

Abstract
Synopsis
The hyper IgE syndromes (HIES) are rare primary immune deficiencies characterized by elevated serum IgE, rash and recurrent bacterial infections of the skin and lung. Autosomal dominant HIES, the most common disease in this group, results from STAT3 mutations and has a variety of connective tissue and skeletal abnormalities. The genetic etiologies of the more rare autosomal recessive form(s) still need delineation. Treatment of these syndromes has relied on prophylactic and therapeutic antimicrobials and aggressive skin care. The new and evolving genetic and immunologic understandings of this previously elusive set of diseases should lead to more effective disease-specific therapies.
Introduction
The Hyper-immunoglobulin E syndromes (HIES) are rare primary immune deficiencies characterized by elevated serum IgE, dermatitis and recurrent skin and lung infections. There are two forms of HIES: a dominant form caused by mutations in STAT3, and a recessive form, for which a genetic cause is unclear1-4. These two different syndromes have distinct presentations, courses, and outcomes and share very little in terms of pathogenesis other than the IgE elevation. The dominant form is characterized by non-immunologic features including skeletal, connective tissue, and pulmonary abnormalities in addition to recurrent infections and eczema. In contrast, the recessive form lacks the somatic features and has marked viral infections and neurologic complications. We will discuss the diagnostic, laboratory and clinical aspects of these disorders as well as their genetic etiologies.
Autosomal Dominant Hies (STAT3 deficiency)
The disease subsequently identified as Hyper IgE Recurrent Infection syndrome (HIES) was first described as Job's syndrome by Davis et al in 1966,5 referring to the Biblical Job, who was “smote with sore boils”. The syndrome was refined by Buckley et al in 1972, who recognized extremely high serum IgE levels6. Since that time, the classic triad of eczema, recurrent skin and lung infections and high serum IgE has been expanded to include skeletal, connective tissues, cardiac and brain abnormalities7-9. Until 2007, HIES remained the last of the major immune deficiencies without neither a known genetic etiology nor a comprehensive understanding of the associated immune dysfunction. We now know that STAT3 mutations are responsible for most, if not all, cases of autosomal dominant HIES, and these mutations have begun to explain the multi-system nature of the disease2,3. To distinguish this dominant disease due to STAT3 mutation from the recessive forms of hyper IgE syndromes, and to distinguish this disease from other syndromes of IgE elevation, we will also refer to this disease as STAT3 deficiency.
Clinical Manifestations
STAT3 deficiency is a disease of multi-organ dysfunction (Table 1). Although what brings patients to initial attention is usually eczema and recurrent infections, these individuals have abnormalities in vessels, connective tissue and skeleton. Prior to genetic testing, the diagnosis of HIES has typically been difficult to confirm until both immunologic and somatic features exist. A clinical scoring system has been developed which includes both of these categories10.
Table 1.
Clinical Characteristics of STAT3 Deficiency.
| Immunologic Characteristics (% Frequency) |
Non-Immunologic Characteristics (%Frequency) |
|---|---|
| Newborn rash (81%) | Characteristic face (83%) |
| Boils (87%) | Retained primary teeth (72%) |
| Recurrent pneumonias (87%) | Minimal trauma fractures (71%) |
| Pneumatocoeles (77%) | Scoliosis >10 degrees (63%) |
| Eczema (100%) | Hyperextensibility (68%) |
| Mucocutaneous candidiasis (83%) | Focal Brain Hyperintensities (70%) |
| Peak Serum IgE >2000 IU/ml (97%) | Chiari 1 Malformations (18%) |
| Eosinophilia (93%) | Craniosynostosis (unknown) |
| Increased incidence of Lymphoma | Arterial Aneurysms (unknown) |
Skin
A newborn rash is usually the first manifestation of STAT3 deficiency8,9. Pustular and eczematoid rashes usually begin within the first month of life, typically first affecting the face and scalp. In a series of 43 patients, 8 babies (19%) were born with the rash, and 23 (53%) acquired the rash within the first week of life9. Biopsies typically show an eosinophilic infiltrate and bacterial culture usually grows Staphylococcus aureus. The rash can be quite significant, especially in childhood. To achieve and maintain good control, anti-staphylococcal therapies (antibiotics or topical antiseptics, such as bleach) are often essential.
Boils are a classic finding in this disease, and characteristic of the diagnosis. The degree of inflammatory symptoms, such as tenderness and warmth is often quite variable. The “cold” abscesses initially described by Davis et al are common. However, despite the absence of external signs of inflammation, upon aspiration, there is frank pus, and S. aureus is usually cultured. With prophylactic antibiotics, the occurrence of these boils typically substantially diminishes. Trouble areas may persist in intertriginous areas such as the axillae, the inguinal region or under the breasts.
Lungs
Recurrent pyogenic pneumonias are the rule. Pneumonias typically start in childhood, and the most frequent bacteria isolates are S. aureus, Streptococcus pneumoniae, and Haemophilus influenzae (Box 1). Similar to the occurrence of cold abscesses, these pneumonias may present with fewer symptoms (e.g., cough, sputum production) than would be expected in a normal person given the extent of disease. This dearth of symptoms and subsequent delay in clinical presentation may contribute to advanced disease and significant tissue damage prior to identification and initiation of appropriate therapy. Upon sputum inspection or bronchoscopy, pus is clearly present.
