

Abstract
Glaucomatous optic neuropathy causes blindness through the degeneration of retinal ganglion cells (RGCs) and their axons, which comprise the optic nerve. Glaucoma traditionally is associated with elevated intraocular pressure, but often occurs or may progress with intraocular pressure in the normal range. Like other diseases of the CNS, a subset of glaucoma has been proposed to involve an autoimmune component to help explain the loss of RGCs in the absence of elevated intraocular pressure. One hypothesis involves heat shock proteins (HSPs), because increased serum levels of HSP autoantibodies are prominent in some glaucoma patients with normal pressures. In the first direct support of this hypothesis, we found that HSP27 and HSP60 immunization in the Lewis rat induced RGC degeneration and axon loss 1–4 months later in vivo in a pattern with similarities to human glaucoma, including topographic specificity of cell loss. Infiltration of increased numbers of T-cells in the retina occurred much earlier, 14–21 d after HSP immunization, and appeared to be transient. In vitro studies found that T-cells activated by HSP immunization induced RGC apoptosis via the release of the inflammatory cytokine FasL, whereas HSP immunization induced activation of microglia cells and upregulation of the FasL receptor in RGCs. In summary, our results suggest that RGC degeneration in glaucoma for selected individuals likely involves failed immunoregulation of the T-cell-RGC axis and is thus a disturbance of both proapoptotic and protective pathways.
Keywords: autoimmunity, glaucoma, microglia, T-cells, FasL, heat shock proteins
Introduction
By 2020, nearly 80 million people worldwide will suffer from glaucoma, the leading cause of irreversible blindness (Quigley and Broman, 2006). Although glaucoma typically is associated with chronic elevation in intraocular pressure, in many individuals, glaucomatous damage occurs without eye pressure exceeding the normal range. Growing evidence suggests that glaucomatous pathology in some of these cases could arise from an autoimmune response and thus comprise an autoimmune neuropathy. For example, an increased prevalence of monoclonal gammopathy (Wax et al., 1994), retinal Ig deposition (Wax et al., 1998a), elevated serum titers of autoantibodies to many optic nerve (Tezel et al., 1999) and retina antigens (Romano et al., 1995; Tezel et al., 1998; Wax et al., 1998b, 2001; Maruyama et al., 2000; Kremmer et al., 2001; Yang et al., 2001b), and abnormal T-cell subsets (Yang et al., 2001a) have been reported in many patients with normal pressure glaucoma. Increased autoantibodies in the serum of glaucoma patients include heat shock proteins (HSPs), such as HSP60 and HSP27, and α-crystallins (Tezel et al., 1998), expression of which are upregulated in the glaucomatous retina and optic nerve head (Tezel et al., 2000) and are known to induce neuronal apoptosis by attenuating the ability of native HSPs to stabilize the cytoskeleton (Tezel and Wax, 2000).
Ocular immune-privilege functions to regulate immune responses thereby controlling potentially damaging and sight-threatening autoimmune disease (Gregerson, 1998; Streilein et al., 2000). As in the CNS, apoptotic elimination of T-cells is considered an essential protective mechanism to prevent inflammation and antigen encounter in the eye. Despite immune-privilege, however, T-cells are able to enter normal, uninjured brain as part of constitutive immune surveillance (Hickey et al., 1991; Raivich et al., 1998). The site-specific parenchymal recruitment of T-cells could initially play an important role as a protective mechanism, because it allows early contact of the immune system with cellular debris, and removal of pathogenic agents from the CNS. This elicits what has been called “protective immunity,” in that T-cells mediate the protection of neurons from degenerative conditions (Schwartz and Kipnis, 2001; Kipnis et al., 2002). However, once T-cells are presented with the antigen, they can also initiate an immune response leading to neurodegeneration.
As in other types of neurodegeneration (Aloisi, 1999; Carson, 2002), resident microglial cells with increased MHC expression in glaucomatous eyes (Neufeld, 1999) likely participate in the immune-mediated process. However, the precise mechanisms underlying the complex interplay between immune-privilege, protective immunity, and autoimmune neurodegenerative disease are not well understood in glaucoma. Here we provide the first direct evidence that immune system activity can result in the glaucomatous loss of RGCs and their axons. We used an in vivo rat model, in which an autoimmune response was elicited through immunization with HSPs. Considerable evidence implicates the involvement of Fas-ligand (FasL), a member of the tumor necrosis factor (TNF) superfamily of inflammatory cytokines, in the apoptotic elimination of T-cells from the brain (Dowling et al., 1996) and the eye (Griffith et al., 1995). We therefore sought to explore the role of FasL in cellular interactions of activated T-cells and RGCs in vitro. Our results suggest that T-cell-mediated RGC degeneration occurs through Fas/FasL pathway signaling. We further found that cultured retinal microglia express FasL and can induce the apoptotic elimination of T-cells derived from HSP-immunized animals. Thus, microglia may constitute an intrinsic protective mechanism that protects T-cell-induced RGC degeneration in both normal and autoimmune-mediated glaucomatous pathology.
Materials and Methods
Immunization protocol.
Adult male Lewis rats (150–450 g) were obtained from Charles River and housed in a 12 h light–dark cycle environment. All animals were handled according to the regulations of the Institutional Animal Care and Use Committee, and all procedures were adhered to the tenets of the Declaration of Helsinki and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The animals in each control (2–3 months of age, n = 8) and experimental group (2–3 months of age, n = 10) received LPS 250 μg/kg 2 weeks before immunization with HSPs. LPS induces a short-lived uveitis, which resolves spontaneously and results in a permanent breakdown of the blood–eye barrier (Yang et al., 1997, 1998; Pouvreau et al., 1998). This is an important prerequisite to facilitate the likelihood of obtaining a retina/optic nerve specific response, as opposed to a neuropathy elsewhere after HSP immunization. At the time of immunization, the eyes showed no signs of ocular inflammation by slit lamp examination. Animals were immunized with single-dose intraperitoneal injection of purified recombinant HSPs (Stressgene). We prepared 100 μl of stable emulsion containing 100 μg of the antigen by mixing equal volumes of the antigen and the incomplete Freund's adjuvant (IFA) (Sigma-Aldrich) containing purified pertussis toxin (1 μg per animal, Sigma-Aldrich) as an additional adjuvant. Control animals were injected with the IFA mix or IFA mix plus bovine serum albumin (BSA). We examined rat eyes 1 d after immunization, and then every other day to confirm the absence of any damage, infection or hemorrhage inside the eye. Animals were killed 1 or 4 months after immunization. Because our analysis indicated no difference between eyes, retinas or optic nerves for these two time points, animals from the two groups were pooled. Several normal Lewis rats of comparable age did not undergo any treatment and were killed for comparisons.
TCRαβ+ T-cell staining and quantitation in the retina.
Immediately after the animals were killed, they were processed through cardiac perfusion (upper part of body) with 35 ml of BSS followed by 35 ml of 4% paraformaldehyde. The eyes were enucleated and fixed in 4% paraformaldehyde and processed by the following procedures. The retinas were dissected from the eye cup; vitreous and the retinal inner limiting membrane (ILM) were removed completely for sufficient antibody penetration. The retinas were cryoprotected in graded sucrose solutions and repeatedly frozen and thawed to facilitate the penetration of primary antibody into tissue. The retinas were then treated with peroxide to quench the endogenous peroxidase in tissue and incubated in goat serum to block nonspecific binding in retina. The retinas were incubated with primary antibodies [1:100 of mouse to rat TCRαβ monoclonal antibody (PharMingen)] overnight, washed three times with PBS (10 min each time), incubated with biotinylated secondary antibodies [1:400 of goat to mouse without Fc segment (Santa Cruz)] for 2 h. After washing three times in PBS, the retinas were treated in ExtrAvidin solution (1:400) for 1 h. Then, the retinas were stained ABC solutions from mouse ABC staining system (Santa Cruz) until an ideal color was achieved. After immersion in PBS buffer for 2–3 min, the retinas were flat mounted onto glass slides and air-dried overnight. All agents except specifically mentioned are purchased from Sigma. A seamless true color 20× digital image of an entire rat retina was captured. using ScanScope XT, a linear array scanning microscope (Aperio Technology). To count the T-cells, an area of the retinal image was selected and magnified and T-cells (dense, dark and brown positive cells) were identified manually by drawing green circles around them. The entire retina was processed in this manner to count the T-cells. The pattern of T-cell distribution in the entire retina was viewed under low magnification.
Generation of rat T-cell lines.
