

Abstract
Diabetes increases mortality and accelerates left ventricular (LV) dysfunction following myocardial infarction (MI). This study sought to determine the impact of impaired myocardial insulin signaling, in the absence of diabetes, on the development of LV dysfunction following MI. Mice with cardiomyocyte-restricted knock out of the insulin receptor (CIRKO) and wild type (WT) mice were subjected to proximal left coronary artery ligation (MI) and followed for 14 days. Despite equivalent infarct size, mortality was increased in CIRKO-MI vs. WT-MI mice (68 % vs. 40 %, respectively). In surviving mice, LV ejection fraction and dP/dt were reduced by > 40% in CIRKO-MI vs. WT-MI. Relative to shams, isometric developed tension in LV papillary muscles increased in WT-MI but not in CIRKO-MI. Time to peak tension and relaxation times were prolonged in CIRKO-MI vs. WT-MI suggesting impaired, load-independent myocardial contractile function. To elucidate mechanisms for impaired LV contractility, mitochondrial function was examined in permeabilized cardiac fibers. Whereas maximal ADP-stimulated mitochondrial O2 consumption rates (VADP) with palmitoyl carnitine were unchanged in WT-MI mice relative to sham-operated animals, VADP was significantly reduced in CIRKO-MI (13.17 ± 0.94 vs. 9.14 ± 0.88 nmol O2/min/mgdw, p<0.05). Relative to WT-MI, expression levels of GLUT4, PPAR-α, SERCA2, and the FA-Oxidation genes MCAD, LCAD, CPT2 and the electron transfer flavoprotein ETFDH were repressed in CIRKO-MI. Thus reduced insulin action in cardiac myocytes accelerates post-MI LV dysfunction, due in part to a rapid decline in mitochondrial FA oxidative capacity, which combined with limited glucose transport capacity may reduce substrate utilization and availability.
Keywords: insulin signaling, myocardial infarction, energy metabolism, mitochondria, fatty acid metabolism
1. Introduction
Diabetes increases the risk of cardiovascular disease with diabetic subjects having a 2- to 4-fold greater risk of death from myocardial infarction compared with non-diabetic subjects [1-5]. Multiple abnormalities associated with diabetes mellitus such as hyperglycemia, hyperlipidemia and insulin resistance, have been postulated to contribute to adverse outcomes in diabetes following myocardial ischemia [6-11]. The specific contribution of impaired myocardial insulin action per se in the response of the heart to myocardial ischemia is incompletely understood. To examine the role of altered cardiomyocyte insulin signaling in the adaptation to myocardial infarction, independently of confounding systemic factors, we subjected mice with cardiomyocyte-selective knockout of the insulin receptor (CIRKO) [12] to coronary artery ligation. CIRKO mice have normal serum insulin and lipid concentrations and normal systemic glucose homeostasis. Despite a 20% reduction in heart-weight, CIRKO mice develop an equivalent or increased LV hypertrophy following aortic banding or isoproterenol infusion. In addition, CIRKO mice develop decreased LV function in the setting of these stressors [13, 14]. Thus we hypothesized that impaired myocardial insulin action would accelerate adverse LV remodeling following myocardial infarction. The present study revealed that loss of myocardial insulin signaling led to accelerated LV dysfunction that was associated with significant mitochondrial dysfunction due in part to an accelerated decline in mitochondrial fatty acid (FA) oxidative capacity.
2. Materials and methods
2.1. Animals/Myocardial Infarction
Mice with cardiomyocyte-selective ablation of the insulin receptor (CIRKO) were generated as previously described [12]. The animals were fed a standard chow and housed in temperature-controlled facilities with a 12-h light and 12-h dark cycle. Myocardial infarction (MI) was induced in 12 week-old female CIRKO and WT mice. Mice were anesthetized, intubated, and ventilated with room air. A left thoracotomy was performed via the fourth intercostal space. The left coronary artery proximal to the bifurcation of the largest lateral branch was ligated with a 9-0 silk suture. Ligation was considered successful if blanching of the anterior wall of the left ventricle wall occurred and ST segment elevation was observed on the electrocardiogram. Because early studies revealed increased mortality in CIRKO-MI mice, hemodynamic measurements and papillary muscle data were collected 11 days post infarction for WT-MI group and 7 days post infarction for CIRKO-MI group. The goal was to assess potential mechanisms of LV dysfunction during the time period of greatest risk, rather than at the end stage of the process. In later studies, echocardiographic data, mitochondrial respiration, gene expression and capillary density data were collected 14 days post infarction in both groups. All animal experimentation was conducted in accordance with guidelines approved by the Institutional Animal Care and Use Committee of the University of Utah.
2.2. Assessment of in vivo cardiac function
Echocardiography and invasive LV hemodynamic measurements were performed as previously described by us [15].
2.3. Isometric developed tension in isolated LV papillary muscles
LV papillary muscles were dissected in an oxygenated muscle bath. A portion of the mitral valve and chordae tendinae were preserved at one end and a small section of LV wall at the other. These ends were clamped with compression springs, mounted horizontally in a bath perfused with 37°C Tyrode's solution (bubbled with 100% O2, 1mM[Ca2+] and pH 7.4) and electrically stimulated at 1-10 Hz. At the end of the experiment, the length and weight of each muscle was obtained and the average cross sectional area calculated assuming cylindrical geometry. Force was normalized to cross sectional area.
2.4. Capillary density and infarct size
Capillary density was determined by isolectin GS-IB4 staining. Briefly, mice received an intravenous bolus injection of isolectin GS-IB4 Alexa Fluor 488 (20 μg; Invitrogen, Carlsbad, CA) into the jugular vein. Two minutes after injection, mice were killed, the hearts were removed, rinsed and then formalin fixed for 24 hours. Fixed tissues were paraffin-embedded and 8 μm sections cut. Capillary count was then determined using Image J® software and normalized to tissue area. Six sections from each heart that showed the capillaries in cross section were analyzed. In the subset of hearts used for papillary muscle studies, the interventricular septum was cut longitudinally and the left ventricle was opened. The endocardial surface of the heart was photographed and the infarcted area and the non-infarcted area were measured by planimetry. Clear demarcation between infarcted and normal tissue was present in all cases. This method provides similar estimates of infarct size as those obtained using Triphenyltetrazolium chloride (TTC) (Supplemental Figure 1). TTC or other staining techniques were not used in this study because of the need to obtain tissue for papillary muscle and mitochondrial studies.
