine nucleotide translocase, cyclophilin-D, mitochondrial phosphate carrier
1. Introduction
Uncontrolled cell death is a fundamental cause of organ disease in humans. Being primarily post-mitotic, the heart is particularly sensitive to the loss of its constituent cells and even a relatively small increase in cardiomyocyte death can result in pathology. Consequently, there is a desperate need for us to delineate the molecular machinery that underlies cardiomyocyte death, whether it is the acute, extensive damage seen during myocardial infarction or the more insidious, cumulative cell loss that leads to cardiomyopathy. Despite this, our knowledge of these lethal cellular processes is still limited. The discovery that mitochondrial dysfunction is often a common cause of the cardiac cell mortality that underlies the etiology of numerous cardiac diseases has been a first crucial step towards a better understanding of these mechanisms. In particular, research studying the mitochondrial permeability transition pore and its role in cardiac disease has proven fruitful. However, much work remains to be done especially with regards to the actual molecular composition of the mitochondrial pore, and the purpose of this review is to outline what we know and perhaps more importantly what we don't know about the protein components of this fascinating entity.
2. The mitochondrial death pathway
Cardiac stresses, such as ischemia/reperfusion, oxidative stress, and cytotoxic drugs induce a cascade of events at the level of the mitochondrion that, if left unchecked, will kill the myocyte. Initially this involves excessive production of reactive oxygen species, and Ca2+ overload of the mitochondrial matrix [1-4]. This in turn causes permeabilization of the inner mitochondrial membrane. This phenomenon, termed the mitochondrial permeability transition (MPT), dissipates the proton electrochemical gradient (ΔΨm) that drives many mitochondrial functions, leading to ATP depletion, further reactive oxygen species production, and ultimately swelling and rupture of the organelle [1-4]. This in turn releases pro-apoptotic intermembrane space proteins, most notably cytochrome c, Smac/DIABLO, and endonuclease-G (endoG). Cytochrome c binds to the cytosolic protein apaf1 and the resultant “apoptosome” activates the caspase protease system [1,5]. Smac/DIABLO activates caspases by sequestering caspase-inhibitory proteins, whereas endoG mediates DNA fragmentation. Therefore, activation of the mitochondrial pathway may initially induce apoptosis. However, if the stress is severe and/or prolonged, ATP will be depleted and the cell will instead undergo necrosis. These sequelae, plus the ability to inhibit them, indicate the existence of a regulated circuitry within the mitochondrion that underlies the cell death process – one that could potentially be exploited therapeutically.
3. The mitochondrial permeability transition pore and cardiac disease
The MPT pore, a non-specific channel originally thought to span both mitochondrial membranes, mediates the increases in mitochondrial permeability associated with cell death [1-3]. The pore itself is permeable to solutes up to 1.5kDa. This causes equilibration of H+ across the inner membrane, which dissipates ΔΨm and inhibits ATP production. A concomitant influx of water causes swelling of the mitochondria, which stretches the membranes to the point where the outer membrane fails. The mitochondrial pore is redox, Ca2+, voltage, adenine nucleotide, and pH sensitive [1-3]. In particular, increases in matrix Ca2+ and reactive oxygen species induce pore opening, whereas adenine nucleotides inhibit the pore; indeed many cardiac diseases, especially ischemia/reperfusion injury, are associated with increases in MPT activators e.g., Ca2+ and oxidative stress, and reductions in MPT inhibitors, e.g., ATP/ADP [1-4]. Moreover, studies have shown that inhibition of the MPT pore blunts the loss of cardiac myocytes that underlies several cardiac pathologies: myocardial ischemia/reperfusion injury [6-9], Ca2+-induced cardiomyopathy [10], diabetic cardiomyopathy [11], muscular dystrophy [12], and the cardiotoxic actions of anti-cancer agents [13]. Unfortunately, despite these substantial advances regarding our understanding of the MPT pore and its role in cardiovascular disease, the precise molecular architecture of the MPT pore remains unknown.
4. Molecular composition of the MPT Pore: the original paradigm
Based upon biochemical and pharmacological studies, the pore was proposed to consist of the voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocase (ANT) in the inner membrane, plus CypD in the matrix [1-4] (Figure 1). VDAC, ANT, and CypD interact at membrane contact sites and reconstitution of this complex in vesicles yields a Ca2+-sensitive channel reminiscent of the MPT pore [14]. Moreover, pharmacological inhibitors of ANT and CypD, e.g., bongkrekic acid and cyclosporine-A, respectively, can inhibit MPT [6-9,15,16]. However, recent genetic studies have questioned the validity of this paradigm.
Figure 1.
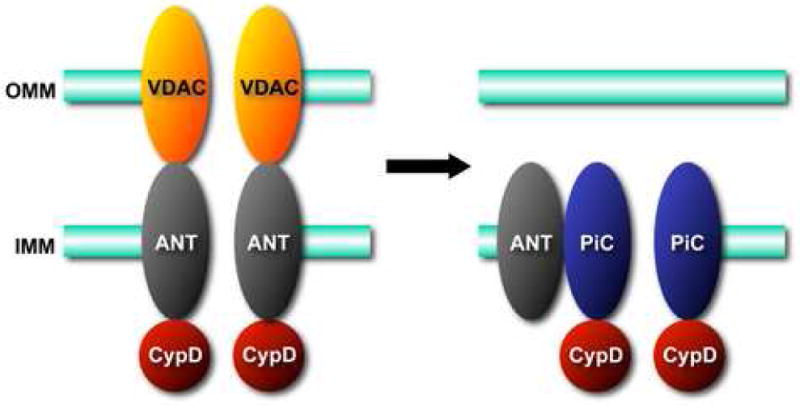
Molecular models for the mitochondrial permeability transition (MPT) pore. A. The original model for the MPT pore, consisting of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM), the adenine nucleotide translocase (ANT) in the inner mitochondrial membrane (IMM), and cyclophilin-D (CypD) in the matrix. B. Revised models in light of recent findings in gene-targeted mice. VDAC is no longer part of the model and it appears that an outer membrane component may not even be necessary for this process. ANT now appears to be more of a regulatory protein, and only CypD remains as an established component. In contrast, the mitochondrial phosphate carrier (PiC) has been added to model as a potential candidate for the pore-unit of the MPT pore.