Cell suspensions were obtained from the inguinal and mesenteric lymph nodes, and spleen of the immunized or control animals. Ten days after the antigen was injected into the rats' hind footpads in 0.1 ml of the emulsion, the rats were killed and their draining lymph nodes and spleen were surgically removed and dissociated. The splenocytes were isolated on Histopaque-1083 (Sigma-Aldrich) from freshly obtained cell suspension. Rat CD3+ T Cell Enrichment Columns from R&D Systems were used to enrich T-cells according to manufacturer's protocol. Purity of the enriched T-cell population was >92%. The cells were washed and activated with the antigen (10 μg/ml) in stimulation medium containing DMEM supplemented with l-glutamine (2 mm), 2-mercaptoethanol (5 × 10−5 m), sodium pyruvate (1 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), nonessential amino acids (1 ml/100 ml), and autologous serum 1% (v/v). After incubation at 37°C and 10% CO2 for 72 h, cells were transferred to propagation medium prepared as described above with the addition of 10% FCS (v/v) and 10% T-cell growth factor derived from the supernatant of Con A-stimulated spleen cells. Cells were grown in propagation medium for 4–10 d before restimulation with their antigen (10 μg/ml) in the presence of irradiated (2000 rad) thymus cells (107 cells/ml) in stimulation medium. The T-cell lines were expanded by repeated stimulation and propagation, as previously described (Schori et al., 2001).
RGC-5 cell culture.
The RGC-5 cell line transformed with Ψ2 E1A virus was developed from postnatal Sprague Dawley rats as described previously (Krishnamoorthy et al., 2001). The “RGC-5” line was found to have numerous characteristics of retinal ganglion cells. The antigenic profile included positive expression of Thy-1, an RGC-specific surface antigen, and another ganglion cell marker, Brn3b. Negative findings for non-RGC markers, such as for glial, amacrine, and horizontal cells, were also confirmed. Other RGC features identified were morphology and sensitivity to glutamate toxicity and neurotrophin withdrawal (Krishnamoorthy et al., 2001). RGC-5 cell cultures were grown in DMEM (Invitrogen Cat #23700–040) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen Cat #26140–079) and 100 U/ml penicillin and 100 μg/ml streptomycin. RGC-5 neurons were plated (10,000/cm2) onto poly-l-lysine-coated glass coverslips in 24-well culture dishes at 37°C in 5% CO2 and air.
Flow cytometry.
For apoptotic cell counts, attached cells in cocultures were separately isolated, stained using APO-BRDU kit (PharMingen), and were counted by flow cytometry [fluorescence-activated cell sorter (FACS) analysis]. Briefly, 2 × 106 cells washed with PBS containing 0.1% BSA were stained with APO-BRDU kit. After washing, cells were measured using a CELLQuest Software system (Becton-Dickinson) equipped with a single air-cooled, argon-ion laser. Incubations omitting the primary antibody served as the control. Data were collected using logarithmic amplification on 10,000 cells, excluding cell debris by a combination of forward and side scatters. Measurement gates were set using the negative controls.
ELISA.
Serum levels of sFasL were measured using 96-well microtiter plates (Costar) coated with 50 μl of 50 ng/ml FasL antibody (Santa Cruz). Negative control wells prepared without antigen or primary antibodies, and positive control wells, in which increasing concentrations of primary antibody were added, were processed simultaneously. Optical density values were recorded at three independent measurements and the average value was calculated.
RT-PCR.
The gene expression of Fas and FasL was evaluated by RT-PCR, using commercially synthesized Fas or FasL specific oligodeoxynucleotide primers. The following primers were synthesized: Fas sense, 5′-AACATGAGAACATCCTGTGCC-3′; Fas antisense, 5′-TCCCTGCTCATGATGTCT-ACC-3′; FasL sense, 5′-TTCTTTATCATGGATGCCAGG-3′; and FasL antisense, 5′-GAACAGTCTTCTTCCCATCTGG-3′. Total cellular RNA was isolated using an RNA kit from Qiagen and was reverse transcribed into cDNA using oligo-dT primers and murine leukemia virus (MLV) reverse transcriptase (Invitrogen) as per the manufacturer's instructions. PCR was performed in a total volume of 50 μl containing PCR buffer, 5 μl of cDNA with 1 U Taq-DNA polymerase, 0.5 mm dNTPs, and 1 μm sense and antisense primers. Amplifications were performed using cycles (32 for Fas, 36 for FasL) of 94°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and an extension cycle of 72°C for 10 min.
Immunoprecipitation.
Pansorbin beads (Calbiochem-Novabiochem) washed with TA buffer (20 mm Tris-HCl, 5 mm sodium azide, pH 7.3) were incubated with rabbit anti-rat FasL antibody (1 mg/ml, Santa Cruz) at 4°C for 25 min. Beads were then centrifuged and the supernatant was decanted. The beads were washed three times with TA buffer (3× the volume of beads), and incubated with the supernatant obtained from cell cultures at 4°C for 2 h, using a ratio of 200 μl of supernatant per 10 μl of beads. At the end of 24 h incubation, supernatant was used to treat the cells.
Labeling and quantification of retinal ganglion cells and axons.
The posterior eyecups of control and immunized rats with a 2–4 mm segment of intact optic nerve were cleared of vitreous and immersion fixed in 4% paraformaldehyde for 2–3 h. An incision was made in the superior vertical meridian of each eyecup for orientation of retinal eccentricity. After rinsing in PBS (pH 7.4), the retina of one eye of each animal was removed and incubated in primary antibodies against Brn3a followed by a biotin-conjugated secondary antibody. The opposing eye was used for other purposes, including vertical sections or whole-mount preparation for additional antibodies not described here. The Brn3a-labeled retina was postfixed for 45 min in 2% glutaraldehyde and the DAB product enhanced following standard protocols (Harvey and Calkins, 2002). The retina was mounted RGC-side up and photographed using an Olympus AX70 Provis light microscope equipped with a motorized X–Y–Z stage, video camera, and a PC workstation interface mediated by ImagePro software (Media Cybernetics). We then assessed all retinas for artifacts caused by excessive vitreous or tearing that would prevent accurate imaging of RGCs. From our original sample of six animals in each group, we included retina from each control animal, but excluded two HSP27 and two HSP60 eyes because of obvious tissue artifacts. We prepared a computer algorithm in which each retina was photographed en montage using a 40× objective with differential interference contrast optics. Each frame of the montage was computed from a collapsed series of images collected within a 20 μm interval optimized in contrast to detect each labeled RGC. This stacking procedure ensures that irregularities in the plane of the RGC layer that arise during the whole-mount procedure do not induce an under-sampling of labeled cells that lie outside the immediate focal plane. The algorithm then applied a shape-size exclusion filter to discriminate labeled RGCs from other high-contrast features, such as tears in the tissue and blood vessels. The sampling area of frame, less the area contained by subtracted features was calculated and used to compute cell density (cells/mm2). All regions with obvious tissue defects that could lead to a null value for RGC density were excluded, as were all edges. We simplified the data analysis by calculating RGC density in radial coordinates, in which the optic nerve head (ONH) serves as the origin and each radius representing retinal eccentricity is separated by a 30° angle. Because RGC density contours in the rat do not show any significant inferior versus superior difference (e.g., see Fig. 2C) (Fukuda, 1977; Schober and Gruschka, 1977), we used the vertical meridian through the ONH to separate data from the temporal and nasal hemifields and averaged across radii and angles for each hemifield. The accuracy of the counting algorithm was confirmed by comparing the computer-generated counts to hand-counts from 10–20 randomly selected frames from each retina; the counts generally differed by <5% with no apparent systematic bias.
Figure 2.
