

1. Introduction
Mitochondria play a critical role in cardiac function, and are also increasingly recognized as end effectors for various cardioprotective signaling pathways. Mitochondria use oxygen as a substrate, so by default their respiration is inhibited during hypoxia/ischemia. However, at reperfusion a surge of oxygen and metabolic substrates into the cell is thought to lead to rapid reestablishment of respiration, a burst of reactive oxygen species (ROS) generation and mitochondrial Ca2+ overload. Subsequently these events precipitate opening of the mitochondrial permeability transition (PT) pore, which leads to myocardial cell death and dysfunction. Given that mitochondrial respiration is already inhibited during hypoxia/ischemia, it is somewhat surprising that many respiratory inhibitors can improve recovery from ischemia-reperfusion (IR) injury. In addition ischemic preconditioning (IPC), in which short non-lethal cycles of IR can protect against subsequent prolonged IR injury, is known to lead to endogenous inhibition of several respiratory complexes and glycolysis. This has led to a hypothesis that the wash-out of inhibitors or reversal of endogenous inhibition at reperfusion may afford protection by facilitating a more gradual wake-up of mitochondrial function, thereby avoiding a burst of ROS and Ca2+ overload. This paper will review the evidence in support of this hypothesis, with a focus on inhibition of each of the mitochondrial respiratory complexes.
2. Mitochondrial Pathologic Events in Cardiac IR Injury
Several recent review articles have addressed this topic in detail [1, 2]. Among the key events which occur in cardiomyocytes during IR injury, is an ionic imbalance precipitated by the metabolic and chemical changes of ischemia, which then triggers mitochondrial dysfunction during reperfusion:
During ischemia, excess protons due to lactic acidosis drive a cytosolic Na+ overload via the Na+/H+ exchanger (NHE) [3]. This Na+ exits via the Na+/Ca2+ exchanger, causing a cytosolic Ca2+ overload. The ischemic lack of mitochondrially derived ATP prevents adequate management of this Ca2+ by the SERCA pump, and subsequently Ca2+ is increased in the cytosol. While some Ca2+ uptake into mitochondria can occur during ischemia (due to the maintenance of membrane potential (ΔΨ) via reverse-mode operation of the ATP synthase, hydrolyzing glycolytic ATP), this Ca2+ alone is not sufficient to trigger opening of the PT pore, which is also held closed during ischemia due to the acidic pH [4]. Upon reperfusion, restoration of mitochondrial ΔΨ facilitates a large Ca2+ influx into mitochondria. Also at this moment a burst of ROS generation occurs, which together with high [Ca2+]m and re-alkalinization triggers PT pore opening [4, 5], leading to cytochrome c release [6] and cell death [2, 7]. In addition to triggering cell death, mitochondrial cytochrome c release also inhibits respiratory function leading to an ATP deficit in the post-ischemic heart [8]. Furthermore, the disruption of Ca2+ and ROS homeostasis that results from PT pore opening is thought to play a role in the development of post-ischemic arrhythmias [9].
Consistent with pathologic roles for Ca2+ overload, ROS generation and PT pore opening in IR injury, several cardioprotective drugs are directed at inhibiting these events: Cariporide and SM-20550 both inhibit NHE [10, 11], diazoxide opens the mitochondrial K+ATP channel to inhibit Ca2+ overload [12, 13], (but may also elicit cardioprotection via K+-independent mechanisms [14]). Cyclosporin A and Sanglifehrin A directly inhibit the PT pore [7, 15], antioxidants scavenge ROS [16], and nitric oxide (NO•) inhibits Ca2+ overload [17]. It was also recently shown that reperfusion with acidic media, thereby extending acidosis into the post-ischemic period, inhibits PT pore opening and is cardioprotective [18].
3. Metabolic Inhibition as a Mechanism of Cardioprotection
The remainder of this review will deal with the concept that reversible inhibition of metabolism, mainly mitochondrial metabolism, can serve to prevent the above mentioned pathologic cascade of events in reperfusion, and may account for the cardioprotective benefit of several pharmacologic agents. This hypothesis was originally forwarded in a previous review on NO• [19], but is presented here in more detail. Figure 1 illustrates several of the mitochondrial and other metabolic inhibitors to be discussed, and their sites of inhibition in the respiratory chain or elsewhere.
Figure 1. Schematic representation of the mitochondrial respiratory chain showing sites of inhibition by various cardioprotective molecules.

For full details, see text.
4. Mitochondrial Complex I Inhibition & Cardioprotection
Complex I (NADH ubiquinone oxidoreductase) is a key site of electron entry into the respiratory chain in cardiac mitochondria, owing to the heavy reliance of the heart on β-oxidation of fatty acids and subsequent generation of NADH from acetyl-CoA in the Krebs’ cycle [20]. In addition to playing a role in cardiac metabolism, complex I is also an important site of ROS generation [21], and a regulator of NADH/NAD+ redox balance in the mitochondrion [22, 23]. Notably, the latter has been shown to be critical in determining the open probability of the PT pore, such that complex I inhibition can directly inhibit pore opening via an increased NADH/NAD+ ratio [24].