2.5. Mitochondrial respiration
Mitochondrial function (oxygen consumption rates) was studied in saponin-permeabilized fibers using previously described methods [16-18]. Substrates used were 20 μM palmitoyl-carnitine with 2 mM malate.
2.6. Total RNA isolation and quantitative PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) and purified using the RNAeasy total RNA isolation kit (Qiagen, Valencia, CA) as previously described by us [15]. Data were normalized relative to the levels of the invariant transcript cyclophilin and expressed as fold change relative to the mean WT sham value. Primer sequences are available upon request.
2.7. Statistics
Data are presented as means ± SEM. Statistical significance was determined by one way ANOVA followed by Fisher's least protected squares test using StatView 5.0.1 software (SAS Institute, Cary, NC) and p < 0.05 was considered significant.
3. Results
3.1. Morphology
Infarct size (% of LV surface) was similar in WT-MI and CIRKO-MI (48 ± 3 % versus 47 ± 3 %, respectively). Body weights, and tibia lengths, were unchanged in sham or infarcted WT and CIRKO mice (Table 1). Heart weights normalized to body weight or tibia length were reduced in sham CIRKO relative to WT shams, as previously described [12], and increased equivalently in WT-MI and CIRKO-MI relative to their respective shams (Table 1).
Table 1. Heart and body weight measurements.
| WT Sham (n=10) |
WT MI (n=6) |
CIRKO Sham (n=10) |
CIRKO MI (n=4) |
|
|---|---|---|---|---|
| Body Weight (BW) (g) | 22.35±0.82 | 20.55±1.07 | 23.00±0.67 | 21.10±1.42 |
| Heart Weight (HW) (mg) | 110±4 | 126±6a | 93±3a,c | 114±7b |
| Tibia Length (TL) (mm) | 16.42±0.09 | 16.20±0.18 | 16.60±0.14 | 16.97±0.25 |
| HW/BW | 4.96±0.14 | 6.31±0.61a | 4.08±0.20a,c | 5.44±0.32b |
| HW/TL | 6.74±0.29 | 7.84±0.46a | 5.61±0.18a,c | 6.73±0.38b |
Data are mean ± SEM.,
p<0.05 vs. WT Sham;
p<0.05 vs. CIRKO Sham;
p <0.05 vs. WT MI
3.2. Increased mortality after MI in CIRKO mice
As shown in Table 2, CIRKO mice undergoing MI surgery had higher acute and late (> 24 hours after surgery) mortality rates compared to WT (68% versus 40%, P <0.05).
Table 2. Increased Mortality after Myocardial Infarction in CIRKO Mice.
| WT Sham (n=11) |
WT MI (n=22) |
CIRKO Sham (n=11) |
CIRKO MI (n=25) |
|
|---|---|---|---|---|
| Perioperative Death | 1 | 5 | 1 | 11 |
| Survival to end of study | 10 | 13 | 10 | 8 |
| Late Death (>24 hours) | 0 | 4 | 0 | 6 |
| Total Mortality Rate | 9% | 40% | 9% | 68%* |
P<0.05 versus WT MI
3.3. Contractile dysfunction in CIRKO MI hearts
LV catheterization and echocardiography were performed to evaluate in vivo cardiac function.
Echocardiography
Systolic and diastolic chamber dimensions and posterior wall thickness were increased in WT hearts following MI, but fractional shortening, ejection fraction and stroke volumes were preserved. CIRKO-MI also developed increased chamber dimensions, but in contrast to WT, adverse LV remodeling was evidenced by thinning of the interventricular septum and 32%, 34% and 28% reduction in ejection fraction, fractional shortening and stroke volume respectively vs. sham operated CIRKO mice (Table 3).
Table 3. Echocardiographic measurements.
| WT Sham Day 0 |
WT MI Day 0 |
CIRKO Sham Day 0 |
CIRKO MI Day 0 |
WT Sham 2 weeks |
WT MI 2 weeks |
CIRKO Sham 2 weeks |
CIRKO MI 2 weeks |
|
|---|---|---|---|---|---|---|---|---|
| LVDd. (cm) | 0.395±0.006 | 0.383±0.009 | 0.374±0.004 | 0.381±0.009 | 0.389±0.006 | 0.419±0.021b,c | 0.391±0.004 | 0.445±0.026b,c,e |
| LVDs. (cm) | 0.288±0.008 | 0.269±0.009 | 0.269±0.007 | 0.271±0.006 | 0.277±0.005 | 0.322±0.032b,c | 0.290±0.005 | 0.374±0.044a |
| IVSd. (cm) | 0.054±0.001 | 0.056±0.004 | 0.056±0.004 | 0.046±0.002 | 0.056±0.003 | 0.063±0.003 | 0.053±0.0008 | 0.048±0.006d |
| IVSs. (cm) | 0.091±0.004 | 0.100±0.007 | 0.081±0.004 | 0.085±0.006 | 0.098±0.005 | 0.098±0.002 | 0.089±0.001 | 0.068±0.014d,c |
| PWd. (cm) | 0.058±0.001 | 0.061±0.003 | 0.064±0.004 | 0.056±0.004 | 0.058±0.003 | 0.080±0.011a | 0.060±0.002 | 0.057±0.005d |
| PWs. (cm) | 0.086±0.003 | 0.091±0.003 | 0.089±0.003 | 0.089±0.004 | 0.091±0.002 | 0.112±0.015a | 0.087±0.002 | 0.080±0.006d |
| % FS | 27.01±1.51 | 29.56±1.02 | 28.17±1.40 | 28.87±0.55 | 28.79±1.10 | 24.09±4.06 | 25.72±0.92 | 16.86±6.01a |
| % EF | 59.86±2.56 | 64.94±1.59 | 62.56±2.15 | 63.98±0.84 | 63.68±1.72 | 54.75±7.03 | 58.85±1.50 | 39.95±10.90a |
| SV. (ml) | 0.037±0.001 | 0.037±0.002 | 0.035±0.001 | 0.035±0.003 | 0.036±0.002 | 0.035±0.001 | 0.035±0.001 | 0.025±0.004a |
| HR (beats/min) | 406±18 | 404±15 | 409±12 | 376±13 | 428±8 | 390±12 | 407±13 | 394±22 |
Data are mean ± SEM. LVDd, LV diastolic diameter; LVDs, LV systolic diameter; IVSd, interventricular septum thickness measured in diastole; IVSs, interventricular septum thickness measured in systole; PWd, posterior wall thickness measured in diastole; PWs, posterior wall thickness measured in systole; FS, fractional shortening; EF, ejection fraction; SV, stroke volume; HR, heart rate. WT sham, n=10; WT MI n=6; CIRKO sham n=10; CIRKO MI n=4.
p < 0.05 vs. all 7 other groups;
p<0.05 vs. same group Day 0;
p<0.05 vs. SHAM same genotype same time point;
p<0.05 vs. WT same surgery same time point;
p <0.05 vs. SHAM same genotype Day 0.
