

1. Introduction
This review is intended to present an overview of synthetic efforts published toward the cyathane diterpenoid natural products. The work summarized here represents an update to the previous review on this topic which was reported in 2000, and as such will only cover cyathane core syntheses described in the literature past this date.1 For the purpose of comprehensive review and comparison, however, all known completed total syntheses will be detailed herein. The focus of this review is centered upon the ring-forming and stereogenic strageties employed within the published strategies toward cyathane natural products.
2. Overview of the Cyathane Diterpenoids
The cyathane diterpene natural products are isolated from a diverse variety of fungi, sponges, and fruiting plants. However, despite their myriad natural sources, are all unified by the presence of a characteristic 5-6-7 tricarbocyclic fused core structure (1, Figure 1). Within this class of natural products can be found the cyathins, the allocyathins, the erinacines, the sarcodonins, the scabronines, the striatals, the cyanthiwigins, and the cyafrins, all of which display the conserved carbon scaffold of the cyathane skeleton (1). Of the twenty carbons that comprise this cyathane framework, the C(6) and C(9) carbons present all-carbon quaternary stereocenters, which bear angular methyl groups at the points of ring fusion. Almost all of the compounds within the cyathane class display these methyl groups with an anti relative stereochemical relationship, though the cyanthiwigin natural products possess a syn arrangement. The cyathane core structure is additionally characterized by the presence of an isopropyl side chain at C(3) and an exocyclic carbon atom connected to C(12)
Figure 1.

Representative cyathane diterpenoid natural products.
2.1 Isolation
In 1970, Ayer and Brodie published a report in which an extract from the bird's nest fungus Cyathus helenae was scrutinized to better understand its antimicrobial activity. Though no full structural assignment was made for any compound within the extract, the active components of the mixture were separated via chromatography. After isolation and elemental analysis, the compounds responsible for the observed antimicrobial activity were named (without structural elucidation) as cyathin A3, A4, B3, B4, C5, and allocyathin A4.2 The first fully characterized cyathane diterpene natural products were subsequently reported by Ayer and coworkers in 1972, when the substance previously identified as cyathin A3 was found to be a mixture of isomeric compounds, which they named cyathin A3 (2) and allocyathin B3 (3).3 Numerous other cyathin and allocyathin natural products were discovered subsequent to this report, including allocyathin B2 (4),4 a cyathane diterpenoid which has since become the focus of numerous synthetic studies.
Following the identification of the primary cyathin diterpenoids, several structurally related compounds were isolated from additional natural sources. It was discovered that the fruiting bodies of Hericium erinaceum contained a number of glycosylated allocyathin B2 analogues, which were eventually named the erinacines. Among the compounds obtained from Hericium erinaceum were erinacine A (5), erinacine B (6), and the more structurally complex erinacine E (7).5
In 1989 Nakayama and coworkers published the isolation and structural assignment of eight new cyathane molecules from the fungus Sarcodon scabrosus which they named the sarcodonins A–H.6 These compounds possess the conserved 5-6-7 tricyclic core found in all other cyathane natural products, but are distinct in that they display additional oxidation at the C(19) position, such as is observed with sarcodonin G (8). An additional class of cyathane diterpene compounds was later identified from the same Sarcodon scabrosus fungus by Oshima in 1998, when isolation and characterization of scabronine A–F were disclosed. Six years later, Liu et al. disclosed the isolation of two additional scabronine molecules from the same source, including scabronine G (9).7 Structural elucidation of these natural products revealed the presence of a carboxyl group at C(17), a feature which marks the scabronines as distinct from the remainder of the cyathanes.
In 1992, Kashman and coworkers published a report detailing the isolation and characterization of the first cyanthiwigin molecules. Initially isolated from the marine sponge Epipolasis reiswigi, cyanthiwigins A–D were fully characterized and assigned absolute configuration via NMR, X-ray, and Mosher ester analysis.8 A decade later the laboratories of Hamann isolated the same four cyanthiwigins, plus an additional 23 compounds of this class, from the Jamaican sea sponge Myrmekioderma styx. This isolation included the products cyanthiwigin F (10) and cyanthiwigin U (11). In years subsequent to their initial report, Hamann and coworkers have detailed the isolation and characterization of the additional cyanthiwigin molecules AB–AG, including the structurally unique spirocyclic cyanthwigin AC (12).9
2.2 Bioactivity
The biological activities of the cyathane natural products vary widely among the different molecules within the class. Many of these diterpene compounds possess antibiotic or antimicrobial activity, such as the previously discussed cyathin A3 (2) and allocyathin B3 (3). Indeed, almost all of the subcategories of cyathane molecules display mild activity in this regard.2,4,9 In addition to serving an antibiotic function, some members of the cyanthiwigin compounds have displayed limited cytotoxic activity against human primary tumor cells, as well as P388 murine leukemia cells and A549 lung tumor cells.9,10
However, the most significant biological activity reported among these diterpenoid molecules is their powerful ability to encourage the synthesis of Nerve Growth Factor (NGF). Both the erinacine and the scabronine natural products have displayed considerable potency in the stimulation of NGF synthesis,5,7b a capacity that implicates their potential as therapeutic agents to treat neurodegenerative ailments such as Alzheimer's or Parkinson's disease.11
2.3 Biosynthesis
The details of cyathane diterpenoid biosynthesis have been covered in Wright's review, and as such will only be briefly summarized here.1 Subsequent to his isolation of the parent cyathins, Ayer conducted an in-depth study to scrutinize the biosynthetic origin of the cyathane diterpenoid core. By growing Cyathus earlei in the presence of 13C-labelled sodium acetate, Ayer was able to isolate and examine the cyathin molecules produced by these fungal bodies. Analysis of these compounds via 13C NMR allowed Ayer to conclude that the biosynthetic pathway toward the cyathane core tricycle involves cascade cyclization and subsequent rearrangement of geranylgeranyl phosphate.12
3. Strategy Summary
Since the isolation of the first cyathane natural products in the early 1970s, numerous research groups have endeavoured to synthesize these compounds. A multitude of differing strategies have been documented in the literature, and many of these approaches are presented here in schematic form. The synthetic efforts reported to date can be roughly categorized by their key transformations, which are further divided as either transition metal-mediated reactions (Scheme 2), or akylation / aldol reactions (Scheme 3).
Scheme 2.
Transition metal and Lewis acid-catalyzed retrosynthetic disconnections toward the cyathane diterpenoid tricyclic core.
Scheme 3.