Box 1. Pathogens of STAT3 Deficiency.
Frequent Pathogens
Staphylococcus aureus (lung and skin)
Streptococcus pneumoniae (lung)
Haemophilus influenzae (lung)
Candida albicans (mucocutaneous)
Secondary Pathogens of Lung
Pseudomonas aeruginosa
Aspergillus species
Scedosporium species
Nontuberculous mycobacteria
Less Frequent Pathogens
Pneumocystis jiroveci (lung)
Histoplasma (gastrointestinal tract)
Cryptococcus (brain and gastrointestinal tract)
Although the pneumonias typically respond promptly to appropriate antimicrobial therapy, the healing of the lungs is aberrant. Pneumatocoeles and bronchiectasis form during the healing process and usually persist once the infection has cleared. These persistent structural abnormalities, which can be quite significant, then predispose to Gram-negative bacterial infection (typically Pseudomonas) and fungal infections (typically Aspergillus or Scedosporium species) in addition to the primary pathogens in STAT3 deficiency (Figure 1). The secondary infections are typically indolent and difficult to clear. These long-term infections are more frequently associated with mortality than the acute pyogenic infections, causing rupture into large pulmonary vessels with life-threatening hemoptysis or fungal dissemination to the brain 13.
Figure 1.
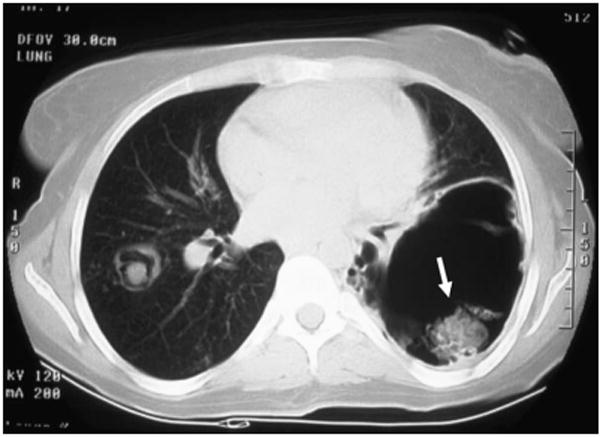
Chest CT of an individual with STAT3 deficiency showing the characteristic pneumatoceles. The pneumatoceles are prone to infection with fungi (arrow indicates an aspergilloma) and Gram-negative bacteria.
Other Infections
Mucocutaneous candidiasis is common in STAT3 deficiency, manifesting typically as oral thrush, vaginal candidiasis or onychomycosis1. Systemic Candida infections are very rare, and most likely nosocomial in origin (such as an indwelling catheter infection). Disseminated Cryptococcus and Histoplasma infections also occur, although less frequently than candidiasis. These uncommon yeast infections are often localized, such as histoplasmosis of the tongue, or intestinal cryptococcal infection14-15. Pneumocystis jiroveci pneumonia (PCP) also occurs, albeit infrequently, and may present in infants in a way similar to the initial presentation of PCP in infants infected with HIV16.
Musculoskeletal Abnormalities
Skeletal abnormalities in STAT3 deficiency include scoliosis, osteopenia, minimal trauma fractures, hyperextensibility, and degenerative joint disease. Scoliosis occurs in about 75% of patients, typically emerging during adolescence in a pattern similar to that of idiopathic scoliosis1. In some patients, there is associated leg length discrepancy; in others scoliosis develops or worsens after thoracotomy for lung infections. The scoliosis varies in severity, and some individuals have required surgical stabilization or correction.
Hyperextensibility of both the large and small joints is frequent, and may be related to the early development of severe degenerative joint disease, particularly of the spine, that we have seen in several patients in their 20s-40s. Several patients have required stabilization procedures as early as in their 3rd decade, and many suffer from chronic pain due to extensive arthritis. Minimal trauma fractures and decreased bone mineral density are also common, but may occur independently of one another1. Fractures tend to be of the long bones, ribs and pelvic bones. Bone resorption has been shown to be abnormally increased in patients with HIES due to abnormalities in the prostaglandin synthetic pathway, and is responsive to non-steroidal agents17,18. Healing appears to be normal following surgery or fractures.
Cranial Abnormalities
Craniosynostosis of varying degrees occurs, but typically does not require surgical repair19,20. Chiari 1 malformations also occur fairly frequently; in one study of 50 individuals, 9 (18%) had Chiari 1 malformation on brain MRI7. The Chiari malformations observed typically do not require surgical repair and are usually incidental findings.