In theory, inhibition of the respiratory chain at sites other than complex I may also result in an increased NADH/NAD+ ratio. However, an interesting but little-known phenomenon which may impact on this is the “cushioning effect”, first described by Chance [25]. In effect, redox events at the upper end of the respiratory chain (complexes I and II) appear to be somewhat insulated from events at the lower end of the chain (complex IV), such that partial reduction of cytochromes a/a3 may not be transmitted up the chain leading to reduction within complex I and an increased NADH/NAD+ ratio. Thus, while it is possible that inhibition in other complexes can affect the NADH/NAD+ ratio, the most effective strategy to increase this ratio is to inhibit the proximal chain at complex I.
Several pharmacologic reagents which are inhibitors of complex I have now been demonstrated to afford protection against IR injury, including amobarbital [26], rotenone [27] and S-nitrosothiols [28, 29]. Furthermore, a wide range of pharmacologic reagents which are known to be cardioprotective have subsequently been found to effectively inhibit complex I (or vice versa). This includes the antianginal agent ranolazine [30, 31], analgesics such as capsaicin [32, 33], volatile anesthetics such as halothane and isoflurane [34, 35], redox-cycling agents such as menadione [36], and anti-diabetics such as metformin [37, 38]. It is not clear whether complex I inhibition per se underlies the protective effects of these reagents, but they are very diverse in structure and their one common feature is their effect on complex I.
The inhibition of complex I by S-nitrosation has emerged as an important potential mechanism of cardioprotection involved in ischemic preconditioning (IPC) [39], although an absolute cause-and effect relationship between S-nitrosation and protection remains to be established. In this regard, S-nitrosothiols at cardioprotective doses (10–20μM) were very poor donors of free NO• and had a negligible effect on complex IV activity. Furthermore, the cardioprotection afforded by these compounds was completely abrogated by exposure to UV light at a time after which all of the original compound had decomposed. UV light destroys SNOs, so this suggests that trans-nitrosation of proteins may be involved in the protective mechanism [29]. Furthermore, cardioprotection by SNOs is independent of classical cGMP/PKG signaling, also arguing against the involvement of simple NO• release as a mechanism [29]. It has also been suggested that complex I S-nitrosation may contribute to the cardioprotective effects of nitrite (NO2−) [40]. More recently (see Nadtochiy et al. manuscript in this issue of JMCC) we have shown that protection elicited by either IPC or SNO-MPG is lost in a mouse harboring a mutation in complex I, underscoring the importance of this complex for cardioprotection.
5. Mitochondrial Complex II – a Dual Role in Metabolism and mKATP Channels
Complex II (succinate dehydrogenase) represents an interesting target for cardioprotection, because recent evidence has suggested that this complex is linked to the function of the putative mitochondrial ATP sensitive channel (mKATP), which is itself implicated in cardioprotection by IPC [41].
First regarding simple complex II inhibition, the inhibitor 3-nitropropionic acid (3-NP) has been shown to protect against injury in both heart and brain models of IR [42, 43]. We have also shown that the commonly used complex II inhibitor malonate is mildly cardioprotective [44, 45]. Somewhat related to nitric oxide, the single electron reduction product of NO•, nitroxyl (HNO or NO−), has also been shown to interact with complex II [46], and interestingly HNO donors such as Angeli’s salt are known to be cardioprotective [47], and are under investigation as potential human therapeutics [48].
Recently, research on complex II in the context of cardioprotection has taken an interesting turn, with the proposal that this complex may be related to the putative mKATP channel [41]. There are several lines of evidence supporting a link between these two proteins:
ATP, an inhibitor of KATP channels, stimulates complex II activity [49].
Openers of the mKATP channel (e.g. diazoxide) inhibit complex II activity [50, 51].
Inhibitors of complex II (e.g. malonate) open the mKATP channel [44].
mKATP opening results in fluorescence (oxidation) of the flavoprotein in complex II [12, 41].
In support of a link between complex II and the putative mKATP channel, we recently showed that the potent and specific complex II inhibitor Atpenin A5 (AA5) is also capable of opening the channel, and is potently cardioprotective [45]. Most notably, AA5 was able to open the channel and provide cardioprotection at a concentration of only 1 nM, which is a concentration that does not significantly inhibit complex II. Thus, inhibition of complex II enzymatic activity per se does not appear to be required for channel opening. The precise relationship between complex II and the putative mKATP channel remains to be identified.
6. Mitochondrial Complex III Inhibition & Cardioprotection
Complex III of the respiratory chain represents the convergence point for electrons entering the Q cycle from upstream complexes I and II, and a variety of other dehydrogenases. Therefore, while inhibition at either complex I or II alone could theoretically be by-passed by feeding electrons through the other complex, this is not the case for complex III, with inhibition resulting in complete blockage of the respiratory chain.
Interestingly, a role for complex III in both IPC and volatile anesthetic preconditioning has been proposed, wherein a transient increase in the generation of ROS from this complex serves as a critical upstream signal [53, 54]. Indeed, the commonly used complex III inhibitor antimycin A has been shown to provide protection against IR injury, and notably this protection was abrogated by antioxidants, suggesting a role for ROS [55]. However, the complex III inhibitor myxothiazol, which inhibits ROS generation from the complex, was able to block volatile anesthetic preconditioning, and was not in itself protective [54]. Thus, the inhibition of complex III activity per se does not appear to elicit cardioprotection via the same mechanism as proposed for complexes I and II (i.e. reversal and gradual wake-up at reperfusion), but instead appears to depend on the resulting augmentation of ROS generation from the complex.