LV catheterization
As shown in Figure 1(A-C), invasive hemodynamic measurements showed a significant reduction in LV systolic pressure (LVSP) in CIRKO-MI vs. WT-MI hearts. Peak rates of LV pressure increase (+dP/dt) and decrease (-dP/dt) were significantly lower in CIRKO-MI relative to WT-MI (40 and 45% lower, respectively, P<0.05).
Figure 1. Hemodynamic parameters obtained after LV catheterization (in vivo) and isometric developed tension in isolated LV papillary muscles.
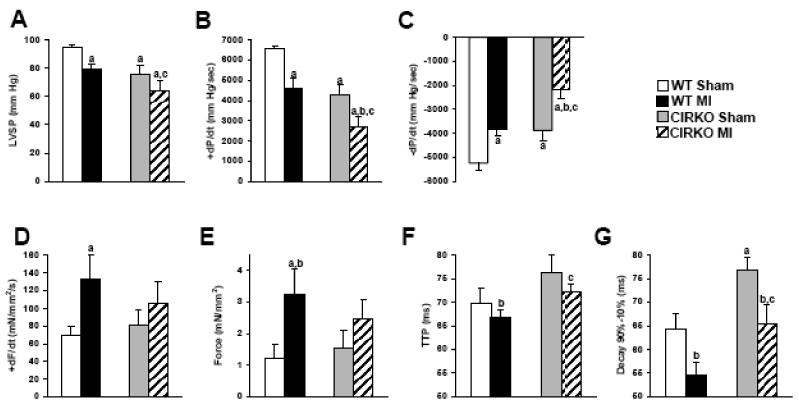
A, LVSP: LV systolic pressure; B, +dP/dt: peak rate of LV pressure increase; C, -dP/dt: peak rate of LV pressure decrease from 4 sham WT, 4 sham CIRKO, 7 WT MI, 4 CIRKO MI. Data represent mean ± SEM., a, p <0.05 vs. WT Sham; b, p <0.05 vs. CIRKO Sham; c, p <0.05 vs. WT MI. D, developed tension; E, dF/dt: rate of change in force development normalized to muscle cross sectional area; F, time to peak force development; G, Decay time from 90% to 10% of maximal force in isolated papillary muscles obtained from 4 sham WT, 4 sham CIRKO, 7 WT MI, 4 CIRKO MI. Buffer Ca2+ concentration was 1mM. Data represent mean ± SEM., a, p <0.05 vs. WT Sham; b, p <0.05 vs. CIRKO Sham; c, p <0.05 vs. WT MI. Hemodynamic measurements and papillary muscle data were collected 11 days post infarction for WT-MI group and 7 days post infarction for CIRKO-MI group.
Papillary muscle function
Isometric developed tension and maximum rate of force development (dF/dt) in isolated LV papillary muscles increased significantly in infarcted WT relative to shams, but did not increase significantly in CIRKO-MI relative to CIRKO-sham (Figure 1D-E). Time to peak tension and the decay time were prolonged in the CIRKO-MI compared to the WT-MI muscles (72.28±1.60 vs. 66.90±1.39 ms and 65.49±4.06 vs. 54.70±2.48 ms, respectively, [Ca2+]=1.0 mM, P <0.05), indicating differences in intrinsic myocardial contractile function (Figure 1F-G).
Gene expression
Consistent with the greater decline in contractile function in CIRKO-MI, mRNA levels of hypertrophy/heart failure markers alpha skeletal actin (ACTA1), brain natriuretic peptide (BNP) and atrial natriuretic peptide (ANP) were disproportionately increased in CIRKO-MI hearts relative to WT-MI. Additionally, CIRKO-MI hearts showed a significant increase in the β-myosin heavy chain isoform while the mRNA expression of the sarcoplasmic reticulum Ca2+ ATPase2 (SERCA2) was significantly repressed relative to the WT-MI group (Figure 2).
Figure 2. Expression of MHC isoforms, SERCA2 and markers of heart failure.
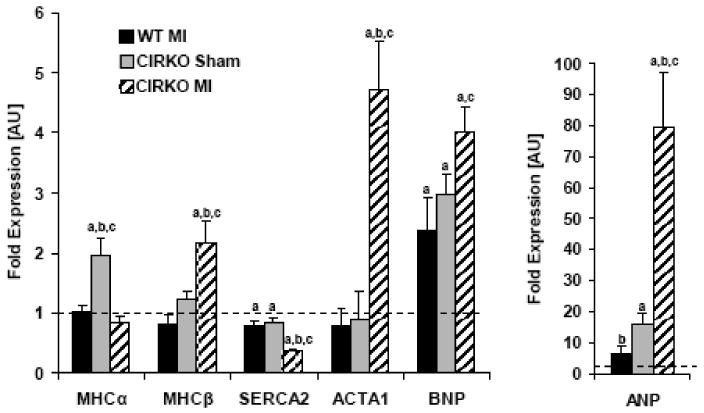
mRNA from 6 sham WT, 6 sham CIRKO, 6 WT-MI and 6 MI- CIRKO hearts were amplified by real-time PCR and normalized to cyclophilin. Values represent fold change in mRNA transcript levels relative to wildtype, which was assigned as 1. Data represent mean ± SEM., a, p <0.05 vs. WT Sham; b, p <0.05 vs. CIRKO Sham; c, p <0.05 vs. WT MI. MHC-α/β: myosin heavy chain isoforms α/β; SERCA2: sarcoplasmic reticulumCa2+ ATPase 2; ACTA1: α-skeletal actin; BNP: brain natriuretic peptide; ANP: atrial natriuretic peptide.