Dental Abnormalities
The majority of individuals with STAT3 deficiency retain at least some, if not all, of their primary teeth past the age of normal primary dental exfoliation1,21. This appears to be a failure of the primary teeth to exfoliate, not of the secondary teeth to develop or to erupt. Once the primary teeth do exfoliate, whether by dental extraction or naturally, the secondary teeth, which are normally formed, emerge. At times, layers of both primary and secondary teeth are simultaneously present when the secondary teeth emerge despite the primaries having not fallen out. There are also characteristic findings of the oral mucosa, tongue, roof of the mouth and cheeks22. Central depressions in the tongue (central rhomboid glossitis) may be caused by or become secondarily infected with candida. The palate typically has a high arch with varying degrees of a central band-type protrusion. Abnormally prominent wrinkles are often observed on the oral mucosa.
Vascular abnormalities
Arterial aneurysms are an important recently appreciated aspect of STAT3 deficiency8. Bilateral berry aneurysms of the internal carotid arteries and mycotic aneurysm have been reported in an autopsy series of HIES13. A large aneurysm in the left anterior descending coronary artery resulted in myocardial infarction in one adult male patient 8. That case prompted us to look more closely at our other adult patients with HIES. We have been surprised at the frequency of coronary artery anomalies, including arterial tortuosity, dilation and aneurysms. Of 18 individuals studied by either cardiac CT or MRI, 14 had one of these abnormalities, with tortuosity and dilation predominating and aneurysms being present in only 4 (unpublished data). It is important to note that there has been a paucity of significant atherosclerosis in the individuals studied by CT and MR angiography, despite their having coronary artery disease risk factors. The fact that one of the patients had a myocardial infarction due to his aneurysm clearly indicates that these can be medically important. Whether and when these aneurysms require therapy, and if so with what, is unclear. Whether the coronary and extracoronary aneurysms are reflections of the same underlying pathophysiology also remains to be determined.
Lacunar infarcts have been reported at a younger age than is typical, and have had varying clinical consequences, ranging from thalamic infarction to no symptoms7. T2 weighted hyperintensities seen on brain MRI are similar to incidental findings that accumulate in otherwise healthy adults as they age, but seen at much younger ages than in the general population7. Similar hyperintensities in elderly otherwise healthy adults are thought to reflect small vessel abnormalities, which may be signs of subtle vascular abnormalities, or demyelination. Gross neurologic abnormalities are not detected in the majority of patients with these findings.
Face
The characteristic facial appearance that individuals with STAT3 deficiency share may make patients look more like one another than their family members1,12,23. Facial asymmetry, broad nose, and deep-set eyes with a prominent forehead are common. The facial skin has a rough appearance with exaggerated pore size. This characteristic appearance typically develops during childhood and adolescence.
Malignancies
There is an increased risk of malignancy associated with STAT3 deficiency24-26. Both Hodgkin's and non-Hodgkin's lymphoma (NHL) have been described, with the majority of the NHL being of B cell origin with aggressive histology. Individuals have been successfully treated and apparently cured with chemotherapy. In one study of 11 individuals, 7 died, but two for reasons other than lymphoma24. The increased mortality may reflect a delay in diagnosis. Other cancers described in HIES have included leukemia and those of the vulva, liver and lung26.
Laboratory abnormalities of STAT3 Deficiency
The two most consistent laboratory findings of Job's syndrome are elevated serum IgE and eosinophilia; otherwise there is a lack of pathognomic laboratory signs. Therefore, the diagnosis has historically been more syndromic than laboratory-based. The serum IgE typically peaks above 2,000 IU/ml, and is usually elevated even at the time of birth. It is important to keep in mind the natural change in IgE levels over time: they are usually undetectable in cord blood, and rise to the adult range slowly over the years. In adulthood the IgE level may diminish over time in some individuals and can actually normalize, despite persistence of the clinical abnormalities of STAT3 deficiency (in one report 20%)1. Eosinophilia is almost always present in these patients at least at some point, but is not correlated with the serum IgE.
Other laboratory findings are variable in STAT3 deficiency. Total white blood cell counts are typically normal, but may not increase appropriately during acute infection. Neutropenia has been reported, but is uncommon. Serum IgG, IgA, IgM are typically normal, although there are some individuals with deficiencies in one or more of these immunoglobulins.
Immunology of STAT3 deficiency
The mechanisms of the immune deficiencies in STAT3 deficiency remain elusive. Multiple reports with small numbers of patients have conflicted regarding whether a neutrophil chemotactic defect is present, and whether there is a T helper 1 (Th-1)/Th2 cytokine imbalance27-32.
STAT3 is a key regulator of many immunologic pathways. Mouse Stat3 homozygous knock-outs die in utero, reflecting the absolute necessity of some Stat3 function for survival. Therefore, most experiments in mice have used organ-specific or conditional knock-out animals. Animals with a myeloid-specific deletion of Stat3 had upregulation of TNF-alpha and interferon-gamma, and down-regulation of IL-6 and IL-1033-34. Consistent with these mouse data, microarray analysis of leukocytes from STAT3 deficient humans showed significant upregulation of pro-inflammatory genes both at rest and after stimulation2. Similarly, patients showed excessive proinflammatory cytokine production in response to innate immune agonists, consistent with the impaired regulation of these cytokines by STAT32. After incubation with lipopolysaccharide (LPS), levels of TNF-alpha and Interleukin (IL)-12 were elevated. Interferon-gamma was also increased after the mitogen phytohemagglutinin (PHA). As expected for a defect in STAT3, upon which the signaling of IL-6 and IL-10 depend, there are impaired responses to these cytokines. Monocyte chemoattractant protein-1 (MCP-1) levels are diminished after IL-6 stimulation of HIES patient leukocytes2.