7. Mitochondrial Complex IV & Cardioprotection – Gases Abound
Complex IV (cytochrome c oxidase) is the major site of O2 consumption in the heart, and thus the modulation of the activity of this enzyme has significant implications for cardiomyocyte oxygenation. One of the most important physiologic and pathologic regulators of complex IV activity is NO•, and there exist several excellent reviews on the intricacies of NO• interactions with the complex [56–58]. Given the central role of NO• in IPC and several cardioprotective phenotypes [59], and the importance of complex IV as a sensitive target of NO•, it is inherently assumed that one of the mechanisms by which NO• may protect the heart is by reversibly inhibiting metabolism at the level of complex IV. The NO-complex IV interaction will not be further considered herein, other than to comment that some of the cardioprotective benefit of nitrite may result from the generation of NO• in mitochondria and the augmentation of such inhibition. In this regard, it is interesting that complex IV is evolutionarily related to bacterial nitrite reductases, and it has been suggested that the complex may act as a nitrite reductase to generate NO• from NO2− during hypoxic conditions [60]. The relative importance of the various proposed nitrite reductases (including hemoglobin, myoglobin, cytochrome c, and complex IV) in the bio-transformation of NO2− in hypoxia/ischemia, is a subject of some debate.
In addition to NO•, it has long been known that carbon monoxide (CO) is an excellent inhibitor of complex IV [61, 62], and CO is also known to be cardioprotective [63]. Furthermore, there is some debate regarding the site within the respiratory chain which is the target of gaseous second messenger hydrogen sulfide (H2S), with some speculation that this gas may also inhibit complex IV [64, 65]. In agreement with the protective benefit of this type of metabolic shutdown, it was recently shown that H2S protects hearts from IR injury [66]. A recent review article discusses the interplay between NO•, CO and H2S at the level of mitochondrial complex IV in detail [64].
8. Inhibition of Glycolysis
The critical role of glycolysis in cardiac metabolism is beyond the scope of this review (for reviews see [20, 67]), although studies have highlighted a role for the regulation of glycolysis in IPC. For example, IPC down-regulates the enzymatic activity of several glycolytic enzymes [68].
A key glycolytic enzyme regulated by IPC is glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which can be inhibited by NO• [69]. This inhibition may proceed via S-nitrosation [70]. More recently, the finding that NO• derived nitro-lipids (nitro-oleate and nitro-linoleate) can inhibit GAPDH via reversible post-translational modification of a critical active site cysteine [71] suggests another mechanism of regulation. This mechanism may be relevant to IPC, since we recently found that such nitro-lipids are endogenously generated during IPC [72]. In addition, nitro-lipids increase protein hydrophobicity which may facilitate translocation of GAPDH to membranes [71, 72], although the functional significance of such protein traffic remains to be elucidated.
Inhibition of GAPDH facilitates a build-up of fructose 1,6-bisphosphate (F1,6-BP) which might be beneficial to the ischemic heart. Exogenous administration of F1,6-BP during early reperfusion improved cardiac recovery after ischemia in isolated hearts [73], and had positive hemodynamic effects in humans with acute myocardial infarct [74]. In addition, it has been reported that F1,6-BP build-up occurs in preconditioned hearts at the time of reperfusion, which may facilitate improved glycolytic flux and functional recovery in the post-ischemic myocardium [75].
Information about phosphofructokinase (PFK) activity in cardiac preconditioning is limited, but a detailed study in hepatic tissue revealed less post-ischemic inhibition of PFK in preconditioned liver [76]. Preservation of PFK function was associated with lower phosphorylation of the enzyme by cAMP dependent protein kinase [76]. In contrast, up-regulation of PFK protein was found in neonatal rat brain after hypoxic preconditioning, suggesting a role in tolerance against hypoxia [77].
The end-product of anaerobic glycolysis is lactate, generated from pyruvate by lactate dehydrogenase (LDH). While IPC does not change the activity of LDH in the cytosol [78], lactate levels during early reperfusion are reported to be 9-fold lower in preconditioned vs. non-preconditioned hearts [75]. This indicates that IPC inhibits glycolysis upstream of LDH, thus preventing acidosis. Furthermore, it has been demonstrated that lactate can be transported into mitochondria by a lactate shuttle [79, 80], and can be oxidized by LDH-1 (isoform H4), possibly providing a valuable substrate for post-ischemic recovery. However, it is unclear whether IPC can up-regulate mitochondrial LDH-1 and facilitate enhanced lactate utilization.
Aside from their metabolic roles in glycolysis, many glycolytic enzymes have alternative roles in cell function which may be important in IPC. For example, the translocation of hexokinase from the cytosol to mitochondria is known to regulate cytochrome c release, BAX translocation and ROS production [81]. Notably, such hexokinase redistribution has been shown to occur in IPC [78].
Somewhat intertwined with glycolysis is the function of the pentose phosphate pathway (PPP), which is the major cellular pathway for generation of NADPH, which in turn is required for the maintenance of cellular glutathione redox state. In contrast, NADPH is the critical substrate for ROS generation by the NADPH dehydrogenase (Nox) family of enzymes, which may play a detrimental role in IR injury [82]. Although it has been reported that inhibition of PPP protected heart from IR injury, IPC did not cause any change in the activity of the PPP [83].