(a) Alkylation and aldol retrosynthetic disconnections of the cyathane diterpenoid tricyclic core. (b) Reddy's retrosynthetic disconnection of the cyanthiwigin AC core.
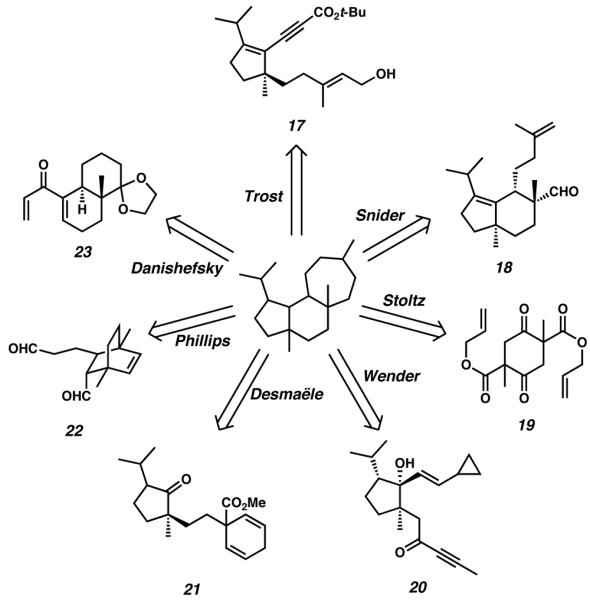
A majority of the cyathane syntheses published to date leverage various metal-catalyzed transformations to accomplish difficult or complicated ring-forming reactions. For example, the strategy employed by Trost et al. relies upon Ru-catalyzed cycloisomerization to close the central B-ring of the cyathane core, an approach which leads back to allylic alcohol 17. Similarly, Desmaële's route relies upon a Pd-catalyzed intramolecular Heck cyclization to construct the core B-ring, invoking diene 21 as a critical retrosynthetic precursor. Danishefsky's approach to the tricycle implements an Fe-catalyzed Nazarov cyclization for construction of the five-membered A-ring, allowing for strategic disconnection back to dienone 23. Beyond the A-and B-rings, metal-catalyzed methodology has also been used to target the seven-membered C-ring of the cyathane core. For example, Snider's method toward these diterpene molecules employs an Al-catalyzed carbonyl-ene reaction for C-ring construction from bicyclic aldehyde 18. The power of transition-metal catalysis has also enabled routes that employ a strategy of simultaneous multicyclic construction. For example, Phillips employs an efficient Ru-catalyzed ringopening ring-closing metathesis strategy to build both the A-and C-rings of the cyathane core in tandem, thus invoking bridged bicycle 22 as a retrosynthetic precursor. Wender's [5 + 2] cycloaddition approach toward the cyathane skeleton allows for cascade construction of both the B- and C-rings simultaneously, thus retrosynthetically disconnecting the core backward to ynone 20. The strategy employed by Stoltz relies upon Ru-catalyzed C-ring construction, but more imperatively, implements Pd-catalyzed alkylation for quaternary stereocenter formation. This invokes bis-b-ketoester 19 as a critical retrosynthetic precursor.
Another unifying approach by which the cyathane tricycle has been targeted is that of an aklyation strategy, often specifically in the form of an aldol reaction. In order to construct the C-ring of the tricyclic diterpene core, Ward and his group employ an ozonolysis and aldol sequence. This ring-expanding strategy invokes tricycle 28 as an important synthetic precursor (Scheme 3a). Tori implements a similar technique to C-ring formation in his synthesis, in which bisaldehyde 26 is invoked via a disconnection of the cyathane core by means of an intramolecular aldol reaction. The cyathane C-ring has also been targeted via a unique [3 + 4] annulation reaction developed by Takeda, which employes 25 as a bicyclic precursor to the larger tricyclic skeleton. A convergent strategy targeting the B-ring of the cyathane core is carried out in Nakada's approach toward these natural products. By disconnecting the central ring of the tricycle, Nakada envisions tethered system 27 as the critical substrate for an intramolecular aldol reaction. Non-aldol alkylation procedures have also played a role in cyathane synthetic design. Reddy disconnects the smaller six-membered C-ring of cyanthiwigin AC via an enolate spiro-alkylation strategy to invoke bicycle 29 as a precursor (Scheme 3b). An anionic alkylation approach has also been employed with a focus upon construction of the five-membered cyathane A-ring. Piers' retrosynthetic analysis opens the A-ring to vinyl iodide 24, a structure, which after lithium-halogen exchange and ketone trapping, is closed to give a tricyclic system.
4. Cyathane Core Syntheses
The section detailed below is intended as an update to the review published in 2000. As such, only cyathane core syntheses published after this date will be summarized here.
Several groups have devised efficient strategies toward the cyathane tricyclic core, with possible extensions toward multiple completed cyathane natural products. These efforts have focused upon preparation of the tricyclic framework found in the typical cyathane molecules.
4.1 Wender's Cyathane Core Synthesis
A general route toward the construction of the 5-6-7 tricyclic diterpene core was reported by the laboratories of Wender in 2001 by implementation of a [5 + 2] Rh-catalyzed cycloaddition reaction.13 Beginning with (−)-limonene (30), hydrogenation, oxidative olefin cleavage, and intramolecular aldol condensation afforded enal 31 (Scheme 4). After reduction and vinyl ether formation, a thermal Claisen rearrangement yielded aldehyde 32 as a 10:1 mixture of inseparable diastereomers. Cyclopentane 32 was thereafter advanced along four steps, including ozonolysis of the exocyclic methylene and addition of lithium cyclopropylacetylide, to furnish cyclopentanol 33 as a mixture of diastereomers. Stereoselective reduction of cyclopropyl alkyne 33 allowed access to diol 34, which was oxidized, then exposed to 1-propynylmagnesium bromide to generate propargyl alcohol 35. Attempts to execute a [5 + 2] cycloaddition reaction using alcohol 35 as the starting material were unfortunately unsuccessful, and yielded only a complicated mixture of products. For this purpose, enyne 35 was oxidized to conjugated ketone 20 before executing the [5 + 2] cycloaddition reaction.
Scheme 4.

Preparation of the critical [5 + 2] cycloaddition precursor.
Upon exposure of vinyl cyclopropane 20 to 5 mol% of [Rh(CO)2Cl]2, the desired cycloaddition reaction proceeded in high yield to provide tricycle 38 as a single diastereomer (Scheme 5).14 The critical [5 + 2] cycloaddition reaction initiates with complexation of the rhodium catalyst to both the alkyne moiety and the vinyl cyclopropane group. This is then followed by oxidative cyclometallation to form an intermediate metallocyclopentane (36), which in turn undergoes strain-driven cyclopropane ring-opening and ringexpansion to generate a transient metallocyclooctadiene species (37). Reductive elimination of rhodium from 37 thereafter yields tricyclic structure 38, representing a completed cyathane core. The structure of tricyclic enone 38 was verified by single-crystal X-ray crystallography. Overall, this strategy allows access to a completed 5-6-7 tricyclic structure in 14 steps and 13% overall yield.
Scheme 5.

Wender's [5 + 2] cycloaddition reaction to construct the cyathane core tricycle.
4.2 Desmaële's Cyathane Core Synthesis
A generalized synthetic route toward the tricyclic cyathane core structure was developed by Desmaële and coworkers in 2002, and an updated version of this strategy was later published in 2005.15 When addressing the challenges present in constructing the tricyclic cyathane framework, Desmaële states that establishing the anti relationship between the methyl groups of C(6) and C(9) represents the most significant obstacle. In order to solve this stereochemical issue, the strategy envisioned employs late-stage construction of the central B-ring of the tricycle via intramolecular Heck reaction between tethered A- and C-ring fragments.
The synthesis was initated from known, enantioenriched keto-ester 39 (Scheme 6).16 Mukaiyama aldol reaction with acetaldehyde, followed by dehydration and isomerization, yielded enone 40. After cuprate addition to and saponification of ester 40, the intermediate keto-acid obtained was then subjected to the Kochi modification of the Hunsdiecker reaction to afford primary iodide 41. Displacement of iodide 41 with the lithium enolate of methyl ester 42 then provided tethered intermediate 21, which was subjected to a four step sequence to access the critical Heck cyclization precursor 43 (Scheme 7). Notably, prior attempts at an intramolecular Heck cyclization involving a sevenmembered ring dieneone proved quite difficult to advance along this synthetic path, and for this reason Desmaële opted to employ a six-membered ring to serve as a surrogate for the cyathane C-ring.
Scheme 6.

Synthesis of Desmaële's alkyl iodide coupling partner.
Scheme 7.