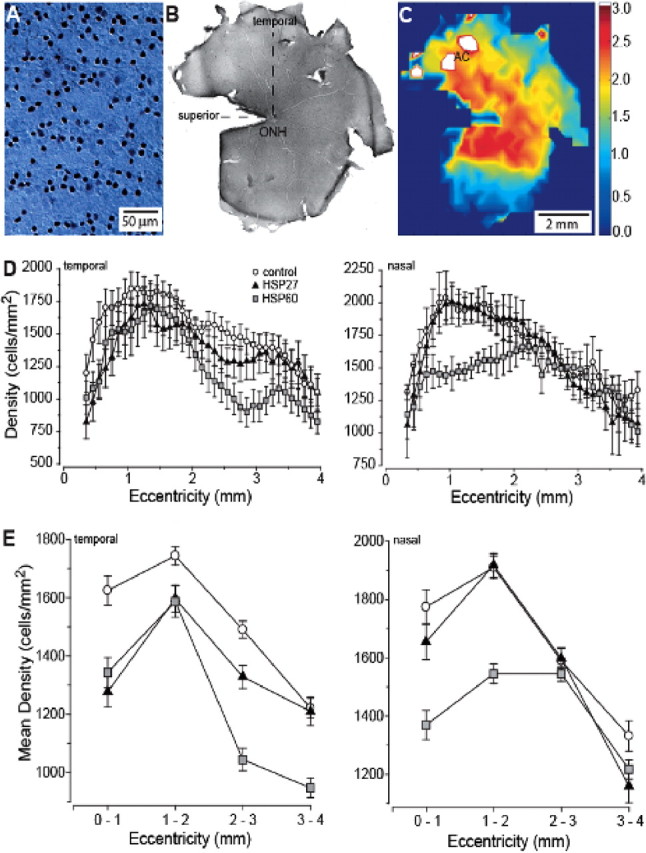
RGC density measurements in the rat retina after immunostaining against Brn3a. A, An individual frame focused through the RGC layer from an actual montage from control retina used in counting. B, Low-magnification view of entire whole-mount montage of control retina showing orientation and location of the optic nerve head (ONH), superior vertical meridian and temporal horizontal meridian. C, False-color representation of the montage of the whole retina shown in B, with area centralis indicated (AC). White areas near AC correspond to region of highest RGC density on the color scale (cells/mm2 × 1000). D, Density of Brn3a-labeled RGCs averaged across animals as a function of eccentricity in the temporal (left) and nasal (right) hemifields of the retina, measured at 0.1 mm intervals. E, Density of Brn3a-labeled RGCs from D pooled within 1 mm intervals as a function of eccentricity in temporal (left) and nasal (right) hemifields. For the temporal hemifield, t test comparison of the means at each interval indicated significance (p ≤ 0.01) for differences between control and HSP27 animals at each eccentricity except 3–4 mm (p = 0.85). The same comparison between control and HSP60 animals yielded significance at all eccentricities (p ≤ 0.02). For the nasal hemifield, t tests indicated significant differences between control and HSP27 retina only at 3–4 mm eccentricity (p < 0.01) and between control and HSP60 retina at 0–1 mm (p < 0.01) and 1–2 mm (p < 0.001).
For quantifying RGC axons, optic nerve segments were isolated from the animals proximal to the eye, stained with osmium tetroxide, and embedded in epoxy resin as previously described (Sappington et al., 2003). We cut 1–2 μm semithin sections from each sample and collected 20–25 high-magnification light micrographs from random locations on each section. An automated algorithm counted each axon that had an intact myelin sheath, as previously described, and assigned an axon density for each micrograph as previously described (Inman et al., 2006). For this analysis, we included 6 control optic nerves and 4 from each HSP group. A post hoc Bonferroni power analysis of the data obtained from both our Brn3a and axon counts indicate our sample sizes were sufficient to reach 95% significance.
Fluorescent immunocytochemistry.
Vertical retinal sections were prepared for fluorescent immunocytochemical labeling as described previously (Harvey and Calkins, 2002). Microglia were labeled using mouse anti-rat RT1a/OX18 (1 μg/ml; Chemicon) and positively identified by amoeboid or ramified morphology. For labeling of the FasL receptor (FasR/CD95), we used a goat anti-mouse monoclonal antibody (TNFRSF6 at 10 μg/ml; R & D Systems). We used secondary antibody solutions containing either goat anti-mouse or donkey anti-goat IgG (10 μg/ml; Molecular Probes). Controls conducted with no primary antibody or without the appropriate IgG isotype demonstrated no specific labeling.
Results
HSP immunization induces retinal ganglion cell and axon loss, in vivo
To simulate the autoimmune component of RGC degeneration in glaucoma, we immunized Lewis rats with HSP60 or HSP27 and harvested tissue a short period (1 or 4 months) later. Examination of cross sections of retinas from each group revealed no discernable changes in any retinal layer (Fig. 1A). Because the retinas appeared so similar in cross section, we did not compare the numbers of photoreceptors or inner retinal neurons, although we cannot rule out the possibility of differences.
Figure 1.
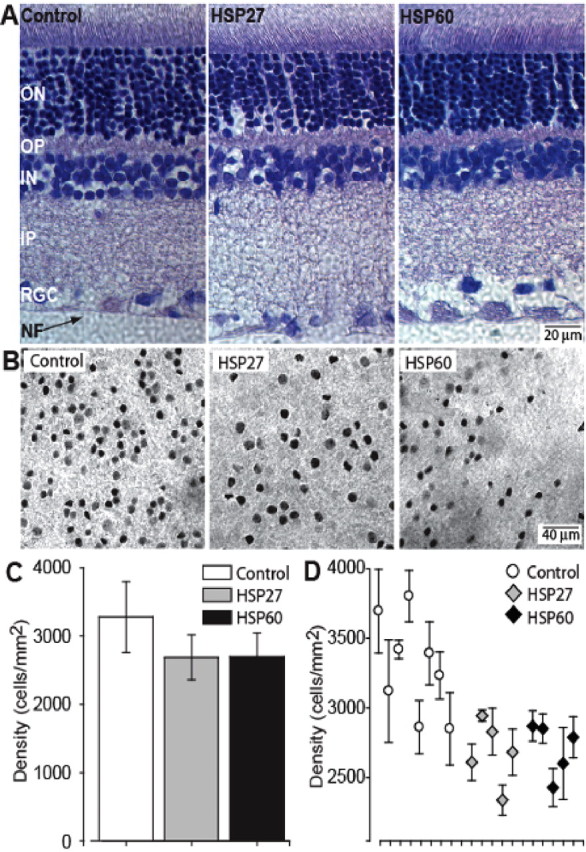
Apparent RGC loss in retinas of HSP-immunized rats. A, Sections of retina from control and HSP27- and HSP60-immunized animals demonstrating similarity of retinal layers: nerve fiber (NF), retinal ganglion cell (RGC), inner plexiform (IP), inner nuclear (IN), outer plexiform (OP), and outer nuclear (ON). Scale is the same for all three panels. B, Whole-mount preparations of retina from control (left), HSP27-immunized (middle), and HSP60-immunized (right) rats labeled with immunocytochemistry against the RGC-specific marker Brn3a. Plane of focus is the RGC layer. Scale is the same for all three panels. C, Quantification of RGC density near the neuroretinal rim in three animal groups. The difference between the mean cell counts of control and HSP-immunized groups was highly significant (p < 0.001). D, Mean peak RGC density plotted for individual retinas in our three groups of animals.
To assess whether HSP immunization could induce RGC loss similar to that found in human glaucoma, we immunolabeled whole-mount preparations using the RGC-specific marker Brn3a (Xiang et al., 1995; Quina et al., 2005). We observed differences in RGC density in two regions that each correspond to peak RGC density in the normal rat. The first is in the rudimentary area centralis, which lies 1.5–2 mm eccentric of the optic disk in the upper temporal quadrant; the second lies along an isodensity contour just outside the neuroretinal rim where the optic nerve forms (Fukuda, 1977; Schober and Gruschka, 1977; Dreher et al., 1985). In the retina of HSP-immunized animals, the distribution of Brn3a-labeled RGCs in the region just flanking the area centralis appeared patchy compared with control retinas (Fig. 1B). This trend was more dramatic in HSP60 retina than in HSP27 retina, as the quantification described below demonstrates.
We observed a similar patchiness in HSP-immunized retina in the area of high RGC density near the optic nerve head. We scanned this area at high magnification to identify small regions of highest RGC density in each Brn3a-labeled retina and then averaged counts from 25–50 high-magnification micrographs of these regions (Fig. 1C). In control animals receiving only the LPS dose, peak RGC density was 3276 ± 516 cells/mm2, comparable to the density in normal rats (3112 ± 506 cells/mm2; p = 0.2). However, the density was markedly lower in both HSP27 (2688 ± 328 cells/mm2) and HSP60 retina (2695 ± 351 cells/mm2). This decrease of nearly 20% from control was significant for both HSP groups (p < 0.001). To assess individual variability, we next plotted the mean peak RGC density from each retina analyzed (Fig. 1D). The lowest peak density in the control group was 2850 cells/mm2 and eight of 10 of the HSP retinas were below this value. Similarly, six of eight of the control retinas had a peak RGC density of >3000 cells/mm2, whereas no HSP retina peaked above 2950 cells/mm2. These data indicate that although there is variability in the effect of HSP immunization in individual animals, there is a pervasive trend toward considerable RGC loss.
To determine more precisely RGC distribution in HSP-immunized animals and whether these retinas contain additional pockets of cell loss, we developed an objective means to quantify RGC density across different spatial locations of the retina. In our computer-aided algorithm, high-magnification images are collected from at each retinal location by collapsing stacks of individual slices through the retinal ganglion cell and nerve fiber layer (Fig. 2A). The high-magnification images are compiled into a complete montage covering each retinal location (Fig. 2B). Our algorithm then steps across the montage and counts frame-by-frame the number of labeled cells; cell density at each location is calculated and represented in a false-color image of the whole mount (Fig. 2C).