9. How to Measure Gradual Wake-up of Metabolism at Reperfusion?
When considering the effect of mitochondrial inhibition on cardioprotection, herein we assume that the mechanism of action may include gradual wake-up of metabolism at reperfusion. However, this has never actually been measured. One of the difficulties of such measurements is that to assess mitochondrial function, it is often necessary to isolate mitochondria or to wait until a certain amount of reperfusion time has passed before a measurement can be made. To address this problem, we have recently developed an experimental system which allows real-time measurement of mitochondrial respiration rate throughout IR. As previously reported by several groups [40, 84], this system uses a sealed oxygen electrode chamber to expose isolated mitochondria to simulated hypoxia, with reperfusion attained by un-capping the chamber and allowing O2 from room air to diffuse in. By applying the calculation principles of open-flow respirometry [85–87] to such a system, the instantaneous respiration rate of mitochondria can be calculated even with the lid of the chamber removed.
This method relies on the principle that the rise in liquid phase [O2] in the chamber following removal of the lid (reoxygenation) represents an equilibrium between O2 diffusion into the chamber and mitochondrial O2 consumption. By measuring the [O2] and knowing the diffusion rate, the 3rd parameter (respiration) can be calculated, as illustrated in Figure 2. The universal equation for the calculation of respiration rate (Q) in an open flow respirometry system is: Q = m(C*−C1) − dC1/dt, where m is the mass transfer coefficient, C* is the theoretical [O2] based on equilibration of the liquid and gas phases (from Henry’s law), and C1 is the measured liquid phase [O2] from the O2 electrode. The mass transfer coefficient m is the first order rate constant for the diffusion of O2 between the gas and liquid phases. To calculate m, mitochondria are incubated in the chamber with substrates plus ADP and their respiration brings [O2] to zero. Cyanide is then added to block respiration and the chamber is uncapped, resulting in O2 diffusion into the liquid phase. The natural log of liquid phase [O2] is plotted vs. time, and the slope of this line is m (i.e. O2 diffuses in, but is not consumed because mitochondrial respiration is completely blocked).
Figure 2. Theoretical principles of application of open-flow respirometry to hypoxia-reoxygenation of isolated mitochondria in a Clark O2 electrode chamber.

Mitochondria are incubated in respiration buffer + respiratory substrates & ADP, in a respiration chamber fitted with an O2 electrode. (A): Where indicated, the lid is closed and mitochondria respire to bring [O2] to zero (i.e. hypoxia). 1mM CN− is then added to block respiration and the lid is opened. The resulting rise in [O2] is due to O2 diffusion into the chamber, and equilibrium with room air is eventually obtained. The first-order rate constant for O2 diffusion (from Ln[O2] vs. time) is the mass transfer coefficient, m. C* is the [O2] of air saturated buffer alone (~200μM). C1 is the [O2] measured by the O2 electrode (solid line). Respiration rate (Q, lower dotted trace) is zero throughout “reoxygenation”, due to the CN− inhibition. (B): Mitochondria are incubated as in panel A, without CN−. Opening the lid results in a slower rise of [O2] (C1) due to the equilibrium between O2 diffusing in and O2 consumed by the mitochondria. The shaded gray area represents the range of possible [O2] recovery curves, resulting from zero or maximal respiration. Respiration rates (Q) are calculated instantaneously during reoxygenation, using the equation Q=m(C*−C1)/dC1/dt. (C): Mitochondria are incubated as in B, but with hypoxia. Respiration (Q) recovers rapidly at reoxygenation, but Ox-Phos damage prevents later full recovery.
In Figure 2, C* is the [O2] of air saturated buffer alone, and in this case is ~200μM (based on salinity, solubility constants). C1 is the [O2] measured by the O2 electrode (solid line). Respiration rate (Q, lower dotted trace) is zero throughout “reoxygenation”, due to the CN− inhibition. In panel B, mitochondria are incubated as in panel A, without CN−. Opening the lid results in a slower rise of [O2] (C1) due to the equilibrium between O2 diffusing in and O2 consumed by the mitochondria. The shaded gray area represents the range of possible [O2] recovery curves, resulting from zero or maximal respiration. Theoretical respiration rates (Q) are shown in the lower traces. In panel C, the anticipated result with mitochondria exposed to hypoxia is shown. Respiration recovers rapidly at reoxygenation, but damage to the oxidative phosphorylation machinery prevents later full recovery.
Figure 3A shows data from such experiments. Mitochondria (2.5mg/ml) were incubated in respiration buffer in a Clark O2 electrode chamber [29]. Their respiration bought the [O2] down to a level below 1 μM, and this O2 level (defined herein as hypoxia) was maintained for 30 min. Two minutes prior to the end of hypoxia, additional substrates and ADP were added to the chamber, and at the end of hyoxia the chamber was uncapped and mitochondria were reoxygenated. The rise in liquid phase [O2] in the chamber was measured prior to hypoxia (with the lid closed) and calculated every 5 s. for a period of 200 s. during reperfusion. Respiration rate prior to hypoxia was 18±3 nmol O2/min/mg protein. During reoxygenation, mitochondria exhibited a respiration rate of 10 nmol O2/min/mg within the first five seconds of reperfusion, and this rate declined to 6 nmol O2/min/mg by 200 s. (Fig. 3). These data support that, unchecked, mitochondrial respiration rapidly re-starts at reperfusion. It is anticipated that application of these same principles of open flow respiration to whole cell and perfused organ models will yield important information about the effects of IPC and other protective strategies on the gradual wake-up of respiration at reperfusion.