Preparation of the crucial Heck cyclization precursor.
Several attempts to cyclize precursor 43 via intramolecular Heck reaction under standard conditions were met with difficulty, with most attempts yielding either undesired acetate addition products or incorrect relative stereochemistry.15a Eventually, further optimization of this reaction lead Desmaële to discover that exposure of triflate 43 to 20 mol% of Pd(OAc)2 in the presence of PPh3 and n-Bu4NBr could execute the desired Heck reaction to yield tricycle 44 in 73% yield and a 19:1 diastereomeric ratio based on the newly formed stereocenter at C(6) (Scheme 8). This transformation constructed the central B-ring of the cyathane core while simultaneously establishing the necessary all-carbon quaternary stereocenter at C(6) via desymmetrization of the pendent C-ring precursor.15b,c
Scheme 8.

Heck cyclization and aluminum-promoted ring expansion reactions to target the cyathane tricycle.
Elaboration of tricyclic dienone 44 toward the cyathane core structure proceeded via hydrogenation of the disubstituted olefin using Wilkinson's catalyst to give enone 45. This reduction was then followed by treatment of tricycle 45 with trimethyl aluminum and trimethylsilyl diazomethane to effect an organoaluminium-promoted ring expansion.17 This reaction afforded the completed 5-6-7 tricyclic framework as a 6:1 mixture of ketone (46) and enone (47) isomers. By obtaining these tricyclic structures, Desmaële accomplished the construction of the cyathane core tricycle over 15 steps and an overall combined yield of 1.4% for both isomers obtained.
4.3 Takeda's Cyathane Core Synthesis
A synthesis of the cyathane core leveraging a [4 + 3] annulation strategy was disclosed by Takeda and coworkers in 2000.18 Starting from known racemic enone 48, addition of ethynyl Grignard was followed by a Rupe rearrangement to give extended conjugated enone system 25 (Scheme 9). Formation of the lithium enolate of 25 was followed by addition to acyl silane 49. Initial nucleophilic addition of the enolate of 25 to 49 produces intermediate alkoxide 50, which undergoes Brook rearrangement and subsequent nucleophilic addition to form cyclopropane 51. Divinyl cyclopropane species 51 subsequently undergoes spontaneous [3,3] sigmatropic rearragement, a mechanism which is further accelerated by the presence of an alkoxide. After rearrangement occurs, the completed cyathane core structure 52 is afforded as a single diastereomer.19
Scheme 9.

Takeda's key [4 + 3] annulation to target the C-ring of the cyathane core
Continued functionalization of tricycle 52 was accomplished by diastereoselective DIBAL reduction, a process which provided the isomerically pure silyl enol ether 53 (Scheme 10). Notably, the stereochemistry imparted by this reduction afforded the alcohol epimer analogous to allocyathin B2 at C(14).19 Takeda and coworkers thereafter concluded their efforts with oxidation of the enol silane present in 53 and subsequent cleavage of the C-bound trimethylsilyl group. This provided 54 as the final product of the synthetic sequence.
Scheme 10.

Advancement of Takeda's cyathane core structure.
Beginning from known enone 48, the des-methyl cyathane core was established in three steps and 19% yield, while the more elaborated cyathane analog 54 was produced in 11% yield over five steps.
5. Cyathane Total Syntheses
The following section is intended as a comprehensive review of all disclosed total syntheses of cyathane diterpenoid natural products published to date.
5.1 Snider's (±)-Allocyathin B2 and (−)-Erinacine A
The first total synthesis of any cyathane diterpenoid natural product was accomplished by Snider and coworkers in 1996 with their preparation of allocyathin B2.20 Snider's synthetic plan invoked the use of a carbonyl-ene reaction to target the cyathane core, and this strategy was later extended beyond allocyathin B2 in order to achieve the synthesis of (+)-erinacine E via glycoslyation.
Beginning with known racemic enone 48,21 triflate formation, palladium-catalyzed carbonylation, and oxidation state manipulation allowed access to enal 55 (Scheme 11). Conjugate addition of a cuprate speices generated from Grignard reagent 56 to the β-position of extended unsaturated system 55 provided aldehyde 57, which was subsequently methylated at the α-position to afford bicycle 18. At this point in the synthesis, Snider's route called for construction of the C-ring via intramolecular carbonyl-ene reaction of aldehyde 18. In the event, treatment of 18 with Me2AlCl initiated rearrangement to give a single isomer of alcohol 58 in excellent yield, thus completing the final ring of the tricyclic natural product.22
Scheme 11.

Snider's carbonyl-ene strategy toward the cyathane tricycle.
The synthesis was completed over ten additional transformations, which involved protecting group manipulation, oxidation state modification, and palladiumcatalyzed carbonylation starting from tricycle 58 (Scheme 12). The completed natural product (±)-allocyathin B2 (4) was thus attained from precursor 48 in 17 steps and 6.4% overall yield. Because allocyathin B2 represents an aglycone substrate for the erinacine natural products, Snider and coworkers were well equipped at this point to address the total synthesis of the erinacine compounds. As such, glycosylation of allocyathin B2 with 2,3,4-tri-O-acetyl-α-D-xylopyranosyl (59) bromide and successive global deprotection generated the natural product (−)-erinacine A (5) and isomeric structure 60 as a 1:1 mixture of diastereomers.
Scheme 12.

Completion of allocyathin B2 and glycosylation to achieve erinacine A.
Erinacine A was prepared in 19 steps and 1.0% yield. The cyathane core framework was constructed in seven steps and 38% overall yield.
5.2 Tori's (±)-Allocyathin B2
In a report published by Tori and coworkers in 1998, an intramolecular aldol strategy targeting the synthesis of the natural product allocyathin B2 was described.23
Starting from 3-methyl cyclohexenone (61), conjugate addition, ozonloysis, and oxidation yielded diketone 62 after five steps (Scheme 13). Intramolecular aldol condensation of cyclohexanone 62 afforded bicyclic enone 63, a structure containing the completed five-membered A-ring of allocyathin B2, including the requisite isopropyl group. Subsequent acylation of 63 with acid chloride 64 was followed by methylation and a highly optimized diastereoselective reduction employing Zn(BH4)2 to afford keto-alcohol 65.24 Additional transformations over seven steps provided access to allylic alcohol 66, which was readily oxidized under Swern conditions to give bis-aldehyde 26. Upon exposure of 26 to methanolic KOH, a final intramolecular aldol condensation completed the cyathane C-ring and produced allyocyathin B2 (4). Overall, both allocyathin B2 and the cyathane core structure were synthesized in 19 steps and 0.5% yield, starting from 61.
Scheme 13.

Tori's strategy toward allocyathin B2.
Subsequent to the preparation of allocyathin B2, Tori also disclosed a different strategy toward the construction of the cyathane tricyclic core via ring-closing metathesis.25 Modification of their route toward allocyathin B2 allowed access to bicyclic intermediate 67 (Scheme 14). Upon treatment of this material with 20 mol% of Grubbs secondgeneration metathesis catalyst (69), completed cyathane tricycle 68 was obtained as the sole product of reaction.
Scheme 14.

Tori's ring-closing strategy toward construction of the seven-membered C-ring and completion of the cyathane core.
5.3 Piers' (±)-Sarcodonin G
The first total synthesis of sarcodonin G was described by Piers in 2000.26 Their approach to this cyathane diterpenoid targeted the tricyclic core with an alkylation and ring-expansion strategy, and employed late-stage installation of the periperhal functionality. Piers' synthesis began from known ketone 70 (introduced as a mixture of diastereomers),27 which was subject to hydrazone formation, epimerization at the ring fusion, and nucleophilic attack on alkyl iodide 71 to afford vinyl germane 72 (Scheme 15). Further transformation of germane 72 eventually produced the vinyl iodide species 24.
Scheme 15.