3.4. Mitochondrial oxygen consumption
To explore potential mechanisms for accelerated LV dysfunction in CIRKO-MI we examined mitochondrial function by determining oxygen consumption in fibers that were exposed to the FA substrate palmitoyl carnitine (Figure 3). Basal mitochondrial respiration rates (V0) were unchanged in all fiber preparations. Maximal ADP-stimulated mitochondrial respirations (VADP) in fibers from non-infarcted LV of WT-MI were similar to those of sham operated WT mice. VADP in fibers from sham CIRKO although lower than sham WT, did not achieve statistical significance (p=0.174). VADP was significantly decreased in CIRKO-MI (9.14 ± 0.88 nmol/min/mg dry weight) relative to WT sham and WT-MI groups (14.07 ± 0.97 and 13.17 ± 0.94 nmol/min/mg dry weight, respectively; P <0.05) and the reduction in VADP in CIRKO-MI relative to sham CIRKO (9.14 ± 0.88 versus 12.11 ± 0.72 nmol/min/mg dry weight) approached statistical significance (P=0.057, Figure 3). VOligo, (oxygen consumption in the presence of the ATP synthase inhibitor oligomycin) and the respiratory control (RC) ratio were not different in any group.
Figure 3. Mitochondrial function.
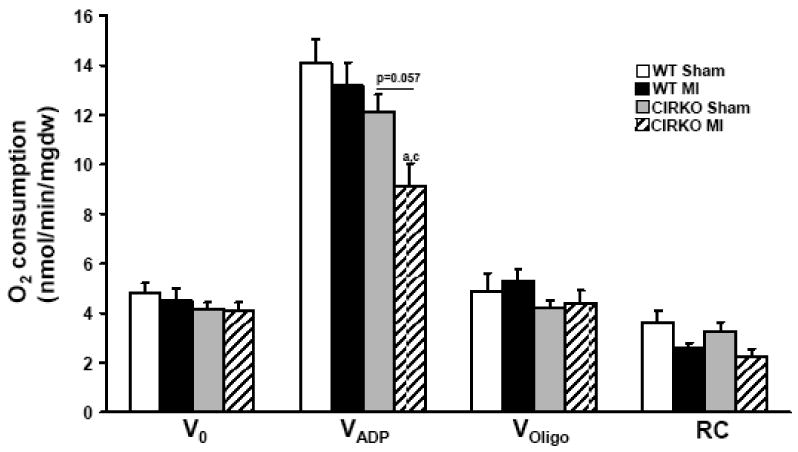
Mitochondrial oxygen consumption in the presence of palmitoyl-carnitine (20μmol/l) and malate (2mmol/l) as substrate, in permeabilized fibers obtained from sham-operated WT and CIRKO mice, and WT and CIRKO mice, 14- days after MI as indicated. a p<0.05 vs. WT Sham; c p<0.05 vs. WT MI. V0 – basal rates of O2 consumption, VADP – maximal ADP stimulated rates of O2 consumption, VOligo – O2 consumption rates in the presence of substrate and oligomycin (1μg/ ml); RC – respiratory control ratio (VADP/Voligo).
3.5. Expression of metabolic genes
To explore mechanisms that could account for mitochondrial dysfunction, expression levels of genes that regulate mitochondrial function and metabolism were determined (Figure 4). In WT-MI but not in CIRKO hearts, expression levels of PGC-1α were reduced relative to their respective shams. In contrast, PPAR-α expression was selectively repressed in CIRKO-MI relative to all other groups. PGC-1β expression was reduced in WT-MI hearts relative to WT sham. Interestingly loss of insulin receptors reduced PGC-1β expression that was not further changed in CIRKO-MI. The expression of fatty acid oxidation (FAO), FA transporter and OXPHOS genes was reduced in the hearts of WT mice following MI. These genes were already lower in CIRKO mice at baseline and expression levels of most, did not change further following MI. However, there was additional repression of the complex I subunit gene NDUFA9, the electron transfer flavoprotein dehydrogenase (ETFDH), the FAO genes for long and medium-chain acyl CoA dehydrogenases (MCAD and LCAD), and carnitine palmitoyl transferase 2 (CPT2) in CIRKO–MI. UCP2 expression was increased in WT-MI. Levels of UCP2 mRNA were already increased in Sham CIRKO and did not increase further after MI. With regards to genes that regulate glucose uptake and glycolysis, MI reduced expression levels of the glucose transporter GLUT4, pyruvate dehydrogenase kinase (PDK4) and hexokinase 2 (HK2) in WT mice. Loss of insulin receptors lowered the expression levels of GLUT4, PDK4 and HK2 genes in non-stressed hearts. In CIRKO-MI, PDK4 and HK2 expression did not decline further, however there was additional reduction in GLUT4 expression. Of interest, expression levels of the hypoxia-inducible factor (HIF-1α) were significantly induced in CIRKO-MI hearts.
Figure 4. Expression levels of genes that regulate mitochondrial metabolism and substrate utilization.