Figure 3. Mitochondrial respiration rate during early reoxygenation.
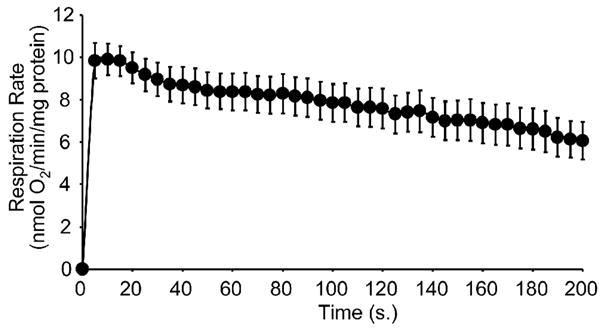
Respiration rate upon reoxygenation was calculated every 5 s. throughout a 200 s. reoxygenation. Data are means ± SEM; N≥3.
10. Clinical Application of the Gradual Wake up Hypothesis
No discussion of post-ischemic metabolism would be complete without mentioning ischemic post-conditioning (IPoCo). In IPoCo (also termed “staccato reperfusion”), blood flow to the heart is gradually restored, for example by careful control over pump rates following removal of a CABG patient from bypass [88]. It seems rational that IPoCo may achieve many of the same biochemical events at the mitochondrial level as discussed above regarding reversible inhibition, i.e. the slow reintroduction of electrons to the respiratory chain, thereby avoiding a surge of ROS generation and Ca2+ overload.
Recently, the pH hypothesis of IPoCo has been proposed [18], in which it is thought that IPoCo may work in part by extending the period of metabolic acidosis from ischemia into reperfusion. Acidic pH is known to inhibit the opening of the mitochondrial PT pore. Interestingly, it was shown that reperfusion with acidic buffers mimics the cardioprotective benefit of IPoCo, while reperfusion with alkaline buffers blocks this benefit [18]. It is possible that reversible mitochondrial inhibition may provide cardioprotective benefit in part by bringing about a state of metabolic acidosis during reperfusion, thereby mimicking IPoCo.
One molecule that has received much attention within the milieu of cardioprotection is nitrite (NO2−) [40]. A strong debate surrounds the mechanisms of nitrite mediated cardioprotection, with a central focus on the identification of the nitrite reductase responsible for the biotransformation of this molecule in hypoxia. Candidates for this activity include deoxyhemoglobin, deoxymyoglobin, and various components of the respiratory chain itself (see discussion in section 7). In addition the mechanism by which the hypoxic metabolites of nitrite bring about protected phenotype is also debated, but interestingly it is thought that mitochondrial S-nitrosation and inhibition may play a role [40]. Notably, regardless of these mechanisms, nitrite is now entering clinical trials for acute myocardial infarction.
Much attention in the preconditioning field has also focused on the effects of volatile anesthetics (VAs), which are broadly shown to be cardioprotective in many species including humans [89]. It has been shown that some VAs can inhibit mitochondrial respiration [35], while others are thought to activate K+ channels in the mitochondrial membrane [90]. Irrespective of the exact mechanism, VAs currently represent one of the most widely used, safest, and most clinically applicable preconditioning agents.
One interesting aspect that has arisen from studies of VA cardioprotection, is the discovery that both IPC and anesthetic preconditioning (APC) are not universally applicable to all patient populations. Factors which are known to abrogate the cardioprotective benefits of IPC/APC include age [91], diabetes [92], gender [93], and the use of drugs such as beta blockers [94]. It is not yet clear if any of this variability in patient response originates at the mitochondrial level.
Although reversible mitochondrial inhibition may indeed be beneficial in cardiac IR injury, current methods of bringing about such a phenotype, i.e. the use of mitochondrial inhibitors, are subject to some very severe side effects. Several respiratory chain inhibitors are neurotoxic: Administration of rotenone elicits a Parkinsonian like syndrome in animals [95], similar to the classical Parkinson’s inducer and complex I inhibitor MPTP [96]. Similarly, administration of 3-NP or malonate to animals creates striatal lesions akin to Huntington’s disease [97, 98]. Thus, while several mitochondrial inhibitors are widely available and cheap, it is strongly emphasized that their use in humans should be avoided due to such devastating side effects. The development of novel mitochondrial inhibitors which do not cross the hematoencephalic barrier may aid in limiting such effects, but even this cannot guarantee the absence of effects on other critical organs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 2.Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc Res. 2006;70:191–9. doi: 10.1016/j.cardiores.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Karmazyn M. The myocardial sodium-hydrogen exchanger (NHE) and its role in mediating ischemic and reperfusion injury. Keio J Med. 1998;47:65–72. doi: 10.2302/kjm.47.65. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307 (Pt 1):93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ. Nitric oxide: a signaling molecule against mitochondrial permeability transition- and pH-dependent cell death after reperfusion. Free Radic Biol Med. 2004;37:1943–50. doi: 10.1016/j.freeradbiomed.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong JS. Mitochondrial membrane permeabilization: the sine qua non for cell death. Bioessays. 2006;28:253–60. doi: 10.1002/bies.20370. [DOI] [PubMed] [Google Scholar]