Vinyl iodide construction in Pier's sarcodonin synthesis.
Upon treatment of 24 with n-BuLi, lithium-halogen exchange and intramolecular trapping of the ketone moiety construct the five-membered A-ring of the natural product (Scheme 16). After deprotonation with KH and addition of Bu3SnCH2I, ether 73 was isolated as the major product. From this ether intermediate, a Still-Mitra [2,3]-sigmatropic rearrangement provides tricycle 74, which contains the primary hydroxyl group at C(19) required for sarcodonin G.28
Scheme 16.

Still-Mitra [2,3] sigmatropic regarrangement to construct the A-ring of sarcodonin G.
Additional synthetic transformation of 74 over four steps yielded β-ketoester 75, which bears an α-alkyl iodide group well suited for ring-expansion methodology developed by Hasegawa (Scheme 17).29 Upon exposure to SmI2 in THF, alkyl iodide 75 is converted in 71% yield to the one-carbon ring expanded product 76. This process smoothly forms the seven-membered C-ring, and thus completes the tricyclic cyathane core. The total synthesis is thereafter concluded in six steps to yield sarcadonin G (8).
Scheme 17.

Ring expansion and endgame for sarcodonin G.
Overall the synthesis of sarcodonin G (8) was accomplished in a total of 21 steps and 4.0% yield. The cyathane tricyclic core was attained in 15 steps and 7.0% overall yield.
5.4 Ward's (±)-Allocyathin B3
The total synthesis of allocyathin B3 was achieved by Ward and coworkers in 2000 by leveraging an interesting cycloaddition strategy for rapid construction of the central B-ring.30 The synthesis was initiated with a Diels–Alder cycloaddition between 2,5-dimethyl-p-benzoquinone (78) and 2,4-bis(trimethylsilyloxy)-1,3-pentadiene (77, Scheme 18). Subsequent [2 +2] cycloaddition with allene and exposure to acidic conditions thereafter afforded a 4-6-6 tricyclic system as a 4:1 mixture of structural isomers (28 to 79), wherein each was produced as a single diastereomer.30a
Scheme 18.

Ward's cycloaddition stragety toward tricyclic formation.
Though only isomer 28 was desired, the 4:1 mixture of 29 and 79 was epoxidized, reduced at the enone moiety, and then treated with benzenethiol at reflux under basic conditions to effect closure of the cyathane core A-ring. This furnished α-thiophenyl enone 80 in excellent yield, in a sequence that required only a single purification.30b
Desulfurization, deoxygenation, protection, and epimerization over nine steps then allow access to 5-6-6 allylic benzoyl ester 81 (Scheme 19). The six-membered ring olefin of this intermediate provided the reactivity required to construct the seven-membered C-ring of the natural product. Ozonolysis of enone 81 in the presence of Sudan III indicator generated a sensitive keto-aldehyde intermediate which was subjected to aldol reaction, transacylation, and successive trapping via O-methylation to afford 5-6-7 tricycle 82. This three step sequence completed the core framework and simultaneously established the trans-annular ketal bridge present in allocyathin B3.
Scheme 19.

Ring expansion via ozonolysis and aldol reaction to target the cyathane core.
In order to install the necessary isopropyl side-chain appended to the A-ring of the natural product, intermediate 82 was then transformed to propargyl ether 83 (Scheme 20). Treatment of alkyl bromide 83 with AIBN in the presence of Ph3SnH and subsequent hydrogenation produced the cyclized tetrahydrofuran 84. After exposure to mild acid, tetracycle 84 opens to tricycle 85, a structure which contains all of the carbon atoms required for completion of the A-ring. From this point, the total synthesis was completed in eleven steps to yield allocyathin B3 (3).
Scheme 20.

Ward's allocyathin endgame strategy.
Overall, Ward's strategy toward allocyathin B3 comprises 34 steps and is concluded in 0.1% yield. The tricyclic cyathane core was attained in 18 steps and 11% overall yield.
5.5 Ward's (+)-Cyathin A3
A variant of Ward's allocyathin A3 strategy was later rendered enantioselective to achieve the total synthesis of the related diterpene natural product cyathin A .30,31
The initial Diels–Alder reaction between 2,5-dimethyl-p-benzoquinone (78) and 2,4-bis(trimethylsilyloxy)-1,3-pentadiene (77) was made asymmetric by employing Mikami's titanium-based BINOL catalyst (Scheme 21).33 After extensive optimization of this cycloaddition, Ward and coworkers found that addition of Mg powder and silica gel afforded cycloadduct 86 in 90% yield and 93% ee. With 86 in hand, [2 + 2] cycloaddition and acidic hydrolysis proceeded as in the case of Ward's allocyathin B3 synthesis to furnish 28 and 79 as a 4:1 mixture of structural isomers.
Scheme 21.

Catalytic enantioselective Diels-Alder approach to intermediates 86 and 29.
From the isomeric mixture of 28 and 79, eight additional steps lead to enone 87, and thus allow Ward to follow his previously reported route (Scheme 22). Advancement of tricycle 87 along fifteen steps (including ozonolysis and aldol reaction for ring expansion) afforded the completed cyathane core in the form of acetal 88. From this enone, oxidation, triflate formation, reductive deoxygenation, and hydrogenation produced tricycle 89 with the A-ring completed. With this material in hand, Ward was able to conclude the synthesis with four additional reactions, thus furnishing cyathin A3 (2) in 34 steps and 1.0% overall yield. The cyathane core structure was attained in 17 steps and 15% overall yield.
Scheme 22.

Endgame synthesis for cyathin A3. (TTBP = 2,6-di(tert-butyl)-4-methylpyridine.)
5.6 Nakada's (+)-Allocyathin B2
The first enantioselective total synthesis of allocyathin B2 was described by the laboratories of Nakada in a report published in 2004.33 In order to target this cyathane diterpenoid molecule, Nakada envisioned a convergent strategy starting with aldehyde 90 and alkyl iodide 91 (Scheme 23). Both fragment 90 and fragment 91 were prepared in enantioenriched form based on previous work published by Nakada and coworkers.34,35 After lithiumhalogen exchange was performed on iodide 91, the resulting alkyl lithium was added to a solution of aldehyde 90 to provide access to alcohol 92. Deoxygenation, deprotection, and oxidation of this structure afforded diketone 93.
Scheme 23.

Fragment coupling to prepare the tethered aldol precursor.
The tethered diketone 93 was subsequently cyclized via intramolecular aldol reaction upon treatment with potassium tert-butoxide (Scheme 24). This nucleophilic attack served to form the C(5)-C(4) bond, thus constructing the central B-ring of the natural product. After dehydration, tricycle 94 was isolated as the major product of the reaction sequence. In seven additional steps ketone 94 was transformed into alkyl iodide 95, an intermediate designed to undergo the samarium-mediated one-carbon ring expansion developed by Hasegawa and similar to that employed by Piers for the synthesis of Sarcodonin G (Scheme 17, Section 5.3).29 When 95 was exposed to SmI2 in the presence of HMPA, the expected migratory ring-expansion occurred in excellent yield to generate the completed cyathane core structure in the form of γ-keto ester 96.
Scheme 24.