4.1 VDAC is not a component of the MPT pore
The most abundant protein in the outer mitochondrial membrane, VDAC (also known as porin) facilitates the efficient transport of ATP/ADP across the outer leaflet [17,18]. The VDAC family consists of 3 gene products (VDAC1, 2, and 3) that exhibit structural and functional homology [17-19]. In their open state the VDAC channel is huge, being permeable to solutes up to 5kD in size. Interestingly even when in the closed state, the channel is still 1.5kD permeable and becomes more selective for cations [18,20].
As discussed above, VDAC has always been considered a key component of the MPT pore. This was based on an original hypothesis by Zoratti [21,22], who suggested that the electrophysiological properties of the MPT pore were reminiscent of those of the VDAC channel. However, there is only limited data substantiating this hypothesis. Shimizu et al first reported that anti-VDAC antibodies, which were able to block VDAC channel activity, were effective in preventing Ca2+-induced MPT and cytochrome c release in isolated liver mitochondria [23]. Such “inhibitory” antibodies were also reported to block arsenic trioxide-induced MPT [24]. By means of a drug library screen Bernardi's laboratory discovered a novel VDAC1-binding compound, Ro 68-3400, which blocked Ca2+-induced MPT in isolated mitochondria [25]. Biochemically, Crompton's group was able to pull-down VDAC along with ANT when mitochondrial lystates were incubated with a GST-CypD fusion protein [14]. Moreover, reconstitution of this VDAC-ANT-CypD complex yielded a Ca2+-dependent, cyclosporine-sensitive channel similar to the MPT pore [14]. Together, these studies indicated that VDAC was indeed a critical part of the MPT pore's molecular architecture.
However, there is now considerable evidence to indicate that the conclusions of these original studies were in fact incorrect. An oft forgotten point is that even its “closed” form, the channel of VDAC is still permeable to solutes 1.5kD in size, i.e., the same size as when the MPT pore is open [18]. From this alone it is hard to reconcile how closing VDAC would close the pore. Moreover, Colombini has shown that closure of VDAC has been shown to increase Ca2+ influx into mitochondria [20], which would have the net effect of inducing MPT rather than inhibiting it. In addition, the specificity of the VDAC “inhibitors” described in the previous studies has been seriously questioned. VDAC antibodies are known to recognize other non-VDAC proteins and therefore cannot be considered totally specific [18]. Moreover, despite the original postulation that Ro 68-3400 was able to bind VDAC1, a subsequent study by the same group found that radioactive Ro 68-3400 was still able to label a ∼32kDa protein in VDAC1-/-mitochondria and was still retained on an hydroxyapatite column when all VDACs had been eluted [26]. Therefore, Ro 68-3400's ability to inhibit MPT is independent of VDAC. Finally, Halestrap's group (at the same time as Crompton's) was able to reconstitute a mitochondrial pore-like channel that only contained ANT and CypD and was devoid of VDAC [27], and MPT can still be observed in mitoplasts that are devoid of the outer membrane [28].
With the pharmacological and biochemical evidence in dispute, it was left to genetic approaches to try and answer the question of whether VDAC is truly a component of the MPT pore. Such an approach was particularly essential, as it would potentially enable us to differentiate between the 3 different VDAC isoforms; something the previous studies had not been able to address. Shoshan-Barmatz's laboratory has published several studies showing that overexpression of VDAC1 can induce MPT and apoptosis in transformed cell lines [29-31]. However, although the authors also knocked down VDAC1 with shRNA they did not look at whether reduced VDAC1 altered MPT [29]. Moreover, in our hands a 2-3-fold increase of VDAC1 in cardiomyocytes, rather than being toxic, was actually cytoprotective (unpublished data). A cytoprotective action has also been described for VDAC2, which appears to act via sequestering the pro-apoptotic protein Bak [32].
It is still surprising that although VDAC1-/- and VDAC3-/- mice, originally developed by Bill Craigen [33,34], have been available for nearly a decade, it is only in the past couple of years that they have been specifically studied in the context of MPT and cell death. Bernardi's group first demonstrated that mitochondria from VDAC1-/- still exhibited a normal cyclosporine-sensitive MPT response [26]. Similarly, we found that Ca2+-induced MPT was unaffected in either VDAC1-/- or VDAC3-/- mitochondria [35]. While this suggested that VDAC1 and VDAC3 were not necessary for MPT, it was still possible that the remaining VDAC isoforms functionally compensated for the lack of VDAC1. Unfortunately, although VDAC1/3 double-null mice can be generated, their numbers are few due to a combination of partial embryonic lethality and breeding issues [36]. Coupled with the fact that VDAC2-/- embryos die very early in gestation [32], generating a VDAC triple-null mouse is not genetically possible at the current time. To circumvent this problem, we isolated fibroblasts from VDAC1/3 double-null mice and subsequently knocked-down VDAC2 in these cells using siRNA [35]. This resulted in a cell that was essentially deficient in all 3 isoforms. Interestingly, these cells were still sensitive to both oxidative stress and Ca2+ induced MPT and cell death [35]. Together, these data conclusively proved that VDAC is dispensable for MPT, and is not an essential component of the MPT pore. In fact, the fibroblasts lacking all VDAC were actually more sensitive to oxidative stress-induced cell death [35], suggesting that VDAC more likely functions as a protective protein rather than a pro-death channel. This is consistent with the proposal that opening of VDAC during toxic stress is needed to maintain critical metabolite flux across the mitochondrial outer membrane, thereby preserving cellular function [37,38]. Moreover, VDAC appears to act as an important scaffold that enables the crucial mitochondrial targeting of cytoprotective proteins such as Akt [38,39], protein kinase Cε [40,41] and hexokinase [39,41,42].