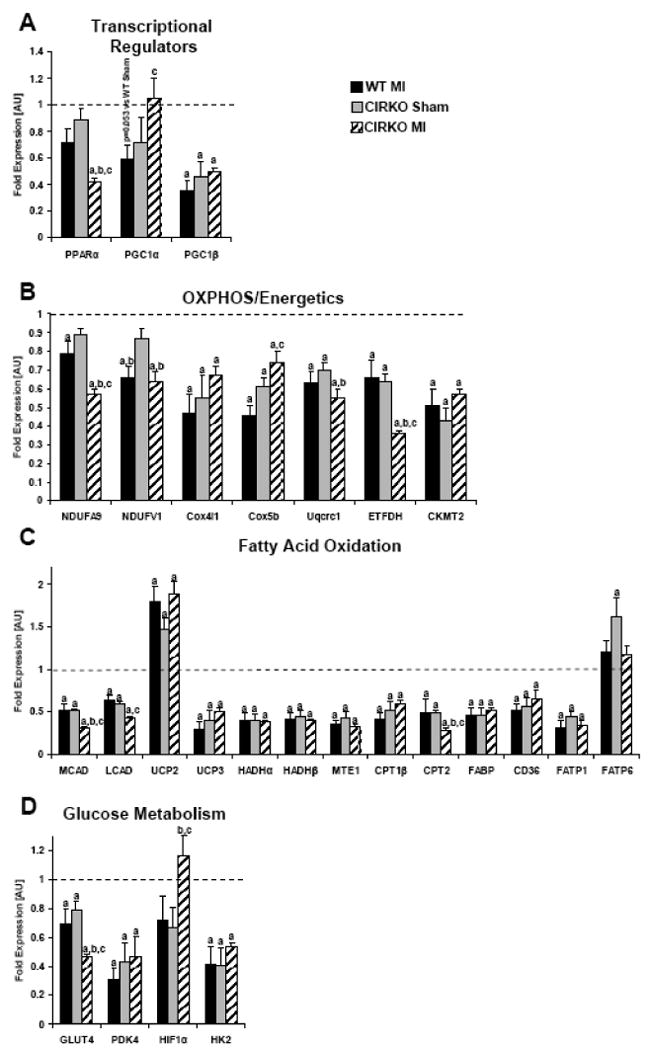
mRNA from 6 sham WT, 6 sham CIRKO, 6 WT-MI and 6 CIRKO-MI hearts were amplified by real-time PCR and normalized to cyclophilin. Values represent fold change in mRNA transcript levels relative to wildtype, which was assigned as 1. A, Transcriptional regulators of mitochondria, B, Regulators of mitochondrial oxidative phosphorylation (OXPHOS), C, Regulators of FA transport and mitochondrial FA oxidation, D, Regulators of glucose metabolism. Data represent mean ± SEM., a, p <0.05 vs. WT Sham; b, p <0.05 vs. CIRKO Sham; c, p <0.05 vs. WT MI. PPARα: peroxisome proliferator-activated receptor α; PGC1α/β: peroxisome proliferator-activated receptor gamma co-activator 1 α/β; NDUFA9: NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 9; NDUFV1: NADH dehydrogenase (ubiquinone) flavoprotein 1; Cox4i1: cytochrome c oxidase subunit IV isoform 1; Cox5b: cytochrome c oxidase subunit Vb; Uqcrc1: ubiquinol-cytochrome-c reductase complex core protein 1; ETFDH: electron transfer flavoprotein dehydrogenase; CKMT2: mitochondrial creatine kinase 2; MCAD: medium chain acyl-CoA dehydrogenase; LCAD: long chain acyl-CoA dehydrogenase; UCP2/3: uncoupling protein 2/3; FABP: fatty acid binding protein; HADHα/β: 3-hydroxyacyl-Coenzyme A dehydrogenase α/β; CPT1β: carnitine palmitoyl transferase 1β; CPT2: carnitine palmitoyl transferase 2; MTE1: mitochondrial thioesterase 1; CD36: fatty acid translocase; GLUT4: glucose transporter 4; PDK4: pyruvate dehydrogenase kinase 4; HIF1α: hypoxia inducible factor 1α; HK2: hexokinase 2; FATP1/6: fatty acid transport protein 1/6.
3.6. Capillary density
We previously reported that LV dysfunction following isoproterenol treatment was associated with increased fibrosis and decreased capillary density [14]. Thus to determine if decreased vascularity of the heart following myocardial infarction could account for contractile dysfunction and increased mortality in the CIRKO mice, we determined capillary density using isolectin staining. Capillary density was equivalently decreased in both WT-MI and CIRKO-MI groups compared to their respective sham animals (Figure 5), ruling out the possibility that decreased capillary density in CIRKO hearts following MI accounted for contractile dysfunction.
Figure 5. Capillary Density.
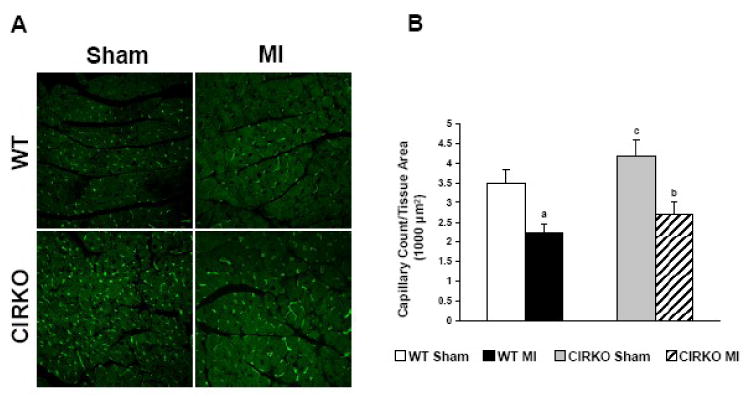
A, Representative micrographs of isolectin staining on tissue sections obtained from WT Sham, WT MI, CIRKO Sham and CIRKO MI hearts. B, Quantification of capillary number in the 4 groups. WT Sham (n=4), WT MI (n=7), CIRKO Sham (n=4), CIRKO MI (n=4). a, p <0.05 vs. WT Sham; b, p <0.05 vs. CIRKO Sham; c, p <0.05 vs. WT MI.
4. Discussion
The present study shows that genetic loss of insulin signaling leads to accelerated LV dysfunction following myocardial infarction. LV dysfunction is associated with reduced rates of mitochondrial oxygen consumption in response to the FA substrate palmitoyl carnitine. Although there is a general repression of FAO genes in the post-MI LV in control mice, loss of insulin signaling is associated with reduced expression of many FAO and OXPHOS genes at baseline. However, there is progressive reduction in levels of the transcriptional regulator of FAO, PPAR-α and additional declines in expression levels of MCAD, LCAD, CPT2 and ETFDH. Thus loss of insulin signaling in the heart may limit FA utilization during remodeling of the LV following myocardial infarction. In addition, there is further repression of GLUT4 expression in the LV of CIRKO mice post-MI. GLUT4 is an important mediator of basal myocardial glucose uptake in vivo [12, 19, 20], thus reduced GLUT4 expression might further limit substrate availability thereby contributing to impaired LV function following MI. Although we focused primarily on the potential role of mitochondrial dysfunction in impairing LV function in CIRKO hearts following MI, our studies revealed additional pathogenic mechanisms. Invasive LV catheterization revealed reduced −dP/dt and papillary muscle preparations revealed impaired relaxation kinetics. Although diastolic relaxation is an energy requiring process, which could be impaired in the context of reduced mitochondrial energetics, defects in calcium re-uptake or myosin isoform switching could also contribute to impaired diastolic relaxation, as was confirmed by increased expression of the slow-twitch β-myosin isoform and reduced expression of SERCA2 in CIRKO hearts following MI.