Many of the specific immune defects of STAT3 deficiency remain somewhat unexplained. The predominance of infections of the lung and skin may be because STAT3 is a key regulator of beta-defensins of the skin and lung through IL-22 signalling35. The high IgE may result from defects in STAT3 mediated IL-21 receptor signaling, as heterozygous IL-21 receptor knock-out mice have increased IgE36. HIES is not exclusively a disorder of too little inflammation and resulting inability to control invading microorganisms. It is also surprisingly a disorder of too much inflammation. The increased inflammation is evident in the lung, where tissue breakdown leading to pneumatocoeles may be a consequence of exuberant inflammation. In contrast, there are aspects of STAT3 deficiency that are more consistent with too little inflammation, such as the frequent “cold” abscesses, and the relative paucity of symptoms compared to the extent of disease. Exploration and charting of these areas of aberrant regulation, both too little and too much, should be highly informative about the nature of the early and late aspects of inflammation.
Genetics of STAT3 Deficiency
STAT3 mutations cause most, if not all, cases of autosomal dominant HIES. So far, mutations have been found in only 2 regions of the STAT3 gene: the SH2 domain (mediating protein-protein interaction) and the DNA binding domain (mediating the interaction of protein with DNA). There are mutational hot spots in both the SH2 and DNA binding domains, as shown by multiple independent families carrying the same mutation2,3. Many of the same mutations have been found in multiple ethnic groups among Caucasians, Africans, and Asians, indicating recurrent de novo mutations. All mutations so far have been missense mutations or in frame deletions, allowing the production of full-length mutant STAT3 protein able to exert a dominant negative effect. The importance of the production of mutant protein with dominant negative effect is shown by the fact that mice with heterozygous deletions of STAT3 have no apparent phenotype, suggesting that having a single copy of a functional gene is adequate for most functions. This suggests that the development of human HIES requires an inhibition of function below that afforded by a single allele. The mutations have no intrinsic function on their own, but act in a dominant-negative manner to inhibit the function of the normal allele3. Consistent with this in vitro phenotype, HIES is transmitted in an autosomal dominant manner. Importantly, somatic mosaicism has been recognized in a male who had an intermediate HIES score but had 2 HIES affected children. His peripheral blood showed evidence for 2 populations of cells: one population with the mutant allele and one population without it2. Efforts to define the existence of somatic mutations that cause intermediate types of disease are underway. Despite the existence of only 2 mutated domains of STAT3 that are thought to mediate such different effects as DNA and protein binding, genotype/phenotype correlations have yet to be determined.
STAT3 is a major signal transduction protein involved in diverse pathways including wound healing, angiogenesis, immune pathways, and cancer. Homozygous STAT3 knock-out mice are not viable, but organ- and tissue-specific knock-outs are and have been informative. In mice with STAT3 deficiency of the pulmonary epithelium, there is excessive inflammation and airspace enlargement when exposed to hyperoxia, reminiscent of the pneumatoceles that form in HIES patients following bacterial pneumonia37. Mice with hematopoiesis-specific STAT3 deficiency develop osteopenia and increased osteoclast generation, reminiscent of the osteopenia and fractures of HIES patients 38. Mice with brain-specific STAT3 have increased inflammation, demyelination, and astrocytosis, reminiscent of the hyperintensities seen in HIES patient brain MRIs7,39. STAT3 is also involved in vascular remodeling and atherosclerosis, which may be relevant to the coronary artery aneurysms and lack of atherosclerosis in HIES patients40.
Therapy of Hyper IgE syndromes
Specific therapies for STAT3 deficiency are only now being developed but successful supportive care has been well honed. Effective skin care often depends on control of both superficial and invasive S. aureus infection. Bleach baths (120 ml bleach in a tub of water, soaked in for 15 minutes 3 times weekly) and swimming in chlorinated pools are highly effective. Systemic immune suppression (such as corticosteroids) to treat the eczema are usually not necessary, as there is typically an excellent response to antimicrobials, but topical steroids help in difficult cases. Antimicrobial prophylaxis to prevent S. aureus skin and lung infection (such as trimethoprim-sulfamethoxazole 2.5 mg/kg twice daily), may be broadened if Gram-negative lung infections occur. Antifungal prophylaxis to prevent pulmonary aspergillosis remains attractive but unproven, but it is highly effective to treat and prevent mucocutaneous candidiasis. Treatment of pneumonia is ideally guided by the etiologic agent. Bronchoscopy is helpful to recover the pathogen and to assist with clearance of mucus and pus, as these patients often do not have an adequate cough response. Since STAT3 deficient patients often feel well and have minimal fever despite significant infection, it is good to have a low threshold to investigate slight changes, like new cough, chest discomfort, or fatigue, even in the absence of fever. The decision to resect the large pneumatoceles that sometimes form following pneumonia is complex. These large cysts may become secondarily infected and be a source of infection, bleeding, and possibly death. On the other hand, thoracic surgery can be complicated by poor expansion of the remaining lung after surgery, often resulting in thoracoplasty.