4.2 ANT may only be a regulatory component of the MPT pore
The ANT family of transporters mediates the exchange of ATP and ADP across the inner mitochondrial membrane [38]. The ANT proteins belong to the large, conserved SLC25 family of 6 transmembrane inner mitochondrial membrane carriers [43,44]. Two highly homologous isoforms, ANT1 and ANT2, are present in mammals, which are encoded for by the Slc25a4, and Slc25a5 genes, respectively. ANT1 is predominantly localized to cardiac and skeletal muscle, whereas ANT2 is found in all tissues [43,44]. An additional ubiquitously expressed isoform, ANT3 (Slc25a6), has been described in humans, but appears to be lacking in rodents [43].
Unlike VDAC, the pharmacological evidence supporting a role for ANT in MPT has, until recently, been far more convincing. Combined with the fact that ADP/ATP are excellent inhibitors of MPT, one of the first clues that ANT might play a role in this process was the discovery by Halestrap's group that a specific inhibitor of ANT, bongkrekic acid, blocked Ca2+-induce MPT in isolated cardiac and liver mitochondria [45]. Somewhat paradoxically, another ANT inhibitor, atractyloside, was able to sensitize the pore to Ca2+ [16,46]. However, this is reconciled by the fact that bongkrekate and atractyloside fix the ANT protein in different conformations – the ‘m (matrix)’ and ‘c (cytosolic)’ conformations, respectively, and treatment of mitochondria with MPT-inducing doses of Ca2+ was associated with increasing amounts of the ‘c’ conformer of ANT [15]. Together, all these data suggested that ANT was indeed part of the MPT pore and that conformational changes were required to change its function from adenine nucleotide carrier to lethal pore. Several studies have extended these pharmacological findings to the cardiac myocyte. Xu and colleagues found that atractyloside was able to reverse the cardioprotective effects of preconditioning in isolated cardiomyocytes [46]. Similarly, atractyloside was able to block the infarct-sparing effects of nitric oxide donors [47] and cardiac specific PKCε transgenesis [40] in mice. Conversely, bongkrekic acid protected cardiomyocytes against oxidative stress- [48,49] and Gαq- [50] induced MPT and cell death.
The postulation that ANT is an essential component of the MPT pore proposition has been supported by various biochemical studies. Brdiczka's labaroatory demonstrated that ANT interacted with CypD at contact sites between the inner and outer mitochondrial membranes, where the MPT pore is believed to reside [51]. Moreover, several groups found that reconstitution of purified ANT in lipid vesicles can form a non-specific pore under certain conditions [52-54]. The simultaneous finding of Crompton and Halestrap that reconstitution of an ANT-CypD complex in liposomes yielded an MPT-like pore [14,27], was even more impressive. Genetic gain-of-function studies in cell culture have also tended to corroborate the in vitro findings. Bauer et al first demonstrated that overexpression of ANT1 could dominantly induce mitochondrial dysfunction and cell death in 293 cells [55], and other groups have observed similar results in other non-cardiac cell lines [56,57]. Interestingly, these studies found that the homologous ANT2 isoform was not cytotoxic which maybe related to the finding that only ANT1 interacts with CypD at contact sites whereas ANT2 does not [51].
However, there are several findings that cast a considerable amount of doubt over whether ANT an essential component of the MPT pore complex. The data outlined above would suggest that it is ANT1 that acts as the pore protein, not ANT2. This, however, raises the issue of localization. ANT1 is primarily restricted to cardiac and skeletal muscle, whereas MPT occurs in all major organs suggesting that ANT1 may not in fact be a bona fide component of the MPT pore. In addition, we have found that overexpression of ANT1 in cardiac myocytes, although cytotoxic, appears to act through a Bax-dependent mechanism rather than through activation of of MPT [58]. Finally, as with VDAC, studies in gene-modified mice have indicated that ANT is not essential for MPT. Using a gain-of-function approach, Walthier's group has demonstrated that, rather than being detrimental, cardiac-specific overexpression of ANT1 in the rat and mice actually ameliorates renin- and streptozotocin-induced cardiomyopathies, respectively [59,60]. Antithetically, Wallace's laboratory has simultaneously targeted both the Slc25a4 and Slc25a5 genes to create ANT1/2 double-null mice [61]. Surprisingly, mitochondria from these mice still exhibit a normal cyclosporine-sensitive MPT response, and hepatocytes derived from these mice still respond normally to various cell death stimuli [61]. Closer inspection of these data, however, does reveal a decrease in Ca2+ sensitivity of the MPT pore in the ANT1/2 double-null mice. Moreover, the abilities of adenine nucleotides and ANT ligands such as bongrekekic acid and atractyloside to modulate pore opening are completely abolished in the ANT-deficient mitochondria [61]. Together, these observations in gene-modified animals would indicate that, rather than being a channel-forming unit of the MPT-pore as was originally proposed, ANT acts more as a peripheral regulatory protein that confers sensitivity of the MPT pore to adenine nucleotides, ANT ligands, and, to a certain extent, Ca2+.