Peak density in individual frames ranged as high as 4000 cells/mm2, always in the isodensity contour including the area centralis or rim of the optic nerve (Fig. 2C). In the rat, all Brn3a-expressing RGCs project to the superior colliculus and lateral geniculate nucleus (Quina et al., 2005), which account for the projections of ∼85% of all RGCs (Linden and Perry, 1983; Dreher et al., 1985). This is consistent with qualitative estimates of the fraction of Brn3a-expressing RGCs (80–85%) (Xiang et al., 1995). Assuming that Brn3a-labeled cells in our material represent a similar fraction, the peak densities we measured are consistent with the known total RGC density in the rat of ∼4500–6000 cells/mm2 (Fukuda, 1977; Schober and Gruschka, 1977; Dreher et al., 1985). We simplified analysis of the whole-mount maps by averaging data across different radii emanating from the optic nerve head for both the nasal and temporal hemifields as described in Materials and Methods. Because of this averaging, the peak density in both temporal and nasal fields was naturally lower. We found an average peak density of Brn3a-labeled RGCs across all samples of 2000–2200 cells/mm2 at ∼1 mm eccentricity for control retina (Fig. 2D). We found that HSP60 or HSP27 immunization significantly reduced the number of Brn3a-labeled RGCs in a spatially specific manner, consistent with our previous observations. When assessed as a function of eccentricity in 0.1 mm intervals from the optic nerve head (ONH), in the temporal retinal hemifield (Fig. 2D, left), both HSP27- and HSP60-immunized retina demonstrated a general reduction in RGCs at most eccentricities compared with control. This loss was again most dramatic in the regions flanking those of highest RGC density in the controls, ∼0–1 mm and 2–3 mm eccentric from the ONH. This includes the areas around the area centralis (Fig. 2C). In the nasal retina (Fig. 2D, right), RGC density in HSP27-immunized animals was generally similar to that in control animals. However, density in HSP60-immunized animals was dramatically lower, especially 1–2 mm eccentric from the ONH.
To further generalize these trends, we averaged RGC density within 1 mm intervals for both the temporal and nasal hemifields (Fig. 2E). When plotted this way, the loss in the temporal hemifield was most similar in HSP27- and HSP60-immunized animals 0–1 mm from the ONH (Fig. 2E, left). This is consistent with the data in Figure 1, C and D. However, the most dramatic reduction in RGCs compared with control animals occurred for HSP60-immunized animals at 2–4 mm from the ONH, where the retinas exhibited a 25% loss. This region includes the flank of the area centralis, as indicated in Figure 1B. In the nasal retina (Fig. 2E, right), the most significant loss of RGCs for these retinas was again a 20–25% decrease near the ONH at 0–2 mm eccentricity (Fig. 2D). Thus, HSP60-immunized animals demonstrated the most profound loss of RGCs. The pattern of loss in the temporal retina with HSP60 immunization in particular exhibited prominent similarities to that detected in glaucomatous human eyes (Quigley, 1993; Araie, 1995), where typical lesions involve the loss of RGCs in mid-peripheral and parafoveal zones surrounding the region of highest density.
Because retinal degeneration in animal models of glaucoma is accompanied by loss of RGC axons in the optic nerve (e.g., Inman et al., 2006), we next turned our attention to examination of optic nerves from HSP-immunized animals. The optic nerve from control eyes demonstrated tight packing of individual axons within bundles or fascicles contained by thin processes of astrocyte and oligodendrocyte glia (Fig. 3A). Axons from these nerves demonstrated intact myelin sheaths, a well rounded morphology, and a variety of diameters, which we did not attempt to quantify in this study. Optic nerve from HSP27-immunized animals demonstrated a modest decrease in axonal packing, as indicated by greater filling of the interfascicular space with glial processes and irregularities within individual fascicles (Fig. 3B). Axons from these nerves often showed signs of degeneration, including multiple myelin sheathes. Finally, optic nerve from HSP60-immunized animals showed obvious degeneration (Fig. 3C), with loss of axons, several gliosis, and the presence of degenerated axonal profiles.
Figure 3.
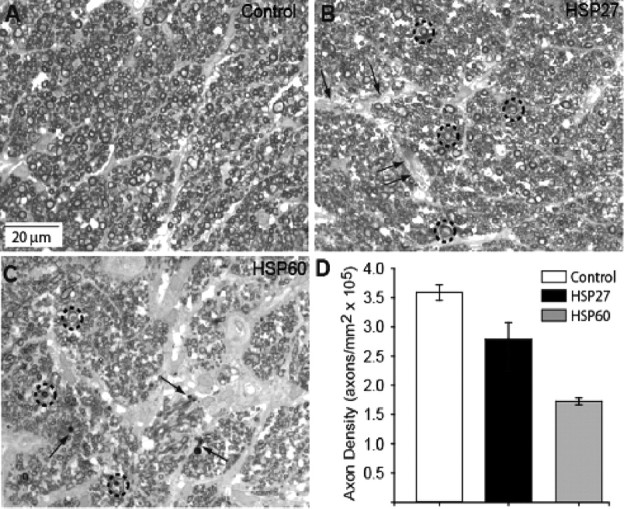
Axon density is reduced in optic nerve of HSP-immunized animals. A, High-magnification light micrograph of control nerve demonstrating typical distribution of RGC axons of a variety of diameters tightly packed into regular fascicles. B, Optic nerve of HSP27-immunized animal has modest decrease in axon density, distension of interfascicular regions (arrows), and appearance of multilaminar myelin sheaths for some axons (circles). C, Optic nerve from HSP60-immunized animal demonstrates severe axonal dropout, obvious gliosis within the interfascicular regions, axons with multilaminar myelin sheaths (circles), and the appearance of small, degenerated profiles (arrows). D, Quantification of axon density in three animal groups. The difference between the mean axon counts of control and HSP-immunized groups was highly significant (p < 0.001).
To support these qualitative observations, we quantified axonal density in our cross sections of the optic nerve. In a random series of 20–25 high-magnification micrographs such as those shown in Figure 3A–C, we found an average axon density in control nerves of 3.59 ± 0.16 × 105 axons/mm2. The cross-sectional area of the optic nerve for these eyes ranged from 0.24 to 0.34 mm2, yielding an extrapolated number of total axons of 76,000–115,000 axons. This is consistent with the published number of RGC axons in the rat nerve of ∼100,000 (Linden and Perry, 1983; Dreher et al., 1985). We found a 22% reduction in axon density for HSP27-immunized animals and a more dramatic 57% reduction for HSP60-immunized animals (Fig. 3D). Because the cross-sectional area for each group of nerves did not differ statistically (0.26 ± 0.03 mm2), these reductions would extrapolate to an average loss of ∼20,000 axons for the HSP27-immunized animals and a loss of ∼53,000 axons for the HSP60-immunized animals.
Detection of retinal TCRαβ+T-cells in HSP27- and HSP60-immunized rat retinas
Although the search for T-cell infiltration of the retina has been unproductive in both human glaucoma postmortem specimens as well as in high IOP animal models (our unpublished observations), we reasoned that an immunization model seeking to demonstrate experimental glaucomatous RGC and axonal loss might be similar to other models of immune-mediated neuropathy in which CD4 T-cell expression occurs only transiently during the disease process in effected parenchymal tissue (Ludowyk et al., 1992; Shikishima et al., 1993; Verhagen et al., 1994; Begolka et al., 1998). We therefore sought to determine whether and when T-cells would appear in the retina in HPS-immunized animals by immunolabeling against TCRαβ+ T-cells. We observed the unequivocal presence of T-cells in the retina parenchyma in both sham (Fig. 4A) and adjuvant-only retina (Fig. 4C). These levels did not differ between 7 and 21 d after immunization (p > 0.05) (Table 1), nor did they differ between the two HSP-immunized groups (p > 0.05). In contrast, the number of labeled T-cells increased nearly fourfold for HSP60 immunization (Fig. 4B) by 21 d compared with adjuvant only (p < 0.001) and by over twofold for HSP27 immunization (Fig. 4D) by 14 d compared with adjuvant only (p = 0.017) (Table 1). In both HSP-immunized groups, the number of T-cells decreased back to adjuvant-only levels by 60 d which suggests that HSP immunization results in the transient penetration of T-cells into retinal parenchymal tissue, a phenomenon we are investigating further. Interestingly, the spatially densest distribution of T-cells in both the sham (normal) and adjuvant-only retinas appears to be confined to a particular quadrant with penetration beginning in adjacent quadrants near the retinal margin (Fig. 4A,C). This tendency increases with HSP27 immunization, where two quadrants are T-cell replete (Fig. 4D), until the entire retina is T-cell enriched with HSP60 immunization (Fig. 4B). This may be significant given that RGC pathology is known to follow a similar sectoral pattern in rodent models of high-pressure glaucoma (Jakobs et al., 2005; Howell et al., 2007; Soto et al., 2008). The pattern is also intimated by our Brn3a quantification, which indicates topographic loss of RGCs (Figs. 1, 2). As a whole, these data strongly suggest that RGC loss in our experimental model may be the direct result of result T-cell activity and interactions with target cells in the retina.
Figure 4.