- 7.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341 (Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 8.Borutaite V, Mildaziene V, Brown GC, Brand MD. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta. 1995;1272:154–8. doi: 10.1016/0925-4439(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 9.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–35. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto S, Matsui K, Ohashi N. Protective Effect of Na+/H+ Exchange Inhibitor, SM-20550, on Impaired Mitochondrial Respiratory Function and Mitochondrial Ca2+ Overload in Ischemic/Reperfused Rat Hearts. J Cardiovasc Pharmacol. 2002;39:569–75. doi: 10.1097/00005344-200204000-00013. [DOI] [PubMed] [Google Scholar]
- 11.Chakrabarti S, Hoque AN, Karmazyn M. A rapid ischemia-induced apoptosis in isolated rat hearts and its attenuation by the sodium-hydrogen exchange inhibitor HOE 642 (cariporide) J Mol Cell Cardiol. 1997;29:3169–74. doi: 10.1006/jmcc.1997.0561. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–9. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 13.Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol (Lond) 1999;519(Pt 2):347–60. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drose S, Brandt U, Hanley PJ. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J Biol Chem. 2006;281:23733–9. doi: 10.1074/jbc.M602570200. [DOI] [PubMed] [Google Scholar]
- 15.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–9. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 16.Horwitz LD, Fennessey PV, Shikes RH, Kong Y. Marked reduction in myocardial infarct size due to prolonged infusion of an antioxidant during reperfusion. Circulation. 1994;89:1792–801. doi: 10.1161/01.cir.89.4.1792. [DOI] [PubMed] [Google Scholar]
- 17.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, et al. Concentration-dependent Effects of Nitric Oxide on Mitochondrial Permeability Transition and Cytochrome c Release. J Biol Chem. 2000;275:20474–9. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 18.Cohen MV, Yang XM, Downey JM. The pH hypothesis of postconditioning: staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation. 2007;115:1895–903. doi: 10.1161/CIRCULATIONAHA.106.675710. [DOI] [PubMed] [Google Scholar]
- 19.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–99. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 20.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 21.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103:7607–12. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, et al. Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry. 2008;47:9636–45. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontaine E, Bernardi P. Progress on the mitochondrial permeability transition pore: regulation by complex I and ubiquinone analogs. J Bioenerg Biomembr. 1999;31:335–45. doi: 10.1023/a:1005475802350. [DOI] [PubMed] [Google Scholar]
- 25.Chance B. Reaction of oxygen with the respiratory chain in cells and tissues. J Gen Physiol. 1965;49:163–95. doi: 10.1085/jgp.49.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther. 2006;316:200–7. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 27.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961–7. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 28.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–34. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–25. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCormack JG, Stanley WC, Wolff AA. Ranolazine: a novel metabolic modulator for the treatment of angina. Gen Pharmacol. 1998;30:639–45. doi: 10.1016/s0306-3623(97)00301-7. [DOI] [PubMed] [Google Scholar]
- 31.Wyatt KM, Skene C, Veitch K, Hue L, McCormack JG. The antianginal agent ranolazine is a weak inhibitor of the respiratory complex I, but with greater potency in broken or uncoupled than in coupled mitochondria. Biochem Pharmacol. 1995;50:1599–606. doi: 10.1016/0006-2952(95)02042-x. [DOI] [PubMed] [Google Scholar]
- 32.D’Alonzo AJ, Grover GJ, Darbenzio RB, Hess TA, Sleph PG, Dzwonczyk S, et al. In vitro effects of capsaicin: antiarrhythmic and antiischemic activity. Eur J Pharmacol. 1995;272:269–78. doi: 10.1016/0014-2999(94)00653-o. [DOI] [PubMed] [Google Scholar]
- 33.Satoh T, Miyoshi H, Sakamoto K, Iwamura H. Comparison of the inhibitory action of synthetic capsaicin analogues with various NADH-ubiquinone oxidoreductases. Biochim Biophys Acta. 1996;1273:21–30. doi: 10.1016/0005-2728(95)00131-x. [DOI] [PubMed] [Google Scholar]
- 34.Bains R, Moe MC, Larsen GA, Berg-Johnsen J, Vinje ML. Volatile anaesthetics depolarize neural mitochondria by inhibiton of the electron transport chain. Acta Anaesthesiol Scand. 2006;50:572–9. doi: 10.1111/j.1399-6576.2006.00988.x. [DOI] [PubMed] [Google Scholar]
- 35.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH: ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687–93. doi: 10.1113/jphysiol.2002.025015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yue Y, Krenz M, Cohen MV, Downey JM, Critz SD. Menadione mimics the infarct-limiting effect of preconditioning in isolated rat hearts. Am J Physiol Heart Circ Physiol. 2001;281:H590–H595. doi: 10.1152/ajpheart.2001.281.2.H590. [DOI] [PubMed] [Google Scholar]
- 37.Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–9. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- 38.Kawabata H, Ishikawa K. Cardioprotection by metformin is abolished by a nitric oxide synthase inhibitor in ischemic rabbit hearts. Hypertens Res. 2003;26:107–10. doi: 10.1291/hypres.26.107. [DOI] [PubMed] [Google Scholar]
- 39.Hill BG, Darley-Usmar VM. S-nitrosation and thiol switching in the mitochondrion: a new paradigm for cardioprotection in ischaemic preconditioning. Biochem J. 2008;412:e11–e13. doi: 10.1042/BJ20080716. [DOI] [PubMed] [Google Scholar]