Before the identification of STAT3 deficiency as the cause of HIES, there were only a few immunomodulator trials. Levamisole is an unusual antihelminthic that also stimulates T cell and NK cell function. In a blinded randomized study it was found to be inferior to placebo41. Interferon-gamma has been used with mixed results. In vitro it improved neutrophil chemotaxis, but in vivo it had inconsistent effects on IgE levels42. Intravenous immunoglobulin (IVIG) may decrease the number of infections for some patients 43. Case reports and small case series have extolled cyclosporine and histamine-2 receptor blockade44,45. Omalizumab (the monoclonal antibody against IgE) has not yet been studied, and it is unclear whether there may be any benefit.
Two bone marrow transplants in HIES patients have been reported 46,47. An adult died 6 months after transplantation 47. His death was thought to be due to complications of the transplant, but he had a decrease in serum IgE and improvement in HIES related symptoms in the post-transplant interval. A 7 year old girl was transplanted because of her severe HIES. Although she initially had a good response, when post-transplant immune suppression was weaned, her serum IgE once again became elevated and infections recurred despite engraftment of the donor cells46. Subsequently she appears to be doing well, leaving open issues of short and long term benefits to transplant (A. Cant, personal communication).
The role of bisphosphonates in treating the osteoporosis and minimal trauma fractures is undefined. Although we have treated several patients with bisphosphonates leading to improved bone mineral density without adverse events, it is unclear whether this will translate into fewer fractures. Possible dental adverse events from bisphosphonates also remain unclear. The proper therapy for coronary artery aneurysms, or other blood vessel abnormalities, if any, is undefined. Coronary artery aneurysms from Kawasaki disease are typically treated with anticoagulation depending on the size of aneurysm (from aspirin to warfarin). However, the development, progression, and significance of the arterial abnormalities in STAT3 deficiency are unknown. The hypothetical benefits of anticoagulation must be weighed against the real risks of pulmonary hemorrhage. Close attention to blood pressure and other cardiovascular risk factors seems sensible.
A complex multisystem disease like STAT3 deficiency requires a sophisticated multidisciplinary approach. In addition to close infectious disease management, other subspecialists are often required. The orchestrated expertise of orthopedists for scoliosis, fractures and degenerative joints; dentists to address the retained primary teeth; and pulmonologists for diagnostic and therapeutic bronchoscopy and pulmonary toilet.
Autosomal Recessive Hies (AR-HIES)
In 2004, Renner et al described 13 patients from six consanguineous families who had a disease similar to, but distinct from, what we now know to beautosomal dominant HIES (STAT3 deficiency).4 Their patients had elevated serum IgE, eczema and recurrent skin and cutaneous viral infections, but lacked the connective tissue and skeletal findings characteristic of STAT3 deficiency. One patient has been described with a recessive disease similar to AR-HIES due to a homozygous mutation in Tyrosine kinase 2 (Tyk2), a major signal-transducing molecule for IL-12, IL-6, and interferon alpha 48. However, Tyk2 deficiency is distinct from AR-HIES, and patients with AR-HIES have normal Tyk2 and STAT3 sequences49.
Clinical Manifestations (Box 2)
Box 2. Clinical Characteristics of AR-HIES.
Eczema
Boils
Recurrent pneumonia without pneumatoceles
Sepsis
Mucocutaneous candidiasis
Skin viral infections
Neurologic symptoms
Vasculitis
Increased serum IgE
Eosinophilia
The predominant clinical manifestations of AR-HIES are severe eczema and recurrent skin and lung infections4. All of the patients described with AR-HIES have had severe eczematoid rashes, starting early in life, although not necessarily in the newborn period. Skin abscesses do occur, and are typically from S. aureus. However, the skin disease of AR-HIES differs from STAT3 deficiency in the much higher incidence of cutaneous viral infections including Molluscum contagiosum, herpes simplex and varicella zoster virus infections. AR-HIES and STAT3 deficient patients share the propensity towards mucocutaneous candidiasis. Sinopulmonary infections are common in AR-HIES, with etiologies including S. aureus, Haemophilus influenzae, Proteus mirabilis, Pseudomonas aeruginosa, and Cryptococcus. However, as opposed to STAT3 deficient patients, in whom pneumatoceles complicate pneumonias, AR-HIES patients heal their lung infections without pneumatoceles. Fatal sepsis occurs in AR-HIES from both Gram positive and negative bacteria.
The AR-HIES patients have more symptomatic neurologic disease than those with STAT3 deficiency4. In the Renner series, seven had neurologic symptoms, ranging from facial paralysis to hemiplegia. The etiology of the neurologic complications was not clear for all patients, but one had a cerebral cryptococcoma with meningitis, while others had severe central nervous system vasculitis.
AR-HIES lacks the connective tissue and skeletal abnormalities of STAT3 deficiency. AR-HIES has normal primary tooth exfoliation, no fractures and normal facies.