Immunolabeling of retinal TCRαβ+ T-cells. The T-cells are marked with green circles in each retinal image (20×) for counting. A, Adjuvant, 21 d; B, HSP60, 21 d; C, adjuvant, 14 d; D, HSP27, 14 d. The images located at the right top corner of each panel represents the TCRαβ+ T-cell distribution patterns of entire retinas. Scale bar at lower left side of each panel, 100 μm.
Table 1.
TCRαβ+ T-cells in HSP27- and HSP60-immunized Lewis rat retinas
| Groups | 7 d | 14 d | 21 d | 60 d |
|---|---|---|---|---|
| Sham | 114 ± 58 | 82 ± 41 | 111 ± 45 | 63 ± 27 |
| Adjuvant | 152 ± 74 | 134 ± 82 | 173 ± 105 | 69 ± 26 |
| Hsp27 + adj. | 136 ± 58 | 313 ± 212* | 268 ± 190 | 78 ± 35 |
| Hsp60 + adj. | 127 ± 98 | 159 ± 118 | 637 ± 192** | 178 ± 236 |
Data are shown as mean ± SD; n = 10–12 retinas/group. *versus adj. p value = 0.017; **versus adj. p value = 0.000002; all adjuvants versus corresponding shams NS (p > 0.05).
HSP-activated T-cells induce retinal ganglion cell apoptosis in vitro via FasL
To assess whether HSP immunization could induce a proapoptotic interaction between T-cells and RGCs, we cocultured the immortalized RGC-5 cell line with T-cells isolated from rats killed 4 months after HSP60 or HSP27 immunization. In control experiments, we found no evidence of apoptosis in RGC-5 or T-cells cultured alone, nor in control cocultures in which T-cells were activated using IFA or BSA (Fig. 5A). However, the rate of RGC-5 apoptosis measured by FACS assessment of BRDU labeling in the presence of T-cells activated by HSP60 or HSP27 immunization was 24.92 ± 1.81% and 21.82 ± 2.12%, respectively (Fig. 5B,C). We examined whether the apoptosis of RGC-5 cells in cocultures is mediated by soluble factors released from T-cells by performing passive transfer experiments in which RGC-5 cells were treated with the conditioned medium obtained from HSP-activated T-cells. The conditioned medium obtained from HSP60-activated T-cells induced apoptosis in RGC-5 cells (Fig. 5D). However, the conditioned medium from HSP27-activated T-cells did not. In addition, the conditioned medium obtained from control cultures, in which T-cells were activated using IFA or BSA, did not induce RGC-5 apoptosis. The apoptosis rate of RGC-5 cells induced by the conditioned medium obtained from HSP60-activated T-cells measured by FACS assessment of BRDU labeling was ∼21% (Fig. 5E). To determine whether there might be a significant role for Fas/FasL in T-cell mediated RGC degeneration, we measured soluble FasL (sFasL) levels in conditioned media obtained from activated T-cells using ELISA. The conditioned medium obtained from HSP60-activated T-cells had a higher level of sFasL compared with the medium obtained from HSP27-activated or control T-cells (Fig. 5F).
Figure 5.

Effect of T-cell activation on RGC-5 cell survival. RGC-5 cells were cocultured with HSP activated T-cells for 24 h in serum-free medium. A, Formation of oligonucleosomal fragments was determined by 2% agarose gel electrophoresis. Lane 1: 100 bp DNA ladder as the size marker; Lane 2: HSP27 activated T-cells; Lane 3: HSP60 activated T-cells; Lane 4: IFA activated T-cells; Lane 5: BSA activated T-cells; Lane 6: RGC-5 cells alone; Lane 7: T-cells alone. B, Rate of RGC-5 cells apoptosis measured by FACS assessment of BRDU labeling as described in Materials and Methods in the presence of HSP60 activated T-cells. C, Rate of RGC-5 cells apoptosis measured by FACS assessment of BRDU labeling as described in Materials and Methods in the presence of HSP27 activated T-cells. D–F, Effect of conditioned medium obtained from HSP activated T-cells on apoptosis of RGC-5 cells. D, Formation of oligonucleosomal fragments was determined by 2% agarose gel electrophoresis. Lane 1: 100 bp DNA ladder as the size marker; Lane 2: Medium from HSP60 activated T-cells; Lane 3: Medium from HSP27 activated T-cells; Lane 4: Medium from IFA activated T-cells; Lane 5: Medium from BSA activated T-cells; Lane 6: Medium from RGC-5 cells alone; Lane 7: Medium from T-cells alone. E, Rate of RGC-5 cells apoptosis measured by FACS assessment of BRDU labeling as described in Materials and Methods in the presence of conditioned medium from HSP60 activated T-cells. F, Soluble FasL levels in the conditioned medium obtained from the activated T-cells, using ELISA. Similar results were obtained in three independent experiments.
To test whether sFasL contributes to the toxicity observed for RGC-5 cells in Figure 4, we performed immunoprecipitation experiments using FasL antibody to remove sFasL from the condition medium of HSP60-activated T-cells. Incubation of RGC-5 cells with the sFasL-depleted conditioned medium resulted in a significant decrease in apoptosis compared with incubations with the conditioned medium without immunoprecipitation (Fig. 6A). After immunoprecipitation of sFasL, the percentage of apoptotic cells in RGC-5 measured by FACS assessment of BRDU labeling as described in Materials and Methods was 6.32 ± 0.74% (Fig. 6B), which was ∼75% lower than the apoptosis rate obtained (24.92 ± 1.81%) before immunoprecipitation (Fig. 5B). Furthermore, incubation of RGC-5 cells in the presence of increasing concentrations of recombinant human FasL (25, 50, 100, 200 ng/ml) for 5 h resulted in death of RGC-5 cells in a dose-dependent manner (Fig. 6C). The concentrations of recombinant human FasL used in this experiment were much higher than the FasL levels seen after immunization with HSP60 (Fig. 5F). We used the FasL concentrations recommended by the supplier. It is possible that the recombinant protein used for in vitro studies might be less active compared with biological protein found in the serum after immunization. Western blot analysis (Fig. 6D) and RT-PCR (Fig. 6E) confirmed the expression of Fas in RGC-5 cell line; however, no FasL expression was detectable in this cell line by Western blotting using FasL antibody (data not shown). These findings confirm that sFasL is directly involved in the apoptosis of RGC-5 cells induced by HSP60-activated T-cells.
Figure 6.

Effect of soluble FasL from HSP60 activated T-cells' conditioned medium on apoptosis of RGC-5 cells. A, Formation of oligonucleosomal fragments was determined by 2% agarose gel electrophoresis. Lane 1: 100 bp DNA ladder as the size marker; Lane 2–4: RGC-5 cells + medium from HSP60 activated T-cells before immunoprecipitation with FasL antibody; Lane 5: RGC-5 cells + medium alone. Lane 6–8: RGC-5 cells + medium from HSP60 activated T-cells after immunoprecipitation with FasL antibody; Lane 9: RGC-5 cells + medium from IFA activated T-cells; Lane 10: RGC-5 cells + medium from BSA activated T-cells; Lane 11–15: RGC-5 cells treated with IL-2 (50 ng/ml), IL-4 (10 ng/ml), IL-6 (10 ng/ml), TNF-α (50 ng/ml), TNF-β (50 ng/ml), respectively. Lane 16: RGC-5 cells treated with anti-FasL Ab (5 μg/ml) alone. B, Rate of RGC-5 cells apoptosis measured by FACS assessment of BRDU labeling as described in Materials and Methods in the presence of conditioned medium from HSP60 activated T-cells after immunoprecipitation with FasL antibody. Similar results were obtained in three independent experiments. C, Dose-dependent effect of recombinant human FasL on RGC-5 cells death. D, E, Expression of Fas in RGC-5 cells by Western blotting (D) and RT-PCR (E).
To establish the relevance of FasL in our animal model, we measured sFasL in the sera of HSP-immunized animals. The sera from HSP60-immunized rats had a higher concentration of sFasL (7 ± 0.6 ng/ml) than either groups immunized with HSP27 (3 ± 0.8 ng/ml), IFA (0.5 ± 0.2 ng/ml) or IFA + BSA (0.7 ± 0.2 ng/ml). The increase in serum sFasL was significant (p < 0.05), which is consistent with our finding that HSP60 immunization induced greater RGC loss than HSP27 immunization.