- 40.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci U S A. 2004;101:11880–5. doi: 10.1073/pnas.0401703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirata T, Fukuse T, Ishikawa S, Hanaoka S, Chen Q, Shoji T, et al. “Chemical preconditioning” by 3-nitropropionate reduces ischemia-reperfusion injury in cardiac-arrested rat lungs. Transplantation. 2001;71:352–9. doi: 10.1097/00007890-200102150-00003. [DOI] [PubMed] [Google Scholar]
- 43.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, et al. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning”. J Cereb Blood Flow Metab. 1997;17:257–64. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Wojtovich AP, Brookes PS. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta. 2008;1777:882–9. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial K(ATP) channels. Basic Res Cardiol. 2009 doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shiva S, Crawford JH, Ramachandran A, Ceaser EK, Hillson T, Brookes PS, et al. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem J. 2004;379:359–66. doi: 10.1042/BJ20031758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, et al. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 48.Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, et al. The pharmacology of nitroxyl (HNO) and its therapeutic potential: not just the Janus face of NO. Pharmacol Ther. 2007;113:442–58. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutman M, Kearney EB, Singer TP. Control of succinate dehydrogenase in mitochondria. Biochemistry. 1971;10:4763–70. doi: 10.1021/bi00801a025. [DOI] [PubMed] [Google Scholar]
- 50.Dzeja PP, Bast P, Ozcan C, Valverde A, Holmuhamedov EL, Van Wylen DG, et al. Targeting nucleotide-requiring enzymes: implications for diazoxide-induced cardioprotection. Am J Physiol Heart Circ Physiol. 2003;284:H1048–H1056. doi: 10.1152/ajpheart.00847.2002. [DOI] [PubMed] [Google Scholar]
- 51.Schafer G, Wegener C, Portenhauser R, Bojanovski D. Diazoxide, an inhibitor of succinate oxidation. Biochem Pharmacol. 1969;18:2678–81. [PubMed] [Google Scholar]
- 52.Pasdois P, Beauvoit B, Tariosse L, Vinassa B, Bonoron-Adele S, Santos PD. MitoK(ATP)-dependent changes in mitochondrial volume and in complex II activity during ischemic and pharmacological preconditioning of Langendorff-perfused rat heart. J Bioenerg Biomembr. 2006;38:101–12. doi: 10.1007/s10863-006-9016-3. [DOI] [PubMed] [Google Scholar]
- 53.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–8. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 54.Ludwig LM, Tanaka K, Eells JT, Weihrauch D, Pagel PS, Kersten JR, et al. Preconditioning by isoflurane is mediated by reactive oxygen species generated from mitochondrial electron transport chain complex III. Anesth Analg. 2004;99:1308–15. doi: 10.1213/01.ANE.0000134804.09484.5D. [DOI] [PubMed] [Google Scholar]
- 55.Kabir AM, Clark JE, Tanno M, Cao X, Hothersall JS, Dashnyam S, et al. Cardioprotection initiated by reactive oxygen species is dependent on activation of PKCepsilon. Am J Physiol Heart Circ Physiol. 2006;291:H1893–H1899. doi: 10.1152/ajpheart.00798.2005. [DOI] [PubMed] [Google Scholar]
- 56.Brunori M, Forte E, Arese M, Mastronicola D, Giuffre A, Sarti P. Nitric oxide and the respiratory enzyme. Biochim Biophys Acta. 2006;1757:1144–54. doi: 10.1016/j.bbabio.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 57.Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. Am J Physiol Cell Physiol. 2007;292:C1993–C2003. doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 58.Giulivi C, Kato K, Cooper CE. Nitric oxide regulation of mitochondrial oxygen consumption I: cellular physiology. Am J Physiol Cell Physiol. 2006;291:C1225–C1231. doi: 10.1152/ajpcell.00307.2006. [DOI] [PubMed] [Google Scholar]
- 59.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 60.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3:277–87. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 61.D’Amico G, Lam F, Hagen T, Moncada S. Inhibition of cellular respiration by endogenously produced carbon monoxide. J Cell Sci. 2006;119:2291–8. doi: 10.1242/jcs.02914. [DOI] [PubMed] [Google Scholar]
- 62.Pearce LL, Lopez ME, Martinez-Bosch S, Peterson J. Antagonism of Nitric Oxide Toward the Inhibition of Cytochrome c Oxidase by Carbon Monoxide and Cyanide. Chem Res Toxicol. 2008 doi: 10.1021/tx800140y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–e8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 64.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–9. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 65.Nicholson RA, Roth SH, Zhang A, Zheng J, Brookes J, Skrajny B, et al. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J Toxicol Environ Health A. 1998;54:491–507. doi: 10.1080/009841098158773. [DOI] [PubMed] [Google Scholar]
- 66.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–5. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kodde IF, van der SJ, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol A Mol Integr Physiol. 2007;146:26–39. doi: 10.1016/j.cbpa.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 68.Vogt AM, Poolman M, Ackermann C, Yildiz M, Schoels W, Fell DA, et al. Regulation of glycolytic flux in ischemic preconditioning. A study employing metabolic control analysis. J Biol Chem. 2002;277:24411–9. doi: 10.1074/jbc.M201138200. [DOI] [PubMed] [Google Scholar]