Why is the genetic data so discrepant with the biochemical and pharmacological data? One major caveat with ANT knockout study is that these are not healthy mice [62] and there appear to be wholesale changes in the mitochondrial protein profile [63] that could of compensated for the lack of ANT at some level, or sensitized the cells to death through another mechanism. Alternatively, the newly discovered ANT4 isoform in mice [64] may have been able to compensate functionally for the lack of ANT1 and 2. However, the fact that this isoform appears to be restricted to the testes suggests that this was unlikely. Finally, due to issues of embryonic lethality, the ANT2 gene had to be floxed and then excised using a Cre transgene [61]. It is rare that Cre-based recombination is 100% effective, so it is possible that some residual ANT2 was present at sufficient enough quantities to mediate MPT. With regards to the biochemical data, many mitochondrial membrane proteins can form non-specific pores when reconstituted in liposomes [65-67] and it is not certain whether this represents a true property of the protein or whether it is an artefact due to the artificial nature of the system. Moreover, Halestrap's group have recently found that the CypD-binding, pore-forming protein they originally identified (and reconstituted) as ANT [27], may in fact be the mitochondrial phosphate carrier [68], a finding we will discuss further in section 5.3.
4.3 CypD is a critical regulatory component of the MPT pore
The final piece of the original MPT puzzle is CypD. This mitochondrial matrix protein is a member of the large family of peptidylprolyl isomerases, which catalyze the rotation of proline peptide bonds, thereby inducing a conformational change in the target protein [69]. Although the physiological role of CypD remains unknown, its pathological role as a component of the MPT pore is widely accepted. CypD was first identified by Halestrap's group, who used N-terminal sequencing of a peptidylprolyl isomerases purified from mitochondria to identify a novel mitochondrial matrix cyclophilin [70]. Crompton subsequently used photo-labelled cyclosporine-A, known to inhibit the MPT pore, to identify the same cyclophilin [71]. Importantly, cyclosporine-A was able to inhibit CypD's enzymatic activity [70]. Moreover, Ca2+ was able to suppress the binding of cyclosporine-A to CypD, suggesting that CypD could be the Ca2+ sensor of the pore [70]. However, one major caveat with the use of cyclosporine-A as an MPT inhibitor is that it is also a potent inhibitor of the phosphatase calcineurin [72], hence its clinical use as an immunosuppressive. However, other non-immunosuppressive inhibitors of CypD's peptidylprolyl isomerase activity, such as sanglefehrin-A and Debio-25, have since been identified, and have been shown to inhibit MPT in isolated mitochondria [7,9,73].
In the specific context of the heart, a considerable number of studies have demonstrated that pharmacological inhibition of CypD is profoundly cardioprotective. Over 15 years ago, both Halestrap's and Crompton's groups first showed that administration of cyclosporine-A could protect against ischemia/reperfusion-induced injury in perfused rat hearts [74,75]. Since then a large number of studies have reported similar findings using a variety of different heart models (in vitro, ex vivo, in vivo) in a variety of species, including humans [6-9,76]. Importantly, the other CypD inhibitors, sanglefehrin-A and Debio-025, have also been shown to have anti-infarct properties [7,9,77], confirming that cyclosporine-A's protective actions are due to inhibition of CypD and MPT, rather than calcineurin. Moreover, the beneficial effects of pharmacological CypD inhibition have also been extended to several models of cardiomyopathy including diabetes [11], cancer drugs [13], muscular dystrophy [12], and sepsis [78]. Consequently, the pharmacological data supporting a role for CypD as a component of the MPT pore, and a critical regulator of cardiac myocyte death is overwhelming.
In light of all this, it was only a matter of time before mice lacking the CypD gene were generated. Moreover, as there is only one gene for CypD (the Ppif locus), it was a much easier proposition for targeting than either VDAC or ANT. Indeed, 4 independent groups, including our own, simultaneously generated Ppif-/- mice [6,79-81]. All four studies demonstrated that CypD-deficient mitochondria and cells were resistant to Ca2+ and oxidative stress-induced MPT and cell death. Interestingly, cyclosporine-A was unable to confer additional protection in the CypD-null mitochondria, thereby confirming that CypD is indeed the molecular target for the anti-MPT actions cyclosporine-A [6,79,80]. Whether this is also true for sanglefehrin-A or Debio-25 remains to be tested. Importantly, we demonstrated that re-expression of wildtype CypD re-sensitized the knockout cells to MPT and cell death, whereas an isomerase-dead mutant of CypD (R96G) failed to rescue the phenotype [6]. These data further confirmed that the peptidylprolyl isomerase function of CypD is essential for its ability to regulate MPT.
The Ppif-/- mice have also been extensively studied in vivo, especially with regards to cardiac disease. Both Tsujimoto's and our group demonstrated that CypD-deficient mouse hearts were considerably more resistant to myocardial ischemia/reperfusion injury than wildtype hearts. [6,80], thereby confirming the previous studies that showed an infarct-sparing effect with cyclosporine-A and sanglefehrin-A. Building upon this, Derek Yellon's group has reported that preconditioning and postconditioning do not confer additional protection against ischemic injury in the CypD-null mice [82], suggesting that inhibition of the MPT pore is a major component of the cardioprotective mechanism. The positive findings in ischemia/reperfusion have since been expanded to other models of cardiac disease. Nakayama and colleagues examined the role of MPT in a transgenic model of cardiac Ca2+-overload [10]. These mice, which overexpress the β2 regulatory subunit of the L-type-Ca2+ channel, exhibited a profound cardiomyopathy, associated with rampant cardiac myocyte death and replacement fibrosis [10]. However, crossing the transgenics into the CypD-null background almost completely normalized these ultrastructural and functional deficits. Similarly, Ppif ablation has been shown to ameliorate both doxorubicinand muscular dystrophy-induced cardiomyopathies [10,12]. Whether loss of CypD is also protective in other models of cardiomyopathy such as transverse aortic constriction or diabetes remains to be seen, although a recent study has reported increased levels of CypD and an increased MPT response in mitochondria from hypertrophic hearts [83].