HSP immunization induces microglia activation and upregulation of FasR, in vivo
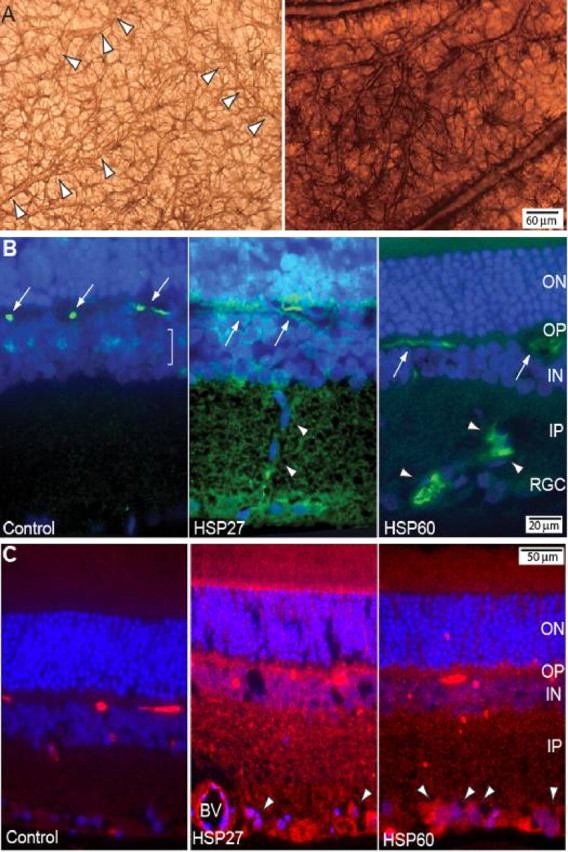
Because macroglial reactivity has been described in human eyes with glaucoma (Tezel and Wax, 2003), we sought to examine whether a similar activated glial phenotype was present in HSP-immunized eyes. A broad comparison of control versus immunized retinas revealed strong indications of large-scale glial activation in the retina that was devoid of lymphocytic infiltrates. In HSP-immunized animals, especially near vascular elements, astrocytes demonstrated greatly increased expression of glial-acidic fibrillary protein, indicative of hypertrophy and reactivity (Fig. 7A, right). Because microglia are the antigen-presenting cells of the retina, we further sought to determine whether HSP immunization induces the activation of microglia cells in vivo, using histocompatibility complex class 1 protein OX18, a standard marker for microglia ramification and hypertrophy (Ashwell et al., 1989; Wang et al., 2000). We distinguished microglia from endothelial cells, which may also be labeled by OX18, by their larger cell bodies, amoeboid or ramified morphology, and exclusion from the inner wall of blood vessels. Immunolabeling of control retina for OX18 was restricted to the cell body of resident microglial cells with very little evidence of ramification (Fig. 7B, left). In contrast, HSP27-immunized retina (Fig. 7B, middle) exhibited increased expression of OX18 in ramified microglial processes throughout the retina as indicated by the diffuse labeling in all layers. Moreover, in this retina, OX18-positive microglia cells were seen migrating through the inner retina, consistent with their activation. HSP60-immunized retina demonstrated a decrease in diffuse OX18 label, whereas levels of OX18 in individual microglia throughout the retina appeared more intense (Fig. 7B, right), particularly in the inner retina and near RGCs. Thus, OX18 labeling in experimental animals reveals microglial activation resembling that seen in the retina of glaucomatous human eyes (Tezel and Wax, 2003).
Figure 7.
Glial activation in the retina by HSP immunization. A, Expression of glial-acidic fibrillary protein (GFAP) in astrocytes of control (left panel) and HSP60-immunized (right panel) rats. Arrowheads in the left panel delineate capillaries outlined by GFAP-labeled astrocyte processes. Scale is the same for both panels. B, Retinal sections from control (left), HSP27-immunized (middle), and HSP60-immunized (right) animals immunostained for OX18 (green) and counter-labeled with the nuclear dye DAPI (blue). In control retina, OX18 labeling is localized discretely to microglia cell bodies (arrows) in the outer plexiform (OP) layer and to a lesser extent to microglia (bracket) in the inner nuclear layer (IN). In HSP27 retina, OX18 label is intensified in ramified microglial processes throughout the retina, most prominently in the outer nuclear (ON), inner plexiform (IP), and retinal ganglion cell (RGC) layers. Migrating microglial cells are identified by their elongated, elliptical cell bodies (arrowheads). In HSP60 retina, levels of OX18 have increased in individual microglia cells in the OP (arrows) and in the RGC and IP layers, where the microglia have become more ramified. Scale is the same for all three panels. C, Retinal sections from control (left), HSP27-immunized (middle), and HSP60-immunized (right) animals immunostained for FasR (red) and counter-labeled with the nuclear dye DAPI (blue). Label is minimal in control retina. In HSP27 retina, FasR is increased throughout the retina and is prominent in endothelial cells of blood vessels (BV) and in the large cell bodies of RGCs (arrowheads). In HSP60 retina, FasR label is less diffuse throughout the retina and is again prominent in RGC cell bodies (arrowheads). Scale is the same for all three panels.
Finally, we examined the localization of the FasL receptor, FasR (CD95) in our animal model. In control retina, localization of FasR was sporadic with very little background staining (Fig. 7C, left). In contrast, levels of FasR in both HSP27-immunized (Fig. 7C, middle) and HSP60-immunized (Fig. 7C, right) retina appeared far higher throughout the entire retina. As with OX18, FasR label in HSP27 retina was more diffuse than in HSP60 retina, but in both cases, FasR was distinguished clearly in the cell bodies of RGCs. This is consistent with our in vitro results, showing a role for FasL in mediating RGC degeneration with HSP immunization.
Discussion
Our primary finding is that short-term immunization of rats with HSP60 or HSP27 results in a reduction of Brn3a-labeled RGCs in the retina, with the most significant loss in the temporal retina flanking the area centralis of HSP60-immunized retina (Figs. 1, 2). Because the rodent area centralis is analogous to the fovea in the primate retina (both are regions of peak RGC density), the pattern of RGC loss in HSP60-immunized rats exhibits similarities to the lesion in human normal pressure glaucoma, which presents clinically as loss of field in the parafoveal zone (Quigley, 1993; Araie, 1995). The distribution of T-cells in the retina also appeared to follow a topographically specific pattern (Fig. 2), suggesting that our model may bear similarities to RGC pathology in rodent models of high-pressure glaucoma (Jakobs et al., 2005; Howell et al., 2007; Soto et al., 2008). We also observed a sharp reduction in RGCs in the retina nearest the optic nerve head (Figs. 1C,D, 2E) typically associated with high RGC density in rodents (Fukuda, 1977; Schober and Gruschka, 1977; Dreher et al., 1985). In human glaucoma, the arcuate lesion following a paracentral scotoma adheres to the pattern formed by the RGC axons as they exit the retina. Thus, the pattern of degeneration indicated by Brn3a quantification may result from a corresponding loss of axons. Our quantification of RGC axon density in the optic nerve (Fig. 3) suggests that loss of axons accompanies the decrease in Brn3a-labeled RGCs, again more severely for HSP60 immunization.
The in vivo findings were supported by in vitro experiments demonstrating that the HSP-activated T-cells can induce apoptosis of RGC-5 cells. Several studies demonstrate that T-cells activated by non-neural antigens may be involved in neurodegenerative diseases with an autoimmune component (Linington et al., 1988; Andersen et al., 1993). Our findings indicate that non-neural antigens like HSPs can also induce RGC degeneration. However, based on our data, we assume that the pathways mediated by HSP27 or HSP60 immunization may be different, because the concentration of sFasL in HSP27-stimulated T-cells was not elevated. Our previous studies support the involvement of HSP27 as an intrinsic cytoprotectant in retinal cells that may be compromised in glaucoma (Tezel and Wax, 2000; Tezel et al., 2000). We propose that in addition to intrinsic neuroprotective functions, HSP27, like other HSPs, may also serve as an antigenic stimulus activating the innate and/or adaptive immune response during glaucomatous neurodegeneration. By serving as a danger signal, upregulation of HSP27 may facilitate detection and elimination of stressed RGCs by the immune system. Thus, uncontrolled immune activity, including T-cell-mediated cytotoxicity to RGCs and their axons, may eventually facilitate the progression of neurodegeneration. In addition to T-cell-mediated injury supported by present in vitro findings, our previous studies support that HSP27 antibody (present in the glaucoma patient sera at increased titers) may also induce RGC death by inactivating the ability of native HSP27 to stabilize actin cytoskeleton (Tezel and Wax, 2000).
Consistent with the immunostimulatory ability of HSPs, glial MHC expression is induced under stress conditions (Maehlen et al., 1989; Molleston et al., 1993) in association with a stress-induced immune response (Young and Elliott, 1989), and HSPs are implicated in the development of a number of human autoimmune diseases (van Noort, 1996). This suggests that tissue stress as evidenced by increased expression of HSPs in glaucomatous human eyes (Tezel et al., 2000), along with a persistent glial activation (Tezel and Wax, 2003), may be an immunostimulatory signal leading to RGC toxicity in human glaucoma similar to the experimental autoimmune model used in this study. Therefore, this HSP immunization model offers a novel experimental tool for further studies of immunoregulation in glaucomatous neurodegeneration.