Many human studies confirm that outcomes of ischemia/reperfusion and adaptations of diabetic hearts to ischemia are impaired [1, 3, 21-25]. Ischemic heart disease accounts for more than 50% of deaths in diabetic patients and mortality from acute myocardial infarction is increased 2-fold in diabetic patients compared with non-diabetic individuals [26-29]. Metabolic disturbances associated with diabetes mellitus such as hyperglycemia, hyperlipidemia and insulin resistance, have been postulated to contribute to the incidence and outcomes of myocardial ischemia in diabetes [6-11], however the specific contribution of insulin resistance or loss of insulin action per se have been difficult to elucidate. Studies in animal models of genetic or diet-induced insulin resistance and obesity, have illustrated adverse outcomes following in vivo coronary artery ligation (reviewed in [30]). And although in some of these models, myocardial insulin resistance has been reported, it remains uncertain the extent to which impaired myocardial insulin action may directly contribute to adverse outcomes following myocardial infarction. The present study utilized a model with a genetic deficiency in insulin signaling that was isolated to cardiomyocytes and suggests that impaired myocardial insulin signaling might independently contribute to adverse LV remodeling following myocardial infarction. Importantly, vascular smooth muscle and endothelial insulin signaling are not affected in this model
The mechanisms by which impaired insulin signaling leads to mitochondrial dysfunction are complex. We have recently reported that in CIRKO hearts, mitochondrial dysfunction in non-stressed hearts is due in part to oxidative stress and a global reduction in mitochondrial content of FAO proteins and critical subunits of the pyruvate dehydrogenase complex, that are due to reduced expression of FAO genes and their regulator PPAR-α and the PDH E1α1 subunit [31]. Moreover, PI3K signaling may play an important role in increasing mitochondrial FAO capacity in the context of physiological cardiac hypertrophy [32]. Thus impaired insulin-PI3K signaling reduces mitochondrial metabolic capacity that likely renders them more susceptible to progressive mitochondrial dysfunction during post-MI LV remodeling. Although our data raises the possibility of a causal relationship between mitochondrial dysfunction and accelerated LV dysfunction in the remodeled LV of CIRKO mice following myocardial infarction, an alternative possibility is that mitochondria in CIRKO mice are sensitized to the increase in oxidative stress and tissue hypoxia that characterizes the remodeling LV. Indeed, we observed a striking increase in expression of HIF-1α mRNA in CIRKO hearts post MI. Oxidative stress has been shown to increase the binding of NFKB to the HIF-1α promoter thereby increasing its expression [33-35]. Moreover, HIF-1α has been shown to directly repress the expression of PPAR-α in epithelial cells [36]. Thus it is possible that an initial impairment in mitochondrial dysfunction in CIRKO hearts could be amplified during the process of LV remodeling, which sets up a vicious cycle leading to an acceleration of mitochondrial and contractile dysfunction following MI in CIRKO hearts.
Some recent studies have examined the mitochondrial adaptations to LV hypertrophy following pressure overload and to LV remodeling following myocardial infarction. In general, compensated pressure overload hypertrophy is associated with reduced expression of PGC-1α and its co-activated genes, leading to reduced FAO and OXPHOS capacity [37-40]. In addition, many studies have reported repression of PPAR-α levels in the failing heart [41-44]. In contrast, in heart failure following coronary artery ligation, mitochondrial function has been reported to be preserved and associated with normal PPAR-α expression [45, 46]. These differences might reflect differences in model systems and duration or severity of heart failure. Our data in WT-MI hearts, in which mitochondrial function was normal, is consistent with these latter reports. It is important to note that in the present study, heart failure had not yet developed in WT mice but there was compensated cardiac hypertrophy, with reduction in expression of PGC-1α, OXPHOS and FAO genes. In contrast, CIRKO-MI mice had significant LV dysfunction and although they shared many of the transcriptional changes observed in WT-MI mice, there was clear evidence of mitochondrial dysfunction. Our transcriptional analyses suggest that this could be the result of a specific reduction in PPAR-α expression, which might have contributed to the additional reduction in expression of the beta oxidation enzymes LCAD and MCAD, CPT2, which would limit mitochondrial FA-acyl CoA uptake, and ETFDH, which might limit the delivery of reducing equivalents generated by beta oxidation to the electron transport chain. These mechanisms could all contribute to the observed defect in palmitoyl carnitine supported mitochondrial oxygen consumption. Given that FA oxidation generates up to 70% of myocardial ATP, the reduction in mitochondrial FA oxidative capacity could plausibly contribute to impaired LV function following MI in CIRKO hearts.
Uncoupling protein (UCP2) expression was increased in hearts of WT-MI, despite repression of PGC-1α. In CIRKO hearts, UCP2 expression was induced in non-stressed hearts and remained increased post MI, despite progressive repression of PPAR-α. Cardiac UCP2 expression is regulated by PPAR-α dependent and independent mechanisms [47]. Thus PPAR-α independent pathways primarily regulate UCP2 expression following ischemia in these models. It is generally accepted that the ischemic myocardium generates reactive oxygen species (ROS) from mitochondrial sources in response to ischemic injury, (reviewed in [48-50]). Activation of uncoupling proteins has been suggested to play a cardioprotective during ischemia and reperfusion [51]. UCP2 expression and activity may be regulated by ROS and a putative mechanism by which this may occur in the heart in the context of ischemia may involve activation of ASK1/JNK-p38/CREB-NFκB signaling pathways [49, 52-54]. We have recently reported that oxidative stress is increased in non-stressed CIRKO hearts [31]. Activation of uncoupling proteins could contribute to reduced mitochondrial energetics [55]. Future studies will be required to determine if mitochondrial uncoupling could also contribute to the reduction in mitochondrial energetics in insulin resistant hearts following myocardial ischemia.