Intramolecular aldol reaction and ring expansion to complete the total synthesis of allocyathin B2.
The route toward allocyathin B2 was thereafter concluded with three additional oxidation state manipulations, finishing the synthesis in a total of 18 synthetic operations and 7.6% overall yield. The tricyclic cyathane core was attained in 15 steps and 13.4% overall yield. Additionally, this synthetic approach has been adapted toward a number of other cyathane natural products.36
5.7 Nakada's (−)-Erinacine B
In 2007 Nakada and coworkers extended their synthetic route targeting allocyathin B2 toward the preparation of erinacine B.37 Following their previous synthetic efforts, it was possible to couple enantioenriched alkyl iodide 91 and aldehyde 90 to furnish, after elaboration, hydroxy ketone 97 (Scheme 25).35 After dehydration and deprotection to generate intermediate enone 98, the envisioned synthetic route called for reduction of the α,β-unsaturation present across the central B-ring. Though numerous Birch reduction methods were attempted, it was discovered that such techniques provided mostly over-reduced diol products instead of the desired hydroxy ketone. Ultimately, this challenge was overcome via diastereoselective olefin reduction with SmI2 in the presence of HMPA, which provided access to the desired tricycle 99. This reduction was found to produce a single diastereomer of product, establishing the critical C(5)α-H stereochemistry required for (−)-erinacine B. From this point Nakada was able to once again draw from his synthesis of allocyathin B2. Over eight steps, including SmI2-mediated ring-expansion, tricycle 100 was prepared.
Scheme 25.

Adaptation of Nakada's allocyathin B2 route to erinacine B.
Continued synthesis from ester 100 over nine additional steps allowed access to the protected allylic alcohol 101 (Scheme 26). From this point, reduction of the ketone moiety of 101 was required to establish the stereochemistry found in (−)-erinancine B. Unfortunately, all achiral reagents employed for the purpose of diastereoselective reduction afforded only the undesired epimer at C(14). Because of this difficulty, the ketone of 101 was reduced using the (R)-CBS catalyst, which set the desired relative stereochemistry with high selectivity to afford allylic alcohol 102.
Scheme 26.

Establishing the desired stereochemistry at C(14).
This material was thereafter glycosylated with a previously prepared xylose analog 103 to furnish glycone 104 (Scheme 27). After full deprotection of the carbohydrate ring, this material was treated with triethylamine and lithium bromide to effect an Sn' addition into the allylic alcohol. This provided (−)-erinacine B (6) as the sole product in enantioenriched form.
Scheme 27.

Completion of (-)-erinacine B.
Overall, the synthesis was accomplished in 33 steps and 3.0% yield from 90 and 91. The cyathane core structure was established in 18 steps and 14% overall yield.
5.8 Nakada's (−)-Erinacine E
One year subsequent to their report of erinacine B, Nakada and coworkers disclosed a beautiful modification of their synthetic strategy to target (−)-erinacine E, arguably the most complex cyathane diterpenoid molecule isolated to date.38 Starting from agylcone molecule 102, glycosylation with thioglycoside 105 and deprotection provided access to intermediate 106 (Scheme 28). After additional protecting group manipulation, Swern oxidation generated ketone 107, an intermediate which spontaneously undergoes conjugate addition-elimination to form pentacyclic structure 108.
Scheme 28.

Synthesis of 108 from allocyathin precursor 102 via conjugate addition-elimination.
Treatment of enal 108 with DBU in benzene at room temperature initiated an intramolecular aldol reaction between the enolate generated at C(4′) and the aldehyde present at C(15) (Scheme 29). This aldol reaction is followed by spontaneous benzoate ester migration, thus producing protected erinacine E analog 109 as the final product of the sequence. Further deprotection, oxidation, and diastereoselective carbonyl reduction thereafter complete the total synthesis of (−)-erinacine E (7).
Scheme 29.

Intramolecular aldol reaction for Nakada's endgame of erinacine E.
Overall, erinacine E was prepared in 39 steps from iodide 91 and aldehyde 90, in a total yield of 0.9%.
5.9 Trost's (+)-Allocyathin B2
Trost and coworkers have recently reported a unique Rucatalyzed cycloisomerization strategy for the total synthesis of the cyathane diterpenoid molecule allocyathin B2.39 The synthesis was initiated with a Pd-catalyzed aysmmetric allylic alkylation. Racemic ketone 110 was alkylated in the presence of [(η3-C3H7)PdCl]2 and chiral ligand (S,S)-112 to afford enantioenriched a-quaternary cyclopentanone 111 (Scheme 30).40 The high-yielding allylation establishes an all-carbon quaternary stereocenter in 95% ee. This transformation not only set the configuration required at C(9) of the natural product, but also provided the stereochemical basis upon which all subsequent diastereoselective transformations were leveraged.
Scheme 30.

Asymmetric allylic alkylation to establish stereochemistry at C(9).
Further elaboration of the allyl side-chain of 111, as well as manipulation of the ketone moiety, eventually produced propargyl ester 17 (Scheme 31). With this material, Trost and coworkers planned to seal the central B-ring of allocyathin B2 via an intramolecular ruthenium-mediated cycloisomerization reaction. In the event, treatment of allylic alcohol 17 with CpRu(CH3CN)3PF6 initiated cyclization of the conjugated alkyne onto the trisubstituted olefin with concomitant oxidation of the primary alcohol.41
Scheme 31.

Ru-catalyzed cycloisomerization to establish the central Bring of allocyathin B2.
Notably, the cycloisomerization reaction proceeds with high diastereoselectivity. Both products obtained from this transformation possessed the desired anti relationship between the two all-carbon quaternary stereocenters, and no products bearing a syn arrangement were observed. Trost hypothesizes that initial ruthenium complexation to both the alkyne and alkene moieties of 17 can occur to from either a syn-coplanar orientation (115) or an orthogonal orientation (117, Scheme 32). Because the orbital overlap of the syncoplanar arrangement is likely more conducive to cycloisomerization, ruthenacycle formation from intermediate 115 to give 116 is expected to be much faster than the alternative formation of 118 from 117. After b-hydride elimination occurs from ruthenacycle 116, reductive elimination forms 113 and 114 as the major products of reaction. Because only 113 was desired, ultimately it was determined that increasing the size of the alkynyl ester to a bulky tert-butyl provided preferential formation of the Z olefin isomer in a 6.7:1 ratio with isomer 114.39b
Scheme 32.

Diastereoselectivity considerations in the ruthenium catalyzed cycloisomerization.
From 113, a hydroxylative Knoevenagel reaction was performed to access lactone 119 as a single diastereomer (Scheme 33).42 The final stages of the synthesis involved hydrogenation and nitrile/lactone reduction, followed by an intramolecular aldol reaction to yield (+)-allocyathin B2 (4). Following this sequence, the natural product allocyathin B2 (and the cyathane core) was synthesized in sixteen steps and 5.2% overall yield.
Scheme 33.

Completion of allocyathin B2 via hydroxylative Knoevenagel and intramolecular aldol reactions.
5.10 Danishefsky's (−)-Scabronine G
In 2005, Danishefsky and coworkers reported the first total synthesis of the cyathane diterpenoid scabronine G (9).43 In order to target this bioactive compound, they approached the synthesis with “the pleasingly simple idea that scabronine G can be viewed as an annulated, one-carbon ring-expanded version of the (−)-Wieland–Miescher ketone”. The initial transformations of the synthesis commenced from the protected ketone 120 (Scheme 34). Over five steps, including kinetic enolate trapping and palladium-catalyzed carbonylation, acetal 120 was converted to dieneone 23. Construction of the five-membered A-ring of scabronine G was then accomplished via Lewis acid-promoted Nazarov reaction. Upon treatment of dienone 23 with FeCl3, cyclization proceeded smoothly to afford enone 121 in good yield. Notably, this tetracyclic product was obtained as a single tetrasubstituted olefin isomer.
Scheme 34.

Danishefsky's Nazarov strategy for cyathane A-ring construction.
The enone of tricycle 121 was then subsequently leveraged in order to install the critical all-carbon quaternary stereocenter present at C(9) of scabronine G. Conjugate addition of Nagata's reagent was observed to occur with high axial diastereoselectivity, and the resulting enolate was trapped with TMSCl (Scheme 35).44 The intermediate silyl enol ether obtained from this sequence was then converted to a vinyl triflate species to provide nitrile 122. Further synthetic elaboration over seven steps allowed for the conversion of vinyl triflate 122 to thiopropylmethylidene 123.
Scheme 35.