The traffic of leukocytes into the CNS is highly regulated process. In some conditions, only a small number of activated T lymphocytes in the periphery are able to penetrate the barriers through the action of their surface enzymes and adhesion molecules as part of their roles in immune surveillance (Xu et al., 2004; Crane et al., 2006). However, several disease models in both the eye (experimental autoimmune uveitis) and brain (experimental autoimmune encephalitis) have successfully demonstrated the transient appearance of T-cells in the effected organ of interest. In the latter, CD4 T-cells typically enter the CNS 10–16 d after immunization (Ludowyk et al., 1992; Shikishima et al., 1993; Verhagen et al., 1994; Begolka et al., 1998). Based on our data, we believe that hsp27 or 60-activated T-cells are destructive once they pass the blood barrier to penetrate the retinal parenchyma. Thus, preventing migration of activated T-cells into the retina could be an important neuroprotective strategy. In addition, detection of TCRαβ+ T-cells in hsp27 or 60-immunized retinas suggests that T-cell subsets, such as CD4+ (Xu et al., 2004; Crane et al., 2006) and CD8+ T-cells (Galea et al., 2007), could be involved.
Elimination of reactive T-cells in the CNS by apoptosis is a particularly important common pathway in preventing the development of neurodegenerative autoimmune diseases, as these cells can recruit and activate other inflammatory cells and initiate an autoimmune process (Pender and Rist, 2001). Although microglia have been associated with traumatic injuries or chronic neurodegenerative diseases of the CNS (Streit, 1996; Stoll and Jander, 1999), they also act as stand-by cells in the service of both the immune system and the CNS (Schwartz, 2003). In addition to their buffering of harmful self-compounds and clearing debris from the damaged CNS site, we speculate that microglia may be involved in the apoptotic elimination of reactive T-cells in the retina. similar to that which occurs in brain (Bauer et al., 1998). This likely provides a native protection mechanism against potential cytotoxicity from a broad array of immune responses that may be injurious to retinal neurons and vision In accordance with our findings of early and transient T-cell appearance after immunization, in many neurodegenerative diseases including glaucomatous optic nerve degeneration, virtually no T-cell invasion is detected at later time points in the retina (Wax et al., 1998b; Schwartz and Kipnis, 2001).
We hypothesize it is also possible that small molecules like sFasL might cross the retinal blood barrier in certain pathological conditions such as inflammation and diabetes. Of course, in pathological conditions, sFasL may also be released from other cells (Ju et al., 2006). Elevated sFasL titers in the sera of HSP-immunized rats suggest that in addition to many other factors, sFasL is likely involved in RGC degeneration similar to that detected, in vitro. However, although sFasL produced by HSP60-activated T-cells induced RGC apoptosis in vitro, induction of RGC apoptosis by HSP27-activated T-cells was not associated with sFasL. This suggests that additional factors (such as membrane-bound FasL, other ligands associated with the TNF death receptor family, or other unknown molecules are involved in RGC apoptosis induced by HSP27-activated T-cells. Fas/FasL interaction has also been identified to be an important mechanism to eliminate T-cells in the brain (D'Souza et al., 1996; Dowling et al., 1996) and the eye (Griffith et al., 1995, 1996).
In summary, the outcome of complex interactions between the immune system and retinal resident immune cells appears to be critical for the development of autoimmune RGC degeneration in the retina as occurred in HSP-immunized animals or in human glaucoma in susceptible patients. T-cell-mediated neurodegeneration likely does not depend only on aberrant activation of autoreactive T-cells, but may also reflect a dysfunction in the termination of T-cell response in this immune-privileged site by apoptosis. Errors during the processing of the native cellular interactions appear to be important for the conversion of protective immunity or self-limiting inflammatory responses into chronic neurodegeneration. Experimental autoimmune glaucoma produced by HSP immunization in this study offers a novel in vivo tool to examine the diverse roles of the immune system in immune-privilege, protective immunity, or induction of an autoimmune neurodegenerative disease. It may also facilitate the identification of treatment strategies to prevent pressure independent RGC degeneration as may occur in selected patients with glaucoma.
Footnotes
This work was supported in part by EY12314 (M.B.W.) from the National Eye Institute (Bethesda, MD), the Glaucoma Research Foundation (San Francisco, CA), and Research to Prevent Blindness (New York, NY). We thank Byron Li, Shutong Cao, Bing Li, and Yoseph Yaacobi for their excellent technical assistance in the preparation of this manuscript.
References
- Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol. 1999;468:123–133. doi: 10.1007/978-1-4615-4685-6_10. [DOI] [PubMed] [Google Scholar]
- Andersen O, Lygner PE, Bergström T, Andersson M, Vahlne A. Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J Neurol. 1993;240:417–422. doi: 10.1007/BF00867354. [DOI] [PubMed] [Google Scholar]
- Araie M. Pattern of visual field defects in normal-tension and high-tension glaucoma. Curr Opin Ophthalmol. 1995;6:36–45. doi: 10.1097/00055735-199504000-00007. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Holländer H, Streit W, Stone J. The appearance and distribution of microglia in the developing retina of the rat. Vis Neurosci. 1989;2:437–448. doi: 10.1017/s0952523800012335. [DOI] [PubMed] [Google Scholar]
- Bauer J, Bradl M, Hickley WF, Forss-Petter S, Breitschopf H, Linington C, Wekerle H, Lassmann H. T cell apoptosis in inflammatory brain lesions: destruction of T cells does not depend on antigen recognition. Am J Pathol. 1998;153:715–724. doi: 10.1016/s0002-9440(10)65615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begolka WS, Vanderlugt CL, Rahbe SM, Miller SD. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998;161:4437–4446. [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane IJ, Xu H, Wallace C, Manivannan A, Mack M, Liversidge J, Marquez G, Sharp PF, Forrester JV. Involvement of CCR5 in the passage of Th1-type cells across the blood-retina barrier in experimental autoimmune uveitis. J Leukoc Biol. 2006;79:435–443. doi: 10.1189/jlb.0305130. [DOI] [PubMed] [Google Scholar]
- Dowling P, Shang G, Raval S, Menonna J, Cook S, Husar W. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J Exp Med. 1996;184:1513–1518. doi: 10.1084/jem.184.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher B, Sefton AJ, Ni SY, Nisbett G. The morphology, number, distribution and central projections of Class I retinal ganglion cells in albino and hooded rats. Brain Behav Evol. 1985;26:10–48. doi: 10.1159/000118764. [DOI] [PubMed] [Google Scholar]
- D'Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y. A three-group classification of rat retinal ganglion cells: histological and physiological studies. Brain Res. 1977;119:327–334. doi: 10.1016/0006-8993(77)90314-6. [DOI] [PubMed] [Google Scholar]
- Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. 2007;204:2023–2030. doi: 10.1084/jem.20070064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregerson DS. Immune privilege in the retina. Ocul Immunol Inflamm. 1998;6:257–267. doi: 10.1076/ocii.6.4.257.4029. [DOI] [PubMed] [Google Scholar]
- Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- Griffith TS, Yu X, Herndon JM, Green DR, Ferguson TA. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity. 1996;5:7–16. doi: 10.1016/s1074-7613(00)80305-2. [DOI] [PubMed] [Google Scholar]
- Harvey DM, Calkins DJ. Localization of kainate receptors to the presynaptic active zone of the rod photoreceptor in primate retina. Vis Neurosci. 2002;19:681–692. doi: 10.1017/s0952523802195137. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Sappington RM, Horner PJ, Calkins DJ. Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:986–996. doi: 10.1167/iovs.05-0925. [DOI] [PubMed] [Google Scholar]