Our study has some limitations. CIRKO mice exhibited increased acute mortality following coronary artery ligation. We do not know the mechanisms for this interesting phenomenon but speculate that the initial early mortality in the immediate peri-MI period could be due to arrhythmias. CIRKO mice have previously been shown to exhibit attenuated cardiac potassium currents, a feature which could promote arrhythmias [56]. Future studies will probe these mechanisms further. Because of challenges in generating large numbers of CIRKO mice, we did not measure substrate metabolism in isolated hearts or activity levels of beta-oxidation enzymes or of carnitine palmitoyl transferase, or activity of intracellular survival or stress-activated kinases. It is possible that absence of additional insulin signaling-mediated pro-survival mechanisms [57-60] could have contributed to the accelerated rate of LV dysfunction or excess mortality. Genetic deletion of insulin signaling is perhaps more severe than the more partial degrees of cardiac insulin resistance that might occur in individuals with diabetes, obesity or insulin resistance. Thus additional studies in models with selective, but partial impairment in myocardial insulin action will be required to determine the consequences of lesser degrees of insulin resistance on the myocardial adaptations following myocardial infarction.
In summary, the present study demonstrates that impaired insulin signaling in cardiomyocytes accelerates adverse post-MI LV dysfunction. These changes are associated with a rapid decline in mitochondrial function that parallels a specific reduction in the expression of the transcriptional regulator PPAR-α and additional reduction in expression of genes whose products determine mitochondrial beta-oxidation capacity. Thus, insulin signaling might play an important role in sustaining LV metabolic capacity in post-MI LV remodeling.
Supplementary Material
Acknowledgments
Supported by grants from the Department of Veterans Affairs to SEL, the National Institute of Health (HL70070, U01-HL70525, U01 HL087947) to EDA who is an Established Investigator of the American Heart Association. SS is supported by a mentor-based post-doctoral fellowship from the American Diabetes Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care. 1993 Feb;16(2):434–44. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974 Jul;34(1):29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 3.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004 Sep 11-17;364(9438):937–52. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 4.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998 Jul 23;339(4):229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 5.Huxley R, Barzi F, Woodward M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: meta-analysis of 37 prospective cohort studies. Bmj. 2006 Jan 14;332(7533):73–8. doi: 10.1136/bmj.38678.389583.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000 Mar 4;355(9206):773–8. doi: 10.1016/S0140-6736(99)08415-9. [DOI] [PubMed] [Google Scholar]
- 7.Cao JJ, Hudson M, Jankowski M, Whitehouse F, Weaver WD. Relation of chronic and acute glycemic control on mortality in acute myocardial infarction with diabetes mellitus. Am J Cardiol. 2005 Jul 15;96(2):183–6. doi: 10.1016/j.amjcard.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 8.Svensson AM, McGuire DK, Abrahamsson P, Dellborg M. Association between hyper- and hypoglycaemia and 2 year all-cause mortality risk in diabetic patients with acute coronary events. Eur Heart J. 2005 Jul;26(13):1255–61. doi: 10.1093/eurheartj/ehi230. [DOI] [PubMed] [Google Scholar]
- 9.Houterman S, Verschuren WM, Hofman A, Witteman JC. Serum cholesterol is a risk factor for myocardial infarction in elderly men and women: the Rotterdam Study. J Intern Med. 1999 Jul;246(1):25–33. doi: 10.1046/j.1365-2796.1999.00525.x. [DOI] [PubMed] [Google Scholar]
- 10.Kromhout D, Bosschieter EB, Drijver M, de Lezenne Coulander C. Serum cholesterol and 25-year incidence of and mortality from myocardial infarction and cancer. The Zutphen Study. Arch Intern Med. 1988 May;148(5):1051–5. [PubMed] [Google Scholar]
- 11.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007 Dec;59(4):418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 12.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002 Mar;109(5):629–39. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003 Sep;285(3):H1261–9. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 14.McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, et al. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol. 2005 Dec;39(6):882–92. doi: 10.1016/j.yjmcc.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Sena S, Rasmussen IR, Wende AR, McQueen AP, Theobald HA, Wilde N, et al. Cardiac hypertrophy caused by peroxisome proliferator- activated receptor-gamma agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology. 2007 Dec;148(12):6047–53. doi: 10.1210/en.2006-1559. [DOI] [PubMed] [Google Scholar]
- 16.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005 Oct 25;112(17):2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 17.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007 Oct;56(10):2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 18.Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochimica et biophysica acta. 1987 Jun 29;892(2):191–6. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- 19.Tian R, Abel ED. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation. 2001 Jun 19;103(24):2961–6. doi: 10.1161/01.cir.103.24.2961. [DOI] [PubMed] [Google Scholar]
- 20.Abel ED. Glucose transport in the heart. Front Biosci. 2004 Jan 1;9:201–15. doi: 10.2741/1216. [DOI] [PubMed] [Google Scholar]
- 21.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study. Lancet. 1997 May 24;349(9064):1498–504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 22.Jaffe JR, Nag SS, Landsman PB, Alexander CM. Reassessment of cardiovascular risk in diabetes. Curr Opin Lipidol. 2006 Dec;17(6):644–52. doi: 10.1097/MOL.0b013e3280106208. [DOI] [PubMed] [Google Scholar]
- 23.Aguilar D, Solomon SD, Kober L, Rouleau JL, Skali H, McMurray JJ, et al. Newly diagnosed and previously known diabetes mellitus and 1-year outcomes of acute myocardial infarction: the VALsartan In Acute myocardial iNfarcTion (VALIANT) trial. Circulation. 2004 Sep 21;110(12):1572–8. doi: 10.1161/01.CIR.0000142047.28024.F2. [DOI] [PubMed] [Google Scholar]
- 24.Stevens RJ, Coleman RL, Adler AI, Stratton IM, Matthews DR, Holman RR. Risk factors for myocardial infarction case fatality and stroke case fatality in type 2 diabetes: UKPDS 66. Diabetes Care. 2004 Jan;27(1):201–7. doi: 10.2337/diacare.27.1.201. [DOI] [PubMed] [Google Scholar]
- 25.Zairis MN, Lyras AG, Makrygiannis SS, Psarogianni PK, Adamopoulou EN, Handanis SM, et al. Type 2 diabetes and intravenous thrombolysis outcome in the setting of ST elevation myocardial infarction. Diabetes Care. 2004 Apr;27(4):967–71. doi: 10.2337/diacare.27.4.967. [DOI] [PubMed] [Google Scholar]