- 69.McDonald LJ, Moss J. Stimulation by nitric oxide of an NAD linkage to glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci U S A. 1993;90:6238–41. doi: 10.1073/pnas.90.13.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mohr S, Stamler JS, Brune B. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J Biol Chem. 1996;271:4209–14. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 71.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–63. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nadtochiy SM, Baker PR, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc Res. 2008 doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takeuchi K, Cao-Danh H, Friehs I, Glynn P, D’Agostino D, Simplaceanu E, et al. Administration of fructose 1, 6-diphosphate during early reperfusion significantly improves recovery of contractile function in the postischemic heart. J Thorac Cardiovasc Surg. 1998;116:335–43. doi: 10.1016/s0022-5223(98)70135-7. [DOI] [PubMed] [Google Scholar]
- 74.Marchionni N, Conti A, De AW, Di BM, Ferrucci L, Lombardi A, et al. Hemodynamic and electrocardiographic effects of fructose-1, 6-diphosphate in acute myocardial infarction. Am J Cardiol. 1985;56:266–9. doi: 10.1016/0002-9149(85)90847-1. [DOI] [PubMed] [Google Scholar]
- 75.Yabe K, Nasa Y, Sato M, Iijima R, Takeo S. Preconditioning preserves mitochondrial function and glycolytic flux during an early period of reperfusion in perfused rat hearts. Cardiovasc Res. 1997;33:677–85. doi: 10.1016/s0008-6363(96)00269-6. [DOI] [PubMed] [Google Scholar]
- 76.Peralta C, Bartrons R, Riera L, Manzano A, Xaus C, Gelpi E, et al. Hepatic preconditioning preserves energy metabolism during sustained ischemia. Am J Physiol Gastrointest Liver Physiol. 2000;279:G163–G171. doi: 10.1152/ajpgi.2000.279.1.G163. [DOI] [PubMed] [Google Scholar]
- 77.Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. 2001;21:1105–14. doi: 10.1097/00004647-200109000-00008. [DOI] [PubMed] [Google Scholar]
- 78.Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496–H499. doi: 10.1152/ajpheart.01182.2004. [DOI] [PubMed] [Google Scholar]
- 79.Brooks GA, Dubouchaud H, Brown M, Sicurello JP, Butz CE. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc Natl Acad Sci U S A. 1999;96:1129–34. doi: 10.1073/pnas.96.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brooks GA. Lactate shuttles in nature. Biochem Soc Trans. 2002;30:258–64. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 81.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol. 2008;28:1007–17. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Borchi E, Parri M, Papucci L, Becatti M, Nassi N, Nassi P, et al. Role of NADPH oxidase in H9c2 cardiac muscle cells exposed to simulated ischemia-reperfusion. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zuurbier CJ, Eerbeek O, Goedhart PT, Struys EA, Verhoeven NM, Jakobs C, et al. Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–53. doi: 10.1016/j.cardiores.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 84.Ozcan C, Holmuhamedov EL, Jahangir A, Terzic A. Diazoxide protects mitochondria from anoxic injury: implications for myopreservation. J Thorac Cardiovasc Surg. 2001;121:298–306. doi: 10.1067/mtc.2001.111421. [DOI] [PubMed] [Google Scholar]
- 85.Cole RP, Sukanek PC, Wittenberg JB, Wittenberg BA. Mitochondrial function in the presence of myoglobin. J Appl Physiol. 1982;53:1116–24. doi: 10.1152/jappl.1982.53.5.1116. [DOI] [PubMed] [Google Scholar]
- 86.Brookes PS, Kraus DW, Shiva S, Doeller JE, Barone MC, Patel RP, et al. Control of mitochondrial respiration by NO*, effects of low oxygen and respiratory state. J Biol Chem. 2003;278:31603–9. doi: 10.1074/jbc.M211784200. [DOI] [PubMed] [Google Scholar]
- 87.Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol. 2007;292:H101–H108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- 88.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 89.Stadnicka A, Marinovic J, Ljubkovic M, Bienengraeber MW, Bosnjak ZJ. Volatile anesthetic-induced cardiac preconditioning. J Anesth. 2007;21:212–9. doi: 10.1007/s00540-006-0486-6. [DOI] [PubMed] [Google Scholar]
- 90.Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: the role of mitochondrial KATP channels. Anesth Analg. 2005;101:1590–6. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- 91.Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol. 2007;103:2120–8. doi: 10.1152/japplphysiol.00647.2007. [DOI] [PubMed] [Google Scholar]
- 92.Tanaka K, Kehl F, Gu W, Krolikowski JG, Pagel PS, Warltier DC, et al. Isoflurane-induced preconditioning is attenuated by diabetes. Am J Physiol Heart Circ Physiol. 2002;282:H2018–H2023. doi: 10.1152/ajpheart.01130.2001. [DOI] [PubMed] [Google Scholar]
- 93.Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2007;75:478–86. doi: 10.1016/j.cardiores.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 94.Riess ML, Stowe DF, Warltier DC. Cardiac pharmacological preconditioning with volatile anesthetics: from bench to bedside? Am J Physiol Heart Circ Physiol. 2004;286:H1603–H1607. doi: 10.1152/ajpheart.00963.2003. [DOI] [PubMed] [Google Scholar]
- 95.Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- 96.Gerlach M, Riederer P, Przuntek H, Youdim MB. MPTP mechanisms of neurotoxicity and their implications for Parkinson’s disease. Eur J Pharmacol. 1991;208:273–86. doi: 10.1016/0922-4106(91)90073-q. [DOI] [PubMed] [Google Scholar]
- 97.Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993;61:1147–50. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 98.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–9. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]