Advancement toward Danishefsky's ring-expansion precursor.
In order to access the 5-6-7 tricyclic cyathane core, vinylogous thioester 123 was first treated with the lithium anion of methoxymethyl phenyl sulfide to produce intermediate tertiary alcohol 124 as a combination of diastereomers (Scheme 36). Subsequent exposure of this mixture to HgCl2 effected a one-carbon ring expansion reaction to furnish tricycle 125 and the seven-membered C-ring, thus completing the cyathane framework.45 With ringexpanded aldehyde 125 in hand Danishefsky and coworkers were able to complete (−)-scabronine G (9) in three additional transformations, accomplishing the total synthesis in 20 steps and 8.2% overall yield. The cyathane core framework was constructed in 17 steps and 11% yield.
Scheme 36.

Ring expansion and endgame to complete (−)-scabronine G.
5.11 Phillip's (+)-Cyanthiwigin U, (+)-Cyanthiwigin W, and (−)-Cyanthiwigin Z
The first report of the total synthesis of any member of the cyanthiwigin sub-class of natural products was published by Phillips and coworkers in 2005.46 Phillips' strategy for targeting cyanthwigin U focused upon construction of the tricyclic cyathane core via simultaneous construction of both the A- and C-rings, and employed late-stage installation of the peripheral functionality.
The synthesis began with an asymmetric Diels–Alder reaction between 1,4-dimethyl cyclohexadiene (127) and (−)borneol-appended enone 126 (Scheme 37).47 This cycloaddition reaction proceeded smoothly to give a single diastereomer of 128, thus establishing both quaternary stereocenters of the natural product in a single synthetic operation. After cleavage of the chiral controller, deprotection, and further functional group manipulation, this sequence provided access to bridged bicyclic bis-aldehyde 22 (Scheme 38).
Scheme 37.

Auxiliary-mediated Diels–Alder reaction to establish the quaternary stereocenters of the natural product.
Scheme 38.

Completion of cyanthiwigin U via ring-opening ring-closeing metathesis.
Addition of vinyl Grignard to bis-aldehyde 22 was followed oxidiation to bis-enone 129. The bicyclo[2.2.2]octene system of 129 was designed to be well suited to the planned synthetic strategy of “two-directional” tandem ring-opening, ring-closing metathesis developed by Phillips in prior reports.48 By treating this bis-enone with 20 mol% of Grubbs' second-generation metathesis catalyst (69) under an atmosphere of ethylene, ring-opening metathesis of the strained bridging olefin was rapidly followed by two sequental ring-closing events. This reaction established both the five- and seven-membered rings of the cyathane core, providing tricycle 130 as the ultimate product of the cascade. From this point, the synthesis of (+)-cyanthwigin U (11) was completed in four steps involving oxidation state manipulation, as well as addition of the isopropyl and methyl groups. Phillip's route produces the cyathane core in eight synthetic operations and 19% yield, and in total, the synthesis comprises only 12 steps and is accomplished in 17% overall yield.
Very recently, the laboratories of Phillips have reported the synthesis of additional cyanthiwigin natural products based on an extension of their route toward cyanthiwigin U.49 Treatment of cyanthiwigin U (11) under Luche reduction conditions afforded the natural product cyanthwigin W in 9:1 dr (131, Scheme 39). Protection of the resulting secondary hydroxyl group was then followed by allylic transposition and alcohol oxidation via PCC. After deprotection, this sequence furnished cyanthiwigin Z (132).
Scheme 39.

Preparation of cyanthiwigin W and cyanthiwigin Z from cyanthiwigin U.
5.12 Reddy's (+)-Cyanthiwigin AC
The structure of cyathane diterpenoid cyanthiwigin AC is unique in comparison to other cyathane molecules in that it does not possess a 5-6-7 fused tricyclic core. Instead, the natural product contains both a 5-6 fused bicycle and a 6-6 spirocyclic junction. The first total synthesis of this natural product was reported in 2006 by Reddy and coworkers.50
Starting from the (+)-Hajos–Parrish ketone derivative 29, double-alkylation with bis-mesylate 133 was effective to install the spiro-annulation of the natural product (Scheme 40). After methylenation of the resulting compound, spirocyclic intermediate 134 was isolated as the major product of this two-step sequence. Deprotection, oxidation, and isomerization then furnished enone 135 in preparation for installation of the isopropyl sidechain to the five-membered A-ring.
Scheme 40.

Spiro-annulation of Hajos–Parrish ketone derivative 29.
Conjugate addition of isopropenyl cyanocuprate to tetracycle 135 was followed by diastereoselective reduction of the exocyclic methylene to set the tertiary methyl stereocenter of the natural product (Scheme 41). This yielded ketone 136 as a 2:1 mixture of epimers at C(6). Further elaboration of this material provided access to thioether 137, which upon exposure to IBX in toluene underwent oxidation to generate a mixture of six-membered ring enones.51 After sulfoxide formation and dehydrosulfenylation of this intermediate mixture, enones 138 and 139 were obtained in a 1:1.2 ratio of isomers.
Scheme 41.

Further oxidation and transformation toward cyanthiwigin AC.
From this point, the addition of methyl lithium to dienone 139 produced the natural product as a 1:2 mixture of epimers at C(12), favoring the generation of (+)-cyanthiwiging AC (12, Scheme 42) over isomer 140. The total synthesis of the natural product was concluded in 13 steps and 2.0% overall yield.
Scheme 42.

Endgame of cyanthiwigin AC.
5.13 Stoltz's (−)-Cyanthiwigin F
The total synthesis of cyanthiwigin F via a double catalytic enantioselective alkylation was published by Stoltz in 2008.52 Tandem Claisen condensation and Dieckmann cyclization of diallyl succinate (141) were followed by methylation of both a positions to yield bis-β-ketoester 19 (Scheme 43). Treatment of this material with a palladium catalyst precursor and (S)-t-BuPHOX (142) as a ligand initiates a double catalytic enantioselective alkylation reaction to generate both quaternary stereocenters present in the natural product.53 This reaction affords enantioenriched diketone 143 in 99% ee, as well as the corresponding meso diastereomer 144, in a 4.4:1 diastereomeric ratio.
Scheme 43.

Catalytic enantioselective double-alkylation for cyanthiwigin F.
Diketone 143 was subequently desymmetrized via triflate formation and Negishi cross-coupling with alkyl iodide 144 to furnish tetraene 145 (Scheme 44). This material was subjected to ring-closing metathesis by the action of modified Grubbs' catalyst 146 accompanied by cross-metathesis with vinyl boronate 147. This transformation served to close the seven-membered C-ring of the core scaffold while subsequently functionalizing the remaining allyl side-chain of 145 toward further elaboration. After mild oxidative work-up with aqueous sodium perborate, bicyclic aldehyde 148 was obtained in a single synthetic operation from 145. Closure of the final ring of the cyathane framework was then accomplished via an acyl radical cyclization in a reaction which yielded a single diastereomer of tricyclic diketone 151.54 A potential explanation for the diastereoselectivity of this reaction involves rapid formation of the five-membered A-ring through intermediate acyl radical 149, followed by external hydrogen atom abstraction by teritary radical 150 to give diketone 151. After vinyl triflate formation from this diketone, palladium-catalyzed cross-coupling with a pregenerated isopropyl cyanocuprate species produced both (−)-cyanthiwigin F (10) and reduced product 152 in a 1.8:1 ratio.
Scheme 44.