Despite all this positive data, however, it is important to note that ablation of the Ppif gene does not abolish MPT completely. Instead, the loss of CypD renders the MPT pore less sensitive to Ca2+ and reactive oxygen species, the main triggers of MPT, and the inhibition of MPT in CypD-deficient mitochondria can be overcome if sufficient Ca2+ (in the mM range) is added [6,79-81]. This demonstrates that while CypD does appear to be a critical regulator of the MPT pore, it cannot be the pore per se. The fact that CypD is a small soluble matrix protein would also rule out any ability for it to act as the pore-forming unit.
In conclusion, the original model of the MPT pore has not stood up particularly well to genetic scrutiny (Figure 1). VDAC has been discarded, and it's not even apparent that an outer membrane component is required for what is essentially an inner membrane phenomenon. In addition, ANT appears to act more as a regulatory subunit rather than the essential pore-forming unit it was originally thought to be. CypD remains as the only defined molecular component of the MPT pore, where it functions in an enzymatic capacity to induce MPT pore formation and cell death by binding and regulating unknown proteins.
5. Molecular composition of the MPT Pore: new candidates
Given that ANT and VDAC do not appear to be the critical channel-forming units of the mitochondrial pore, we must look elsewhere for alternatives. Lemasters has proposed that the pore itself is actually formed from any misfolded inner membrane proteins that would be generated during a toxic stress, and that CypD acts a regulator through its chaperone-like ability to bind to such denatured proteins [84]. However, such a hypothesis is unsatisfying for several more plausible that the MPT pore is composed of a definable inner membrane-spanning unit(s). In this regard, several alternative candidates have been proposed.
5.1 Complex-I
It has been postulated that one of the complexes of the oxidative phosphorylation machinery can act as the MPT pore given that ρ0 cells, which lack the electron transport chain, do not undergo a MPT response [28,85]. Of the five complexes that make up the oxidative phosphorylation machinery, complex-I has received the most attention. Fontaine et al first reported that the amount of Ca2+ required to induce MPT in skeletal muscle mitochondria was considerably less when the mitochondria were energized with complex-I substrates as opposed to complex-II or complex-IV substrates [86]. This ability of complex-I to modulate MPT appeared to be dependent on electron flux and was not associated with changes in pH, H2O2 production, and Ca2+ uptake. Consistent with these data, Fontaine also found that rotenone, an inhibitor of complex-I, could block MPT in isolated cells [87]. These studies raised the intriguing possibility that complex-I directly regulates the MPT pore and may even be the pore itself.
However, the vast majority of studies have reported the opposite, i.e., that complex-I inhibition actually induces MPT and cell death [88-91]. In all these cases, complex-I inhibition leads to an increase in mitochondrial reactive oxygen species production, which in turn activates the MPT pore. From a genetic standpoint, the threshold for MPT pore activation was lower in cell lines that have mutations that reduce complex-I assembly and/or activity [92,93]. Again, this sensitization of the pore in these studies appeared to be due to excess mitochondrial oxidant production. Thus, the majority of the literature supports a paradigm whereby inhibition of complex-I, either pharmacologically or genetically, indirectly regulates the MPT pore through an increase in mitochondrially-derived free radicals. Ultimately, such experiments are difficult to interpret, and therefore reconcile, given the intimate relationship between complex-I and mitochondrial homeostasis. Thus perturbations in complex-I can have profound consequences on mitochondrial function (and vice versa), which would secondarily affect the MPT pore. Moreover, complex-I is made up of at least 42 proteins (plus many accessory proteins required for correct assembly of the complex), which makes “specific” gene targeting problematic.
5.2 Other members of the SLC25 transporter family
The most likely candidates are inner membrane exchangers/ion channels that have the capacity to form a pore-like structure (as was originally thought with ANT). Several candidates have been proposed [1,94]: the aspartate-glutamate carrier (AGC), the ornithine-citrulline carrier (ORC), and the mitochondrial phosphate carrier (PiC). Like ANT, all three belong to the SLC25 family of inner mitochondrial membrane carriers [44] and under normal physiological conditions play critical roles in the TCA cycle (AGC), the urea cycle (ORC), and ATP synthesis (PiC).
The AGC normally transports glutamate and H+ into the mitochondria in exchange for aspartate, although this can be reversed in de-energized mitochondria [44]. Satrústegui's group have demonstrated that AGC consists of two isoforms, aralar and citrin which are encoded for by the Slc25a12 and Slc25a13 genes, respectively [95,96]. Aralar is predominantly found in the brain and striated muscle, whereas citrin is more ubiquitously expressed. In the context of the MPT pore the AGC proteins are quite intriguing. Reconstitution of the AGC in proteoliposomes yields a pore-like channel in the presence of mercury based agents [65], which are known to induce MPT [84]. Moreover, the activities of aralar and citrin are stimulated by binding of Ca2+ [95,96]. However, it should be noted that the AGC proteins are modulated by external (i.e., cytosolic) Ca2+ [44,95,96], whereas the MPT pore is only responsive to increases in internal (i.e., matrix) Ca2+. In addition to this discrepancy, we have recently found that neither overexpression nor knockdown of citrin affects MPT and cell death in fibroblasts [58 and unpublished data]. Therefore, although more testing is needed, it would appear unlikely that aralar or citrin are part of the MPT pore.
The ORC carrier (encoded by the Slc25a15 gene) is a 33kDa inner membrane protein that facilitates the import of ornithine into the mitochondrial matrix in exchange for citrulline and H+ [44]. Relatively little is known about this protein but it is known to be expressed in all tissues, with the liver and pancreas containing the largest amount of transcript [44]. Similar to the AGC, reconstituted ORC can form a non-specific pore when treated with mercurial agents [66]. However, its potential role in MPT has yet to be formally tested.