Immunologic Manifestations
Patients with AR-HIES have high serum IgE, comparable to those with STAT3 deficiency4. Their eosinophilia is typically higher than in STAT3 deficiency. Autoimmune phenomena, including hemolytic anemia may occur. Lymphocyte phenotyping and function are inconsistent in AR-HIES, although decreased lymphocyte proliferation was seen in response to a staphylococcal antigen. Neutrophil chemotaxis and nitroblue-tetrazolium reduction are normal.
Genetics of AR-HIES
The patients with AR-HIES and Tyk2 deficiency were products of consanguineous unions 4, 48. The Tyk2 deficient patient shares features of AR-HIES including high serum IgE, eczematoid rash, recurrent skin and sinopulmonary bacterial and viral infections. However, he had Bacille Calmette-Guerin (BCG) and salmonella infections, which are more commonly seen in defects of the Interferon-gamma/IL-12 axis. Indeed, upon cytokine stimulation of the peripheral blood mononuclear cells (PBMCs), defects were found in IL-12 and IFN-alpha signaling. This Tyk2 deficient patient had a homozygous mutation leading to a four nucleotide deletion resulting and a premature stop codon. His related parents were heterozygous for the same deletion and healthy.
Mutations of Tyk2 are absent in AR-HIES 48. Therefore, although it is possible that Tyk2 mutations may cause some cases of AR-HIES disease, AR-HIES is likely heterogeneous, with more than one gene contributing to its etiology. Since STAT3 is the genetic etiology of autosomal dominant HIES, related genes in these pathways may cause some of these undefined diseases.
Therapy of AR-HIES
Therapy of AR-HIES remains supportive and less explored than STAT3 deficiency. Prophylactic antimicrobials likely help, with anti-staphylococcal agents, antivirals if needed, and antifungals if mucocutaneous candidiasis or invasive fungal disease occur. Aggressive skin care may help to prevent invasive bacterial infection.
Disease severity in AR-HIES is often worse than STAT3 deficiency. Immunomodulatory therapies and bone marrow transplantation need further exploration.
Conclusion
Hyper IgE syndromes were first described in 1966, and until recently remained one of the few primary immunodeficiencies without a genetic etiology. However, now two genetic defects have been described: STAT3 mutations act in a dominant negative manner to cause of autosomal dominant HIES, and Tyk2 deficiency acts in a recessive manner to cause one of the cases of AR-HIES. We now need to focus on understanding the pathogenesis of these complicated diseases. Understanding how STAT3 deficiency leads to the many facets of this disease will hopefully help us understand diseases that are more common, such as idiopathic scoliosis, atopic dermatitis, staphylococcal skin abscesses, and the coronary artery aneurysms of Kawasaki disease. Understanding the pathogenesis of STAT3 deficiency will allow us to create better therapies to prevent the morbidity and mortality of many diseases, including STAT3 deficiency.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O'Connell AC, Dent B, Puck JM. Hyper-IgE syndrome with recurrent infections-an autosomal dominant multisystem disorder. New England Journal of Medicine. 1999;340:692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 2.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, et al. STAT3 mutations in the Hyper-IgE syndrome. New England Journal of Medicine. 2007;18:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 3.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, et al. Dominant negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE sydnrome. Nature. 2007;448:1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 4.Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, Bergmann M, Davis J, Belohradsky BH, Grimbacher B. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr. 2004;144:93–9. doi: 10.1016/S0022-3476(03)00449-9. [DOI] [PubMed] [Google Scholar]
- 5.Davis SD, Scaller SJ, Wedgwood RJ. Job's syndrome: recurrent, “cold”, staphylococcal abscesses. Lancet. 1966;1:1013–1015. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 6.Buckley RH, W B, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. [PubMed] [Google Scholar]
- 7.Freeman AF, Collura-Burke CJ, Patronas NJ, Ilcus LS, Darnell D, Davis J, Puck JM, Holland SM. Brain abnormalities in patients with hyperimmunoglobulin E syndrome. Pediatrics. 2007;119:e1121–5. doi: 10.1542/peds.2006-2649. [DOI] [PubMed] [Google Scholar]