- Jakobs TC, Libby RT, Ben Y, John SW, Masland RH. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol. 2005;171:313–325. doi: 10.1083/jcb.200506099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju KR, Kim HS, Kim JH, Lee NY, Park CK. Retinal glial cell responses and Fas/FasL activation in rats with chronic ocular hypertension. Brain Res. 2006;1122:209–221. doi: 10.1016/j.brainres.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremmer S, Kreuzfelder E, Klein R, Bontke N, Henneberg-Quester KB, Steuhl KP, Grosse-Wilde H. Antiphosphatidylserine antibodies are elevated in normal tension glaucoma. Clin Exp Immunol. 2001;125:211–215. doi: 10.1046/j.1365-2249.2001.01578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy RR, Agarwal P, Prasanna G, Vopat K, Lambert W, Sheedlo HJ, Pang IH, Shade D, Wordinger RJ, Yorio T, Clark AF, Agarwal N. Characterization of a transformed rat retinal ganglion cell line. Brain Res Mol Brain Res. 2001;86:1–12. doi: 10.1016/s0169-328x(00)00224-2. [DOI] [PubMed] [Google Scholar]
- Linden R, Perry VH. Massive retinotectal projection in rats. Brain Res. 1983;272:145–149. doi: 10.1016/0006-8993(83)90371-2. [DOI] [PubMed] [Google Scholar]
- Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- Ludowyk PA, Willenborg DO, Parish CR. Selective localisation of neuro-specific T lymphocytes in the central nervous system. J Neuroimmunol. 1992;37:237–250. doi: 10.1016/0165-5728(92)90008-9. [DOI] [PubMed] [Google Scholar]
- Maehlen J, Olsson T, Zachau A, Klareskog L, Kristensson K. Local enhancement of major histocompatibility complex (MHC) class I and II expression and cell infiltration in experimental allergic encephalomyelitis around axotomized motor neurons. J Neuroimmunol. 1989;23:125–132. doi: 10.1016/0165-5728(89)90031-3. [DOI] [PubMed] [Google Scholar]
- Maruyama I, Ohguro H, Ikeda Y. Retinal ganglion cells recognized by serum autoantibody against gamma-enolase found in glaucoma patients. Invest Ophthalmol Vis Sci. 2000;41:1657–1665. [PubMed] [Google Scholar]
- Molleston MC, Thomas ML, Hickey WF. Novel major histocompatibility complex expression by microglia and site-specific experimental allergic encephalomyelitis lesions in the rat central nervous system after optic nerve transection. Adv Neurol. 1993;59:337–348. [PubMed] [Google Scholar]
- Neufeld AH. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch Ophthalmol. 1999;117:1050–1056. doi: 10.1001/archopht.117.8.1050. [DOI] [PubMed] [Google Scholar]
- Pender MP, Rist MJ. Apoptosis of inflammatory cells in immune control of the nervous system: role of glia. Glia. 2001;36:137–144. doi: 10.1002/glia.1103. [DOI] [PubMed] [Google Scholar]
- Pouvreau I, Zech JC, Thillaye-Goldenberg B, Naud MC, Van Rooijen N, de Kozak Y. Effect of macrophage depletion by liposomes containing dichloromethylene-diphosphonate on endotoxin-induced uveitis. J Neuroimmunol. 1998;86:171–181. doi: 10.1016/s0165-5728(98)00042-3. [DOI] [PubMed] [Google Scholar]
- Quigley HA. Open-angle glaucoma [see comments] N Engl J Med. 1993;328:1097–1106. doi: 10.1056/NEJM199304153281507. [DOI] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quina LA, Pak W, Lanier J, Banwait P, Gratwick K, Liu Y, Velasquez T, O'Leary DD, Goulding M, Turner EE. Brn3a-expressing retinal ganglion cells project specifically to thalamocortical and collicular visual pathways. J Neurosci. 2005;25:11595–11604. doi: 10.1523/JNEUROSCI.2837-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano C, Barrett DA, Li Z, Pestronk A, Wax MB. Anti-rhodopsin antibodies in sera from patients with normal-pressure glaucoma. Invest Ophthalmol Vis Sci. 1995;36:1968–1975. [PubMed] [Google Scholar]
- Sappington RM, Pearce DA, Calkins DJ. Optic nerve degeneration in a murine model of juvenile ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2003;44:3725–3731. doi: 10.1167/iovs.03-0039. [DOI] [PubMed] [Google Scholar]
- Schober W, Gruschka H. Retinal ganglion cells of the albino rat: a qualitative and quantitative study. Z Mikrosk Anat Forsch. 1977;91:397–414. [PubMed] [Google Scholar]
- Schori H, Kipnis J, Yoles E, WoldeMussie E, Ruiz G, Wheeler LA, Schwartz M. Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: implications for glaucoma. Proc Natl Acad Sci U S A. 2001;98:3398–3403. doi: 10.1073/pnas.041609498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M. Macrophages and microglia in central nervous system injury: are they helpful or harmful? J Cereb Blood Flow Metab. 2003;23:385–394. doi: 10.1097/01.WCB.0000061881.75234.5E. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 2001;7:252–258. doi: 10.1016/s1471-4914(01)01993-1. [DOI] [PubMed] [Google Scholar]
- Shikishima K, Lee WR, Behan WM, Foulds WS. Uveitis and retinal vasculitis in acute experimental allergic encephalomyelitis in the Lewis rat: an ultrastructural study. Exp Eye Res. 1993;56:167–175. doi: 10.1006/exer.1993.1024. [DOI] [PubMed] [Google Scholar]
- Soto I, Oglesby E, Buckingham BP, Son JL, Roberson ED, Steele MR, Inman DM, Vetter ML, Horner PJ, Marsh-Armstrong N. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J Neurosci. 2008;28:548–561. doi: 10.1523/JNEUROSCI.3714-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Streilein JW, Okamoto S, Sano Y, Taylor AW. Neural control of ocular immune privilege. Ann N Y Acad Sci. 2000;917:297–306. doi: 10.1111/j.1749-6632.2000.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Streit WJ. The role of microglia in brain injury. Neurotoxicology. 1996;17:671–678. [PubMed] [Google Scholar]
- Tezel G, Wax MB. The mechanisms of HSP27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci. 2000;20:3552–3562. doi: 10.1523/JNEUROSCI.20-10-03552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Wax MB. Glial modulation of retinal ganglion cell death in glaucoma. J Glaucoma. 2003;12:63–68. doi: 10.1097/00061198-200302000-00014. [DOI] [PubMed] [Google Scholar]
- Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 1998;39:2277–2287. [PubMed] [Google Scholar]
- Tezel G, Edward DP, Wax MB. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch Ophthalmol. 1999;117:917–924. doi: 10.1001/archopht.117.7.917. [DOI] [PubMed] [Google Scholar]
- Tezel G, Hernandez R, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 2000;118:511–518. doi: 10.1001/archopht.118.4.511. [DOI] [PubMed] [Google Scholar]
- van Noort JM. Multiple sclerosis: an altered immune response or an altered stress response? J Mol Med. 1996;74:285–296. doi: 10.1007/BF00207506. [DOI] [PubMed] [Google Scholar]
- Verhagen C, Mor F, Cohen IR. T cell immunity to myelin basic protein induces anterior uveitis in Lewis rats. J Neuroimmunol. 1994;53:65–71. doi: 10.1016/0165-5728(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Wang X, Tay SS, Ng YK. An immunohistochemical study of neuronal and glial cell reactions in retinae of rats with experimental glaucoma. Exp Brain Res. 2000;132:476–484. doi: 10.1007/s002210000360. [DOI] [PubMed] [Google Scholar]
- Wax MB, Barrett DA, Pestronk A. Increased incidence of paraproteinemia and autoantibodies in patients with normal-pressure glaucoma. Am J Ophthalmol. 1994;117:561–568. doi: 10.1016/s0002-9394(14)70059-5. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Saito I, Gupta RS, Harley JB, Li Z, Romano C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am J Ophthalmol. 1998a;125:145–157. doi: 10.1016/s0002-9394(99)80084-1. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Edward PD. Clinical and ocular histopathological findings in a patient with normal-pressure glaucoma. Arch Ophthalmol. 1998b;116:993–1001. doi: 10.1001/archopht.116.8.993. [DOI] [PubMed] [Google Scholar]
- Wax MB, Yang J, Tezel G. Serum autoantibodies in patients with glaucoma. J Glaucoma. 2001;10:S22–24. doi: 10.1097/00061198-200110001-00009. [DOI] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15:4762–4785. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Manivannan A, Jiang HR, Liversidge J, Sharp PF, Forrester JV, Crane IJ. Recruitment of IFN-gamma-producing (Th1-like) cells into the inflamed retina in vivo is preferentially regulated by P-selectin glycoprotein ligand 1:P/E-selectin interactions. J Immunol. 2004;172:3215–3224. doi: 10.4049/jimmunol.172.5.3215. [DOI] [PubMed] [Google Scholar]
- Yang J, Patil RV, Yu H, Gordon M, Wax MB. T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol. 2001a;131:421–426. doi: 10.1016/s0002-9394(00)00862-x. [DOI] [PubMed] [Google Scholar]
- Yang J, Tezel G, Patil RV, Romano C, Wax MB. Serum autoantibody against glutathione S-transferase in patients with glaucoma. Invest Ophthalmol Vis Sci. 2001b;42:1273–1276. [PubMed] [Google Scholar]
- Yang P, de Vos AF, Kijlstra A. Macrophages and MHC class II positive cells in the choroid during endotoxin induced uveitis. Br J Ophthalmol. 1997;81:396–401. doi: 10.1136/bjo.81.5.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, de Vos AF, Kijlstra A. Interferon gamma immunoreactivity in iris nerve fibers during endotoxin induced uveitis in the rat. Br J Ophthalmol. 1998;82:695–699. doi: 10.1136/bjo.82.6.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA, Elliott TJ. Stress proteins, infection, and immune surveillance. Cell. 1989;59:5–8. doi: 10.1016/0092-8674(89)90861-1. [DOI] [PubMed] [Google Scholar]