5.3 PiC may be a novel component of the MPT pore
It has long been known that inorganic phosphate sensitizes the MPT pore to induction by Ca2+ and oxidants. Presumably, therefore the pore must possess a Pi-binding site. While there are several Pi-binding proteins in the mitochondrion, the most obvious candidate is the mitochondrial PiC, a ubiquitously expressed ∼30kDa inner mitochondrial membrane protein [44]. PiC is encoded for by a single gene (the Slc25a3 locus) that gives rise to 2 splice variants [44]. The PiC's physiological role is to mediate the re-uptake of Pi back into the mitochondrial matrix, which it co-transports with H+ [44]. However, the mitochondrial PiC is now beginning to be scrutinized as a potential regulator of MPT and cell death. In fact it interesting to note that in the study examining the protein labeled by the Ro 68-3400 compound [26], the unidentified 32kDa protein was eluted by Pi. Indeed, the PiC is attractive as a MPT pore component for several reasons. Firstly, the aforementioned role of Pi in the induction of MPT. Secondly, ANT, which we know is a regulator of the MPT pore, tightly associates with the PiC in the so-called “synthasome” [97]. Thirdly, reconstitution of the PiC in liposomes can yield a non-specific pore under certain conditions [67]. Finally, the PiC is expressed in all tissues. One problem with the ANT hypothesis was that only the muscle-specific ANT1 appeared to be involved in the process, yet MPT has been shown to occur in a variety of non-muscle tissues. The PiC does not suffer from this pitfall and therefore fulfills the postulate that an essential component of the MPT pore must be expressed in all tissues that undergo MPT.
From an experimental point of view, there have been two recent studies that suggest that the PiC could be part of the MPT pore. Using a non-biased genetic screen for death-inducing proteins, Alcala and colleagues reported that overexpression of the PiC dominantly induced mitochondrial dysfunction and apoptosis in non-cardiac cells [98]. Halestrap's group has taken a different approach. Utilizing GST-CypD as bait coupled with proteomic analyzes, they identified the PiC as a bona fide CypD-interacting protein [68]. They then went on to demonstrate that MPT-inducing agents enhanced the PiC-CypD interaction, whereas MPT-blocking compounds reduced it. Most importantly, agents that were able to inhibit mitochondrial Pi transport activity also blocked MPT in isolated mitochondria. Together, these data suggest that the PiC may indeed be a component of the MPT pore. Moreover, the study found that ANT was part of the PiC-CypD complex [68], and Halestrap has suggested that alterations in ANTs conformation could influence the PiC [68]. Such a notion would be consistent with the idea that ANT acts as a regulatory protein rather than one of the actual pore-forming units, and would help reconcile the biochemical and genetic data regarding ANT. Obviously the question still remains as to whether the PiC is in fact the pore-forming channel of the MPT pore or whether it represents yet another regulatory protein such as CypD or ANT. Moreover, the agents used to block PiC activity can affect other mitochondrial transporters thus raising the question of specificity. Genetic analyses using RNAi or gene-targeting technologies to deplete the PiC are clearly needed before a role for PiC in MPT can be conclusively established.
6. Conclusion
In order to truly understand the mechanisms that mediate mitochondrial-driven cell death, it is imperative that we identify the channel-forming units of the MPT pore. It is therefore essential that we continue to screen for novel MPT pore candidates using a variety of proteomic, genetic, and pharmacological technologies. By doing so, we will be able to refine our research into this exciting phenomenon, and, more importantly, provide new therapeutic targets for the clinical treatment of cardiac disease.
Acknowledgments
The author apologizes that due to space constraints he has not been able to cite every important study in this field. Work in the author's laboratory is supported by grants from the National Institutes of Health (HL094404 and HL092327) and by an American Heart Association Scientist Development Grant (0635134N).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kroemer G, Galluzzi L, Brenner C. Mitochondrial permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 2.Di Lisa F, Canton M, Menabò R, Kaludercic N, Bernardi P. Mitochondria and cardioprotection. Heart Fail Rev. 2007;12:249–260. doi: 10.1007/s10741-007-9028-z. [DOI] [PubMed] [Google Scholar]
- 3.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 4.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 6.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 7.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–903479. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 8.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 9.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama N, Chen X, Baines CP, Klevitsky R, Zhang H, Jaleel N, Chua BHL, Zhang X, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 12.Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 14.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 15.de Macedo DV, Nepomuceno ME, Pereira-da-Silva L. Involvement of the ADP/ATP carrier in permeabilization processes of the inner mitochondrial membrane. Eur J Biochem. 1993;215:595–600. doi: 10.1111/j.1432-1033.1993.tb18070.x. [DOI] [PubMed] [Google Scholar]
- 16.Haworth RA, Hunter DR. Control of the mitochondrial permeability transition pore by high-affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. J Bioenerg Biomembr. 2000;32:91–96. doi: 10.1023/a:1005568630151. [DOI] [PubMed] [Google Scholar]
- 17.Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 18.Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Sampson MJ, Decker WK, Craigen WJ. Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochim Biophys Acta. 1999;1452:68–78. doi: 10.1016/s0167-4889(99)00120-2. [DOI] [PubMed] [Google Scholar]
- 20.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabó I, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. I. Binary structure and voltage dependence of the pore. FEBS Lett. 1993;330:201–205. doi: 10.1016/0014-5793(93)80273-w. [DOI] [PubMed] [Google Scholar]