- 8.Ling JC, Freeman AF, Gharib AM, Arai AE, Lederman RJ, Rosing DR, Holland SM. Coronary artery aneurysms in patients with hyper IgE recurrent infection syndrome. Clin Immunol. 2007;122:255–8. doi: 10.1016/j.clim.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimbacher B, Schaffer AA, Holland SM, Davis J, Gallin JI, Malech HL, Atkinson TP, Belohradksy BH, Buckley RH, Cossu F, Espanol T, Garty BZ, Metamoros N, Myers LA, Nelson RP, Ochs HD, Renner ED, Wellinghausen N, Puck JM. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet. 1999;65:735–44. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamlin SL, McCalmont TH, Cunningham BB, Esterly NB, Lai CH, et al. Cutaneous manifestations of hyper-IgE syndrome in infants and children. Journal of Pediatrics. 2002;141:572–5. doi: 10.1067/mpd.2002.127503. [DOI] [PubMed] [Google Scholar]
- 11.Eberting CL, Davis J, Puck JM, Holland SM. Dermatitis and the newborn rash of hyper-IgE syndrome. Archives of Dermatology. 2004;140:1119–25. doi: 10.1001/archderm.140.9.1119. [DOI] [PubMed] [Google Scholar]
- 12.Buckley RH. The hyper-IgE syndrome. Clin Rev Allergy Immunol. 2001;20:139–54. doi: 10.1385/CRIAI:20:1:139. [DOI] [PubMed] [Google Scholar]
- 13.Freeman AF, Kleiner DE, Nadiminti H, Davis J, Quezado M, Anderson V, Puck JM, Holland SM. Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol. 2007;119:1234–40. doi: 10.1016/j.jaci.2006.12.666. [DOI] [PubMed] [Google Scholar]
- 14.Hutto JO, Bryan CS, Greene FL, White CJ, Gallin JL. Cryptococcosis of the colon resembling Crohn's disease in a patient with the hyperimmunoglobulinemia E syndrome. Gastroenterology. 1988;94:808–12. doi: 10.1016/0016-5085(88)90257-0. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs DH, Macher AM, Handler R, Bennett JE, Collen MJ, Gallin JI. Esophageal cryptococcosis in a patient with the hyperimmunoglobulin E-recurrent infection (Job's) sydnrome. Gastroenterology. 1984;87:201–3. [PubMed] [Google Scholar]
- 16.Freeman AF, Davis J, Anderson VL, Barson W, Darnell DN, Puck JM, Holland SM. Pneumocystis jiroveci infection in patients with hyper-immunoglobulin E syndrome. Pediatrics. 2006;118:e1271–5. doi: 10.1542/peds.2006-0311. [DOI] [PubMed] [Google Scholar]
- 17.Cohen-Solal M, Prieur AM, Prin L, et al. Cytokine-mediated bone resorption in patients with the hyperimmunoglobulin E syndrome. Clin Immunol Immunopathol. 1995;76:75–81. doi: 10.1006/clin.1995.1090. [DOI] [PubMed] [Google Scholar]
- 18.Leung DY, Key L, Steinberg JJ, Young MC, Von Deck M, Wilkinson R, Geha R. Increased in vitro bone resorption by monocytes in the hyper-immunologulin E syndrome. J Immunol. 1988;140:84–8. [PubMed] [Google Scholar]
- 19.Hoger PH, Boltshauser E, Hitzig WH. Craniosynostosis in hyper-IgE syndrome. Eur J Pediatr. 1985;144:414–7. doi: 10.1007/BF00441793. [DOI] [PubMed] [Google Scholar]
- 20.Smithwick EM, Finelt M, Pahwa S, Good RA, Naspitz CK, Mendes NF, et al. Cranial synostosis in Job's syndrome. Lancet. 1978;15:826. doi: 10.1016/s0140-6736(78)93028-3. [DOI] [PubMed] [Google Scholar]
- 21.O'Connell AC, Puck JM, Grimbacher B, Facchetti F, Majorana A, Gallin JI, Malech HL, Holland SM. Delayed eruption of permanent teeth in hyperimmunoglobulinemia E recurrent infection syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;89:177–85. doi: 10.1067/moe.2000.103129. [DOI] [PubMed] [Google Scholar]
- 22.Domingo D, Freeman AF, Davis J, Puck JM, Tianxia W, Holland SM, Hart TC. Novel intraoral phenotypes in hyper Immunoglobulin E syndrome. Oral Diseases. 2007:1–9. doi: 10.1111/j.1601-0825.2007.01363.x. [DOI] [PubMed] [Google Scholar]
- 23.Borges WG, Hensley T, Carey JC, Petrak BA, Hill HR. The face of Job. Journal of Pediatrics. 1998;133:303–5. doi: 10.1016/s0022-3476(98)70243-4. [DOI] [PubMed] [Google Scholar]
- 24.Leonard GD, Posadas E, Herrmann PC, et al. Non-Hodgkin's lymphoma in Job's syndrome: a case report and review of the literature. Leuk Lymphoma. 2004;45:2521–2525. doi: 10.1080/10428190400004463. [DOI] [PubMed] [Google Scholar]
- 25.Gorin LJ, Jeha SC, Sullivan MP, Rosenblatt HM, Shearer WT. Burkitt's lymphoma developing in a 7 year old boy with hyper IgE syndrome. J Allergy Clinical Immunol. 1989;83:5–10. doi: 10.1016/0091-6749(89)90471-5. [DOI] [PubMed] [Google Scholar]
- 26.Oztop I, Demirkan B, Tarhan O, et al. The development of a pulmonary adenocarcinoma in a patient with Job's syndrome, a rare immunodeficiency condition. Tumori. 2004;90:132–5. doi: 10.1177/030089160409000126. [DOI] [PubMed] [Google Scholar]
- 27.Borges WG, Augustine NH, Hill HR. Defective Interleukin-12/interferon-gamma pathway in patients with hyperimmunoglobulinemia E syndrome. J Pediatr. 2000;136:176–180. doi: 10.1016/s0022-3476(00)70098-9. [DOI] [PubMed] [Google Scholar]