- 26.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. Jama. 1979 May 11;241(19):2035–8. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- 27.Abbott RD, Donahue RP, Kannel WB, Wilson PW. The impact of diabetes on survival following myocardial infarction in men vs women. The Framingham Study. Jama. 1988 Dec 16;260(23):3456–60. [PubMed] [Google Scholar]
- 28.Rytter L, Troelsen S, Beck-Nielsen H. Prevalence and mortality of acute myocardial infarction in patients with diabetes. Diabetes Care. 1985 May-Jun;8(3):230–4. doi: 10.2337/diacare.8.3.230. [DOI] [PubMed] [Google Scholar]
- 29.Abbud ZA, Shindler DM, Wilson AC, Kostis JB. Effect of diabetes mellitus on short- and long-term mortality rates of patients with acute myocardial infarction: a statewide study. Myocardial Infarction Data Acquisition System Study Group. Am Heart J. 1995 Jul;130(1):51–8. doi: 10.1016/0002-8703(95)90235-x. [DOI] [PubMed] [Google Scholar]
- 30.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008 Apr;88(2):389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, et al. Contribution of impaired insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009 doi: 10.1161/CIRCULATIONAHA.108.792101. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, et al. A Conserved Role for Phosphatidylinositol 3-Kinase but Not Akt Signaling in Mitochondrial Adaptations that Accompany Physiological Cardiac Hypertrophy. Cell Metab. 2007 Oct;6(4):294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arteriosclerosis, thrombosis, and vascular biology. 2007 Apr;27(4):755–61. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 34.Gorlach A, Bonello S. The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. The Biochemical journal. 2008 Jun 15;412(3):e17–9. doi: 10.1042/BJ20080920. [DOI] [PubMed] [Google Scholar]
- 35.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. The Biochemical journal. 2008 Jun 15;412(3):477–84. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001 Jun 15;166(12):7543–8. doi: 10.4049/jimmunol.166.12.7543. [DOI] [PubMed] [Google Scholar]
- 37.Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, et al. Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med. 2008 Sep;86(9):1013–24. doi: 10.1007/s00109-008-0385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner RA, Tabibiazar R, Powers J, Bernstein D, Quertermous T. Genome-wide expression profiling of a cardiac pressure overload model identifies major metabolic and signaling pathway responses. J Mol Cell Cardiol. 2004 Dec;37(6):1159–70. doi: 10.1016/j.yjmcc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Mirotsou M, Dzau VJ, Pratt RE, Weinberg EO. Physiological genomics of cardiac disease: quantitative relationships between gene expression and left ventricular hypertrophy. Physiol Genomics. 2006 Oct 3;27(1):86–94. doi: 10.1152/physiolgenomics.00028.2006. [DOI] [PubMed] [Google Scholar]
- 40.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006 Jun 27;103(26):10086–91. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996 Dec 1;94(11):2837–42. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 42.Lehman JJ, Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart failure reviews. 2002 Apr;7(2):175–85. doi: 10.1023/a:1015332726303. [DOI] [PubMed] [Google Scholar]
- 43.Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cellular & molecular biology letters. 2003;8(1):49–53. [PubMed] [Google Scholar]
- 44.Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, et al. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. 2005 Jul 19;112(3):407–15. doi: 10.1161/CIRCULATIONAHA.105.536318. [DOI] [PubMed] [Google Scholar]
- 45.Rennison JH, McElfresh TA, Okere IC, Vazquez EJ, Patel HV, Foster AB, et al. High-fat diet postinfarction enhances mitochondrial function and does not exacerbate left ventricular dysfunction. Am J Physiol Heart Circ Physiol. 2007 Mar;292(3):H1498–506. doi: 10.1152/ajpheart.01021.2006. [DOI] [PubMed] [Google Scholar]
- 46.Morgan EE, Chandler MP, Young ME, McElfresh TA, Kung TA, Rennison JH, et al. Dissociation between gene and protein expression of metabolic enzymes in a rodent model of heart failure. Eur J Heart Fail. 2006 Nov;8(7):687–93. doi: 10.1016/j.ejheart.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005 Dec;54(12):3496–502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- 48.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochimica et biophysica acta. 2006 May-Jun;1757(56):509–17. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 49.Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovascular research. 2009 Feb 15;81(3):457–64. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 50.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovascular research. 2004 Feb 15;61(3):461–70. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 51.McLeod CJ, Aziz A, Hoyt RF, Jr, McCoy JP, Jr, Sack MN. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. The Journal of biological chemistry. 2005 Sep 30;280(39):33470–6. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- 52.Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, et al. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. The Journal of biological chemistry. 2001 Mar 23;276(12):8705–12. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- 53.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002 Jan 3;415(6867):96–9. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 54.Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999 May;116(5):1184–93. doi: 10.1016/s0016-5085(99)70022-3. [DOI] [PubMed] [Google Scholar]
- 55.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda, Md. 2006 Aug;21:250–8. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 56.Shimoni Y, Chuang M, Abel ED, Severson DL. Gender-dependent attenuation of cardiac potassium currents in type 2 diabetic db/db mice. The Journal of physiology. 2004 Mar 1;555(Pt 2):345–54. doi: 10.1113/jphysiol.2003.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 2001 Dec 7;89(12):1191–8. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- 58.Jonassen AK, Brar BK, Mjos OD, Sack MN, Latchman DS, Yellon DM. Insulin administered at reoxygenation exerts a cardioprotective effect in myocytes by a possible anti-apoptotic mechanism. J Mol Cell Cardiol. 2000 May;32(5):757–64. doi: 10.1006/jmcc.2000.1118. [DOI] [PubMed] [Google Scholar]
- 59.Jonassen AK, Aasum E, Riemersma RA, Mjos OD, Larsen TS. Glucose-insulin-potassium reduces infarct size when administered during reperfusion. Cardiovasc Drugs Ther. 2000 Dec;14(6):615–23. doi: 10.1023/a:1007802630604. [DOI] [PubMed] [Google Scholar]
- 60.Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002 Mar 26;105(12):1497–502. doi: 10.1161/01.cir.0000012529.00367.0f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.