- 22.Szabó I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett. 1993;330:206–210. doi: 10.1016/0014-5793(93)80274-x. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol. 2001;152:237–50. doi: 10.1083/jcb.152.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem. 2003;278:49812–49818. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 26.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1-/- mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J Biol Chem. 2008;283:3476–3486. doi: 10.1074/jbc.M707528200. [DOI] [PubMed] [Google Scholar]
- 29.Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci. 2006;103:5787–5792. doi: 10.1073/pnas.0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 31.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–90. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 32.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–7. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 33.Anflous K, Armstrong DD, Craigen WJ. Altered mitochondrial sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J Biol Chem. 2001;276:1954–60. doi: 10.1074/jbc.M006587200. [DOI] [PubMed] [Google Scholar]
- 34.Sampson MJ, Decker WK, Beaudet AL, Ruitenbeek W, Armstrong D, Hicks MJ, Craigen WJ. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J Biol Chem. 2001;276:39206–12. doi: 10.1074/jbc.M104724200. [DOI] [PubMed] [Google Scholar]
- 35.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weeber EJ, Levy M, Sampson MJ, Anflous K, Armstrong DL, Brown SE, Sweatt JD, Craigen WJ. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem. 2002;277:18891–7. doi: 10.1074/jbc.M201649200. [DOI] [PubMed] [Google Scholar]
- 37.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 38.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 40.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anflous-Pharayra K, Cai ZJ, Craigen WJ. VDAC1 serves as a mitochondrial binding site for hexokinase in oxidative muscles. Biochim Biophys Acta. 2007;1767:136–142. doi: 10.1016/j.bbabio.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 43.Fiore C, Trézéguet V, Le Saux A, Roux P, Schwimmer C, Dianoux AC, Noel F, Lauquin GJ, Brandolin G, Vignais PV. The mitochondrial ADP/ATP carrier: structural, physiological and pathological aspects. Biochimie. 1998;80:137–150. doi: 10.1016/s0300-9084(98)80020-5. [DOI] [PubMed] [Google Scholar]
- 44.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 45.Halestrap AP, Davidson AM. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–60. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu M, Wang Y, Hirai K, Ayub A, Ashraf M. Calcium preconditioning inhibits mitochondrial permeability transition and apoptosis. Am J Physiol. 2001;280:H899–908. doi: 10.1152/ajpheart.2001.280.2.H899. [DOI] [PubMed] [Google Scholar]
- 47.Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, Qiao X, Wang Y, Weiss JN, Ping P. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol. 2005;288:H1290–5. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- 48.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akao M, O'Rourke B, Teshima Y, Seharaseyon J, Marbán E. Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ Res. 2003;92:186–94. doi: 10.1161/01.res.0000051861.21316.e9. [DOI] [PubMed] [Google Scholar]
- 50.Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH. Cardiomyocyte apoptosis induced by Gαq signaling is mediated by permeability transition pore formation and activation of the mitochondrial death pathway. Circ Res. 2000;87:1180–7. doi: 10.1161/01.res.87.12.1180. [DOI] [PubMed] [Google Scholar]
- 51.Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dorner A, Zorov DB, Brdiczka D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J. 2001;358:349–358. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tikhonova IM, Andreyev AYu, Antonenko YuN, Kaulen AD, Komrakov AYu, Skulachev VP. Ion permeability induced in artificial membranes by the ATP/ADP antiporter. FEBS Lett. 1994;337:231–4. doi: 10.1016/0014-5793(94)80197-5. [DOI] [PubMed] [Google Scholar]
- 53.Rück A, Dolder M, Wallimann T, Brdiczka D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett. 1998;426:97–101. doi: 10.1016/s0014-5793(98)00317-2. [DOI] [PubMed] [Google Scholar]
- 54.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- 55.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol. 1999;147:1493–1502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zamora M, Granell M, Mampel T, Vinas O. Adenine nucleotide translocase 3 (ANT3) overexpression induces apoptosis in cultured cells. FEBS Lett. 2004;563:155–160. doi: 10.1016/S0014-5793(04)00293-5. [DOI] [PubMed] [Google Scholar]
- 57.Jang JY, Choi Y, Jeon YK, Aung KC, Kim CW. Over-expression of adenine nucleotide translocase 1 (ANT1) induces apoptosis and tumor regression in vivo. BMC Cancer. 2008;8:160. doi: 10.1186/1471-2407-8-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baines CP, Molkentin JD. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J Mol Cell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.01.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walther T, Tschöpe C, Sterner-Kock A, Westermann D, Heringer-Walther S, Riad A, Nikolic A, Wang Y, Ebermann L, Siems WE, Bader M, Shakibaei M, Schultheiss HP, Dörner A. Accelerated mitochondrial adenosine diphosphate/adenosine triphosphate transport improves hypertension-induced heart disease. Circulation. 2007;115:333–344. doi: 10.1161/CIRCULATIONAHA.106.643296. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Ebermann L, Sterner-Kock A, Wika S, Schultheiss HP, Dörner A, Walther T. Myocardial overexpression of adenine nucleotide translocase 1 ameliorates diabetic cardiomyopathy in mice. Exp Physiol. 2008 doi: 10.1113/expphysiol.2008.044800. [DOI] [PubMed] [Google Scholar]
- 61.Kokoszka J, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–34. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 63.Subramaniam V, Golik P, Murdock DG, Levy S, Kerstann KW, Coskun PE, Melkonian GA, Wallace DC. MITOCHIP assessment of differential gene expression in the skeletal muscle of Ant1 knockout mice: coordinate regulation of OXPHOS, antioxidant, and apoptotic genes. Biochim Biophys Acta. 2008;1777:666–75. doi: 10.1016/j.bbabio.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brower JV, Rodic N, Seki T, Jorgensen M, Fliess N, Yachnis AT, McCarrey JR, Oh SP, Terada N. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J Biol Chem. 2007;282:29658–29666. doi: 10.1074/jbc.M704386200. [DOI] [PubMed] [Google Scholar]