Completion of the total synthesis of cyanthiwigin F.
The total synthesis of cyanthwigin F was achieved in nine steps and 1.9% overall yield. The tricyclic cyathane core was accessed in seven steps and 6.2% overall yield.
6. Conclusions
The cyathane diterpenoid natural products have been the focus of numerous total synthetic efforts. Sixteen completed total syntheses of these compounds have been reported, thirteen of which have emerged in the last eight years alone. Because the cyathane natural products have been implicated as important biologically active molecules, particularly in regard to the stimulation of NGF synthesis, continued investigation into and refinement of their laboratory preparation is undoubtedly forthcoming.
Scheme 1.

Biosynthesis of the cyantane core tricycle from geranylgeranyl phosphate.
7. Acknowledgements
The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, Merck, and Bristol-Myers Squibb for financial support. JAE extends his thanks to Roche for support in the form of the excellence in chemistry award.
Biographies
John Enquist was born in Santa Rosa, CA, USA in 1982. He received his BA in chemistry from the University of California, San Diego in 2004, where he worked in the laboratories of Prof. Yitzhak Tor. He has been a graduate student in the laboratories of Prof. Brian Stoltz since 2004, where his work has focused on the total synthesis of natural product molecules. His research interests include cascade reactions, synthesis of biologically active natural products, transition metal-catalyzed transformations, and asymmetric methodology with a focus on the formation of multiple stereocenters.
Brian M. Stoltz was born in Philadelphia, PA, USA in 1970. After spending a year at the Ludwig Maximilians Universität in München, Germany, he obtained his BS in Chemistry and BA in German from Indiana University of Pennsylvania in 1993. He then earned his Ph.D. in 1997 under the direction of Professor John L. Wood at Yale University specializing in synthetic organic chemistry. Following an NIH postdoctoral fellowship in the laboratories of Professor E. J. Corey at Harvard University (1998-2000), he joined the faculty at Caltech in 2000 where he is currently the Ethel Wilson Bowles and Robert Bowles Professor of Chemistry. His research focuses on the design and implementation of new synthetic strategies for the synthesis of complex molecules possessing important biological properties, in addition to the development of new synthetic methods including asymmetric catalysis and cascade processes. In addition to awards from a number of pharmaceutical companies (i.e., Abbott, Amgen, AstraZeneka, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Johnson and Johnson, Merck, Novartis, Pfizer, Roche), Professor Stoltz is the recipient of the Camille and Henry Dreyfus New Faculty and Teacher-Scholar Awards, a National Science Foundation CAREER Award, the Research Corporation Research Innovation and Cottrell Scholars Awards, an A. P. Sloan Research Fellowship, an Arthur C. Cope Scholar Award from the American Chemical Society, and has received the Presidential Early Career Award in Science and Engineering (PECASE) from the White House. In 2006, he was elected a fellow of the American Association for the Advancement of Science (AAAS) and in 2008 was named a KAUST GRP Investigator. Additionally, Professor Stoltz was recognized by the Caltech Graduate Student Council with both a Classroom Teaching Award and a Mentoring Award in 2001 and by the Associated Students of the California Institute of Technology for their 30th Annual Award for Excellence in Teaching in 2006.
References
- 1.Wright DL, Whitehead CR. Org. Prep. Proced. Int. 2000;32:309–330. [Google Scholar]
- 2.Allbutt AD, Ayer WA, Brodie HJ, Johri BN, Taube H. Can. J. Microbiol. 1971;17:1401–1407. doi: 10.1139/m71-223. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ayer WA, Taube H. Tetrahedron Lett. 1972:1917–1920. [Google Scholar]; (b) Ayer WA, Taube H. Can. J. Chem. 1973;51:3842–3854. [Google Scholar]
- 4.Ayer WA, Ling SP. Can. J. Chem. 1979;57:3332–3337. [Google Scholar]
- 5.(a) Kawagishi H, Shimada A, Shirai R, Okamoto K, Ojima F, Sakamoto H, Ishiguro Y, Furukawa S. Tetrahedron Lett. 1994;35:1569–1572. [Google Scholar]; (b) Kawagishi H, Shimada A, Hosokawa S, Mori H, Sakamoto H, Ishiguro Y, Sakemi S, Bordner J, Kojima N, Furukawa S. Tetrahedron Lett. 1996;37:7399–7402. [Google Scholar]
- 6.Shibata H, Tokunaga T, Karasawa D, Hirota A, Nakayama M, Nozaki H, Taka T. Agric. Biol. Chem. 1989;53:3373–3375. [Google Scholar]
- 7.(a) Ohta T, Kita T, Kobayashi N, Obara Y, Nakahata N, Ohizumi Y, Takaya Y, Oshima Y. Tetrahedron Lett. 1998;39:6229–6232. [Google Scholar]; (b) Kita T, Takaya Y, Oshima Y, Ohta T. Tetrahedron. 1998;54:11877–11886. [Google Scholar]; (c) Ma B-J, Zhu H-J, Liu J-K. Helv. Chim. Acta. 2004;87:2877–2881. [Google Scholar]
- 8.Green D, Goldberg I, Stein Z, Ilan M, Kashman Y. Nat. Prod. Lett. 1992;1:193–199. [Google Scholar]
- 9.(a) Peng J, Walsh K, Weedman V, Bergthold JD, Lynch J, Lieu KL, Braude IA, Kelly M, Hamann MT. Tetrahedron. 2002;58:7809–7819. [Google Scholar]; (b) Peng J, Avery MA, Hamann MT. Org. Lett. 2003;5:4575–4578. doi: 10.1021/ol035592f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng J, Kasanah N, Stanley CE, Chadwick J, Fronczek FR, Hamann MT. J. Nat. Prod. 2006;69:727–730. doi: 10.1021/np050197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sennett SH, Pomponi SA, Wright AE. J. Nat. Prod. 1992;55:1421–1429. doi: 10.1021/np50088a006. [DOI] [PubMed] [Google Scholar]
- 11.Conner B, Dragunow M. Brian Res. Rev. 1998;27:1–39. doi: 10.1016/s0165-0173(98)00004-6. [DOI] [PubMed] [Google Scholar]
- 12.Ayer WA, Ling SP, Nakashima TT. Can. J. Chem. 1979;57:3338–3343. [Google Scholar]
- 13.Wender PA, Bi FC, Brodney MA, Gosseln F. Org. Lett. 2001;3:2105–2108. doi: 10.1021/ol0160699. [DOI] [PubMed] [Google Scholar]
- 14.For previous examples of [5 + 2] cycloadditions, see: Wender PA, Takahashi H, Witulski B. J. Am. Chem. Soc. 1995;117:4720–4721.; Wender PA, Husfeld CO, Langkopf E, Love JA. J. Am. Chem. Soc. 1998;120:1940–1941.; Wender PA, Dyckman AJ, Husfeld CO, Kadereit D, Love JA, Rieck H. J. Am. Chem. Soc. 1999;121:10442–10443.