- 28.Chehimi J, Elder M, Greene J, Noroski L, Stiehm ER, Winkelstein JA, Sullivan KE. Cytokine and chemokine dysregulation in hyper-IgE syndrome. Clin Immunol. 2001;100:49–56. doi: 10.1006/clim.2001.5039. [DOI] [PubMed] [Google Scholar]
- 29.Del Prete G, Tiri A, Maggi E, et al. Defective in vitro production of gamma-interferon and tumor necrosis factor-alpha by circulating T cells from patients with the hyper-immunoglobulin E syndrome. J Clin Invest. 1989;84:1830–5. doi: 10.1172/JCI114368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donabedian H, Gallin JI. The hyperimmunoglobulin E recurrent-infection (Job's) syndrome. A review of the NIH experience and the literature. Medicine (Baltimore) 1983;62:195–208. doi: 10.1097/00005792-198307000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Hill HR, Ochs HD, Quie PG, Clark RA, Pabst HF, Klebanoff SJ, Wedgwood RJ. Defect in neutrophil granulocyte chemotaxis in Job's syndrome of recurrent “cold” staphylococcal abscesses. Lancet. 1974;14:617–9. doi: 10.1016/s0140-6736(74)91942-4. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez MF, Patino PJ, Montoya F, Montoya CJ, Sorensen RU, Garcia DO. Interleukin 4 and interferon-gamma secretion by antigen and mitogen-stimulated mononuclear cells in the hyper-IgE syndrome: no TH-2 cytokine pattern. Ann Allergy Asthma Immunol. 1998;81:443–7. doi: 10.1016/s1081-1206(10)63143-2. [DOI] [PubMed] [Google Scholar]
- 33.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 34.Welte T, Zhang SSM, Wang T, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–84. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. Il-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Ozaki K, Spolski R, Feng CG, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–4. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 37.Hokuto I, Ikegami M, Yoshida M, et al. Stat-3 is required for pulmonary homeostasis during hypoxia. J Clin Invest. 2004;113:28–37. doi: 10.1172/JCI200419491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Welte T, Troiano N, Maher SE, Fu XF, Bothwell ALM. Osteoporosis with increased osteoclastogenesis in hematopoietic cell-specific STAT3-deficienct mice. Biochem Biophys Res Commun. 2005;328:800–7. doi: 10.1016/j.bbrc.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Okada S, Nakamura M, Katoh H, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–34. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 40.Wincewicz A, Sulkowska M, Rutkowski R, et al. STAT1 and STAT3 as intracellular regulators of vascular remodeling. Eur J Intern Med. 2007;18:267–71. doi: 10.1016/j.ejim.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Donabedian H, Alling DW, Gallin JI. Levamisole is inferior to placebo in the hyperimmunoglobulin E recurrent-infection (Job's) syndrome. N Engl J Med. 1982;307:290–2. doi: 10.1056/NEJM198207293070506. [DOI] [PubMed] [Google Scholar]
- 42.King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB. Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent-infection syndrome by IFN-gama. Proc Natl Acad Sci USA. 1989;86:10085–89. doi: 10.1073/pnas.86.24.10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimata H. High dose intravenous gamaaglobulin treatment for hyperimmunoglobulinemia E syndrome. J Allergy Clin Immunol. 1995;95:771–4. doi: 10.1016/s0091-6749(95)70185-0. [DOI] [PubMed] [Google Scholar]
- 44.Etzioni A, Shehadeh N, Brecher A, Yorman S, Pollack S. Cyclosporin A in hyperimmunoglobulin E syndrome. Ann Allergy Asthma Immunol. 1997;78:413–4. doi: 10.1016/S1081-1206(10)63204-8. [DOI] [PubMed] [Google Scholar]
- 45.Thompson RA, Kumararatne DS. Hyper-IgE syndrome and H2 receptor blockade. Lancet. 1989;630 doi: 10.1016/s0140-6736(89)90759-9. [DOI] [PubMed] [Google Scholar]
- 46.Gennery AR, Flood TJ, Abinun M, Cant AJ. Bone marrow transplantation does not correct the hyper IgE syndrome. Bone Marrow Transplant. 2000;25:1303–1305. doi: 10.1038/sj.bmt.1702446. [DOI] [PubMed] [Google Scholar]
- 47.Nester TA, Wagnon AH, Reilly WE, Spitzer G, Kjeldsberg CR, Hill HR. Effects of allogeneic peripheral stem cell transplantation in a patient with Job syndrome of hyperimmunologlobulinemia E and recurrent infections. Am J Med. 1998;105:162–4. doi: 10.1016/s0002-9343(98)00200-9. [DOI] [PubMed] [Google Scholar]
- 48.Minegishi Y, Saito M, Morio T. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–55. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Woellner C, Schaffer AA, Puck JM, Renner ED, Knebel C, Holland SM, Plebani A, Grimbacher B. The hyper IgE syndrome and mutations in Tyk2. Immunity. 2007;26:535. doi: 10.1016/j.immuni.2007.05.007. [DOI] [PubMed] [Google Scholar]