- 65.Dierks T, Salentin A, Kramer R. Pore-like and carrier-like properties of the mitochondrial aspartate/glutamate carrier after modification by SH-reagents: Evidence for a preformed channel as a structural requirement of carrier-mediated transport. Biochim Biophys Acta. 1990;1028:281–288. doi: 10.1016/0005-2736(90)90177-p. [DOI] [PubMed] [Google Scholar]
- 66.Tonazzi A, Indiveri C. Chemical modification of the mitochondrial ornithine/citrulline carrier by SH reagents: effects on the transport activity and transition from carrier to pore-like function. Biochim Biophys Acta. 2003;1611:123–130. doi: 10.1016/s0005-2736(03)00033-6. [DOI] [PubMed] [Google Scholar]
- 67.Schroers A, Kramer R, Wohlrab H. The reversible antiport-uniport conversion of the phosphate carrier from yeast mitochondria depends on the presence of a single cysteine. J Biol Chem. 1997;272:10558–10564. doi: 10.1074/jbc.272.16.10558. [DOI] [PubMed] [Google Scholar]
- 68.Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J. 1992;284:381–5. doi: 10.1042/bj2840381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andreeva L, Tanveer A, Crompton M. Evidence for the involvement of a membrane-associated cyclosporin-A-binding protein in the Ca2+-activated inner membrane pore of heart mitochondria. Eur J Biochem. 1995;230:1125–32. doi: 10.1111/j.1432-1033.1995.tb20664.x. [DOI] [PubMed] [Google Scholar]
- 72.Wilkins BJ, Molkentin JD. Calcineurin and cardiac hypertrophy: where have we been? Where are we going? J Physiol. 2002;541:1–8. doi: 10.1113/jphysiol.2002.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reutenauer J, Dorchies OM, Patthey-Vuadens O, Vuagniaux G, Ruegg UT. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br J Pharmacol. 2008;155:574–84. doi: 10.1038/bjp.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–9. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 75.Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res. 1993;27:1790–4. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- 76.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 77.Gomez L, Thibault H, Gharib A, Dumont JM, Vuagniaux G, Scalfaro P, Derumeaux G, Ovize M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol. 2007;293:H1654–61. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 78.Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, Chopin C, Neviere R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;488:377–85. doi: 10.1016/j.jacc.2006.02.069. [DOI] [PubMed] [Google Scholar]
- 79.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 80.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 81.Schinzel A, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim SY, Davidson SM, Hausenloy DJ, Yellon DM. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. 2007;75:530–535. doi: 10.1016/j.cardiores.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matas J, Tien Sing Young N, Bourcier-Lucas C, Ascah A, Marcil M, Deschepper CF, Burelle Y. Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. J Mol Cell Cardiol. 2008 doi: 10.1016/j.yjmcc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 84.He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- 85.Levraut J, Iwase H, Shao ZH, Vanden Hoek TL, Schumacker PT. Cell death during ischemia: relationship to mitochondrial depolarization and ROS generation. Am J Physiol. 2003;284:H549–58. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 86.Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation By electron flow through the respiratory chain complex I. J Biol Chem. 1998;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- 87.Chauvin C, De Oliveira F, Ronot X, Mousseau M, Leverve X, Fontaine E. Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J Biol Chem. 2001;276:41394–8. doi: 10.1074/jbc.M106417200. [DOI] [PubMed] [Google Scholar]
- 88.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998;809:12–7. doi: 10.1016/s0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- 89.Scorrano L, Petronilli V, Colonna R, Di Lisa F, Bernardi P. Chloromethyl-tetramethylrosamine (Mitotracker Orange) induces the mitochondrial permeability transition and inhibits respiratory complex I. Implications for the mechanism of cytochrome c release. J Biol Chem. 1999;274:24657–63. doi: 10.1074/jbc.274.35.24657. [DOI] [PubMed] [Google Scholar]
- 90.García N, Correa F, Chávez E. On the role of the respiratory complex I on membrane permeability transition. J Bioenerg Biomembr. 2005;37:17–23. doi: 10.1007/s10863-005-4119-9. [DOI] [PubMed] [Google Scholar]
- 91.Fiore C, Salvi M, Palermo M, Sinigaglia G, Armanini D, Toninello A. On the mechanism of mitochondrial permeability transition induction by glycyrrhetinic acid. Biochim Biophys Acta. 2004;1658:195–201. doi: 10.1016/j.bbabio.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 92.Porcelli AM, Angelin A, Ghelli A, Mariani E, Martinuzzi A, Carelli V, Petronilli V, Bernardi P, Rugolo M. Respiratory complex I dysfunction due to mitochondrial DNA mutations shifts the voltage threshold for opening of the permeability transition pore toward resting levels. J Biol Chem. 2009;284:2045–52. doi: 10.1074/jbc.M807321200. [DOI] [PubMed] [Google Scholar]
- 93.Haroon MF, Fatima A, Schöler S, Gieseler A, Horn TF, Kirches E, Wolf G, Kreutzmann P. Minocycline, a possible neuroprotective agent in Leber's hereditary optic neuropathy (LHON): studies of cybrid cells bearing 11,778 mutation. Neurobiol Dis. 2007;28:237–50. doi: 10.1016/j.nbd.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez-Enriquez S, He L, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–2472. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 95.Del Arco A, Satrústegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J Biol Chem. 1998;273:23327–34. doi: 10.1074/jbc.273.36.23327. [DOI] [PubMed] [Google Scholar]
- 96.Del Arco A, Agudo M, Satrústegui J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem J. 2000;345:725–32. doi: 10.1042/0264-6021:3450725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J Biol Chem. 2003;278:12305–12309. doi: 10.1074/jbc.C200703200. [DOI] [PubMed] [Google Scholar]
- 98.Alcalá S, Klee M, Fernández J, Fleischer A, Pimentel-Muiños FX. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene. 2008;27:44–54. doi: 10.1038/sj.onc.1210600. [DOI] [PubMed] [Google Scholar]