- 15.(a) Thominiaux C, Chiarnoi A, Desmaële D. Tetrahedron Lett. 2002;43:4107–4110. [Google Scholar]; (b) Drège E, Morgant G, Desmaële D. Tetrahedron Lett. 2005;46:7263–7266. [Google Scholar]; (c) Drège E, Tominiaux C, Morgant G, Desmaële D. Eur. J. Org. Chem. 2006:4825–4840. [Google Scholar]
- 16.The ketoester 39 can be prepared via Michael addition to the imine obtained after treatment of 1-methylcyclopentanone with (S)-1-phenylethylamine. See: Pfau M, Revial G, d'Angelo J, Guingant A. J. Am. Chem. Soc. 1985;107:273–274.; d'Angelo J, Desmaële D, Dumas F, Guingant A. Tetrahedron: Asymmetry. 1992;3:459–505.
- 17.(a) Maruoka K, Concepcion AB, Yamamoto H. J. Org. Chem. 1994;59:4725–4726. [Google Scholar]; (b) Maruoka K, Concepcion AB, Yamamoto H. Synthesis. 1994:1283–1290. [Google Scholar]; (c) Yang S, Hungerhoff B, Metz P. Tetrahedron Lett. 1998;39:2097–2098. [Google Scholar]
- 18.Takeda K, Nakane D, Takeda M. Org. Lett. 2000;2:1903–1905. doi: 10.1021/ol0059753. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Nakajima A, Takeda M, Okamoto Y, Sato T, Yoshii E, Koizumi T, Shiro M. J. Am. Chem. Soc. 1998;120:4947–4959. [Google Scholar]
- 20.(a) Snider BB, Vo NH, O'Neil SV, Foxman BM. J. Am. Chem. Soc. 1996;118:7644–7645. [Google Scholar]; (b) Snider BB, Vo NH, O'Neil SV. J. Org. Chem. 1998;63:4732–4740. [Google Scholar]
- 21.Snider BB, Rodini DJ, van Straten J. J. Am. Chem. Soc. 1980;102:5872–5880. [Google Scholar]
- 22.(a) Snider BB. Comprehensive Organic Synthesis. Vol. 2. Oxford; Pergamon: 1991. p. 527. [Google Scholar]; (b) Snider BB, Yang KJ. J. Org. Chem. 1990;55:4392–4399. [Google Scholar]
- 23.Tori M, Toyoda N, Sono M. J. Org. Chem. 1998;63:306–313. [Google Scholar]
- 24.(a) Gensler WJ, Johnson F, Sloan ADB. J. Am. Chem. Soc. 1960;82:6074–6081. [Google Scholar]; (b) Crabbé P, Garcia GA, Rius C. J. Chem. Soc., Perkin Trans. 1973;1:810–816. [Google Scholar]
- 25.Nakashima K, Fujisaki N, Inoue K, Minami A, Nagaya C, Sono M, Tori M. Bull. Chem. Soc. Jpn. 2006;79:1955–1962. [Google Scholar]
- 26.Piers E, Gilbert M, Cook KL. Org. Lett. 2000;2:1407–1410. doi: 10.1021/ol0057333. [DOI] [PubMed] [Google Scholar]
- 27.Piers E, Yeung BKA, Fleming FF. Can. J. Chem. 1993;71:280–286. [Google Scholar]
- 28.Still WC, Mitra A. J. Am. Chem. Soc. 1978;100:1927–1928. [Google Scholar]
- 29.Hasegawa E, Kitazume T, Suzuki K, Tosaka E. Tetrahedron Lett. 1998;39:4059–4062. [Google Scholar]
- 30.(a) Ayer WA, Ward DE, Browne LM, Delbaere LTJ, Hoyano Y. Can. J. Chem. 1981;59:2665–2672. [Google Scholar]; (b) Ward DE. Can. J. Chem. 1987;65:2380–2384. [Google Scholar]; (c) Ward DE, Gai Y, Qiao Q. Org. Lett. 2000;2:2125–2127. doi: 10.1021/ol006026c. [DOI] [PubMed] [Google Scholar]
- 31.Ward DE, Shen J. Org. Lett. 2007;9:2843–2846. doi: 10.1021/ol070994z. [DOI] [PubMed] [Google Scholar]
- 32.Mikami K, Motoyama Y, Terada M. J. Am. Chem. Soc. 1994;116:2812–2820. [Google Scholar]
- 33.Takano M, Umino A, Nakada M. Org. Lett. 2004;6:4897–4900. doi: 10.1021/ol048010i. [DOI] [PubMed] [Google Scholar]
- 34.Honma M, Sawada T, Fujisawa Y, Utsugi M, Watanabe H, Umino A, Matsumura T, Hagihara T, Takano M, Nakada M. J. Am. Chem. Soc. 2003;125:2860–2861. doi: 10.1021/ja029534l. [DOI] [PubMed] [Google Scholar]
- 35.Iwamoto M, Kawada H, Tanaka T, Nakada M. Tetrahedron Lett. 2003;44:7239–7243. [Google Scholar]
- 36.An analgous synthetic approach was reported by Nakada for progress toward the synthesis of (−)-scabronine A. See: Watanabe H, Nakada M. Tetrahedron Lett. 2008;49:1518–1522.
- 37.Watanabe H, Takano M, Umino A, Ito T, Ishikawa H, Nakada M. Org. Lett. 2007;9:359–362. doi: 10.1021/ol0628816. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe H, Nakada M. J. Am. Chem. Soc. 2008;130:1150–1151. doi: 10.1021/ja7102795. [DOI] [PubMed] [Google Scholar]
- 39.(a) Trost BM, Dong L, Schroeder GM. J. Am. Chem. Soc. 2005;127:2844–2845. doi: 10.1021/ja0435586. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Dong L, Schroeder GM. J. Am. Chem. Soc. 2005;127:10259–10268. doi: 10.1021/ja051547m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Trost BM, Schroeder GM. J. Am. Chem. Soc. 1999;121:6759–6760. [Google Scholar]; (b) Trost BM, Schroeder GM, Kristensen J. Angew. Chem., Int. Ed. 2002;41:3492–3495. doi: 10.1002/1521-3773(20020916)41:18<3492::AID-ANIE3492>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 41.Trost BM, Toste FD, Pinkerton AB. Chem. Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b. [DOI] [PubMed] [Google Scholar]
- 42.Trost BM, Mallert S. Tetrahedron Lett. 1993;34:8025–8028. [Google Scholar]
- 43.Waters SP, Tian Y, Li Y-M, Danishefsky SJ. J. Am. Chem. Soc. 2005;127:13514–13515. doi: 10.1021/ja055220x. [DOI] [PubMed] [Google Scholar]
- 44.Nagata W, Mitsuru Y, Masayuk M. J. Am. Chem. Soc. 1972;94:4644–4653. [Google Scholar]
- 45.Guerrero A, Parrilla A, Camps F. Tetrahedron Lett. 1990;31:1873–1876. [Google Scholar]
- 46.Pfeiffer MWB, Phillips AJ. J. Am. Chem. Soc. 2005;127:5334–5335. doi: 10.1021/ja0509836. [DOI] [PubMed] [Google Scholar]
- 47.Palomo C, Oiarbide M, García JM, González A, Lecumberri A, Linden A. J. Am. Chem. Soc. 2002;124:10288–10289. doi: 10.1021/ja025906e. [DOI] [PubMed] [Google Scholar]
- 48.Minger TL, Phillips AJ. Tetrahedron Lett. 2002;43:5357–5359. [Google Scholar]
- 49.Pfeiffer MWB, Phillips AJ. Tetrahedron Lett. 2008;49:6860–6861. doi: 10.1016/j.tetlet.2008.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reddy TJ, Bordeau G, Trimble L. Org. Lett. 2006;8:5585–5588. doi: 10.1021/ol062304h. [DOI] [PubMed] [Google Scholar]
- 51.Nicolaou KC, Zhong Y-L, Baran PS. J. Am. Chem. Soc. 2000;122:7596–7597. [Google Scholar]
- 52.Enquist JA, Jr., Stoltz BM. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.(a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem., Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 54.Yoshikai K, Hayama T, Nishimura K, Yamada K, Tomioka K. J. Org. Chem. 2005;70:681–683. doi: 10.1021/jo048275a. [DOI] [PubMed] [Google Scholar]