

Abstract
Objective
High density lipoprotein (HDL) receptor, scavenger receptor class B, type I (SR-BI), mediated cellular uptake of lipoprotein cholesterol controls HDL structure and plasma HDL and biliary cholesterol levels. In SR-BI knockout (KO) mice, an unusually high plasma unesterified-to-total cholesterol ratio (UC:TC) and abnormally large HDL particles apparently contribute to pathology, including female infertility, susceptibility to atherosclerosis and coronary heart disease, and anemia. Here we examined the influence of SR-BI deficiency on platelets.
Methods and Results
The high plasma UC:TC ratio in SR-BI KO mice was correlated with platelet abnormalities, including high cholesterol content, abnormal morphologies, high clearance rates and thrombocytopenia. One day after platelets from wild-type mice were infused into SR-BI KO mice, they exhibited abnormally high cholesterol content and clearance rates similar to those of endogenous platelets. Platelets from SR-BI KO mice exhibited in vitro a blunted aggregation response to the agonist ADP, but a normal response to PAR4.
Conclusions
In SR-BI KO mice abnormal circulating lipoproteins, particularly their high UC:TC ratio - rather than the absence of SR-BI in platelets themselves, induce defects in platelet structure and clearance, together with a mild defect in function.
Keywords: HDL receptor, SR-BI, platelet, thrombocytopenia, clearance
Introduction
The HDL receptor SR-BI1 mediates cellular uptake of lipids, especially cholesteryl esters, from lipoproteins and the bidirectional flux of unesterified cholesterol between cells and lipoprotein (reviewed in2, 3). Hepatic SR-BI controls plasma lipoprotein metabolism, biliary cholesterol secretion, the structure and composition of plasma HDL particles. The cytoplasmic, SR-BI-binding adaptor protein PDZK1 plays a key role in tissue-specific posttranscriptional control of hepatic SR-BI protein expression.4 SR-BI-deficiency in homozygous null knockout (SR-BI KO) mice results in hypercholesterolemia (~2.2-fold increase in total plasma cholesterol, much of which is carried in abnormally large HDL particles).5 The ratio of plasma unesterified cholesterol (UC)-to-total cholesterol (TC, unesterified plus esterified cholesterol) in SR-BI KO mice is about double that of WT mice.6, 7 SR-BI KO mice exhibit enhanced susceptibility to atherosclerosis,8 female infertility,8, 9 and red blood cell (RBC) defects10, 11 (reviewed in2). In SR-BI KO mice, essentially liver-specific expression of an SR-BI transgene (‘KO-Tg’ mice) restores plasma cholesterol levels (TC and UC:TC ratio) and the structure of HDL to those seen in WT mice and corrects several SR-BI-deficiency related pathologies.12
In the current study, we examined the effects of SR-BI deficiency on platelets, which in WT mice circulate in the blood with a lifespan of ~5 days13 followed by clearance via the reticuloendothelial system.14 We found that SR-BI KO mice are thrombocytopenic (reduced platelet count). Their platelets exhibited abnormal morphologies, abnormally high unesterified cholesterol, elevated rates of clearance from the circulation and a minor defect in ADP-induced aggregation. These abnormalities appear to be a consequence of the high UC:TC ratio.
Materials and Methods
Four week-six month old male and female mice fed standard lab chow were used with appropriate institutional approvals. Genotypes (determined by polymerase chain reaction): SR-BI+/+ (WT), SR-BI+/−, SR-BI−/− (SR-BI KO), and SR-BI−/− expressing an essentially liver-specific SR-BI transgene (KO-Tg; the SR-BI transgene is not expressed in bone marrow monocytes/granulocytes (AY, MK, Ching-Hung Shen and Jianzhu Chen, unpublished results), - all on mixed 50:50 C57BL/6:129-S4 backgrounds,5, 12. The ’50:50’ ratio represents the average percent of genes contributed by the indicated pure background (e.g., C57BL/6); however, animal-to-animal variation in the source of any particular gene (C57BL/6 or 129-S4) is possible. It was necessary to use SR-BI KO animals (and their wild-type controls) on mixed genetic backgrounds because we were unable to generate sufficient numbers of viable SR-BI KO mice on a pure C57BL/6 background using standard husbandry conditions (unpublished). Thus, unless indicated otherwise, ‘WT’ refers to control mice on the mixed 50:50 C57BL/6:129-S4 background. We also studied PDZK1−/− (on either a mixed 75:25 129Sv:C57Bl/6 or pure 129Sv background, generously provided by Olivier Kocher) and appropriate wild-type controls (50:50 129Sv:C57Bl/6 or pure 129Sv background),4, 15 SR-BI−/−/apoE−/− (dKO, on a mixed 75:25 C57BL/6:129Sv background, WT controls),8, 16 and apoE−/−17, 18 and matched wild-type controls (on a pure C57Bl/6 background, Jackson Laboratories).
All procedures (e.g., platelet labeling and determination of platelet lifetimes) were performed as described previously or in the online data supplement (see http://atvb.ahajournals.org).
Results
SR-BI deficiency causes thrombocytopenia
Figure 1A shows that there was no difference in the blood platelet count between wild-type (WT, also SR-BI+/+) and heterozygous null SR-BI (SR-BI+/−) mice; whereas there was a substantial reduction (38% of WT) in homozygous null (SR-BI−/−, SR-BI KO or simply KO) mice. This thrombocytopenia suggested an abnormally high rate of platelet clearance or an abnormally low rate of platelet production, or both.
Figure 1. Effects of SR-BI deficiency on platelet count and survival.

A. Platelets in whole blood harvested from SR-BI+/+ (WT, black bar, n=20), SR-BI+/− (gray bar, n=17) and SR-BI−/− (KO, white bar, n=14) mice were stained (anti-GPIIbIIIa antibody), and counted by flow cytometry. Results are averages from three experiments, each containing a minimum of 3 mice/group; ** P<0.001 B. WT (black diamonds) and SR-BI KO (KO, white squares) mice were intravenously infused with biotin to label platelets in vivo. Mice were then bled each day for five days and the fractions of platelets labeled with biotin were determined by flow cytometry. The percentage of biotinylated platelets was defined as 100% on day 0. (determined 30 minutes after infusion of biotin-NHS) (n=4–5, *P<0.05, **P<0.001).
Mechanism underlying thrombocytopenia in SR-BI KO mice
To determine if reduced platelet survival times contributed to the thrombocytopenia, we biotinylated platelets in vivo and determined the percentage of surviving labeled platelets as a function of time. Figure 1B shows that there was a dramatically reduced half-life of platelets in SR-BI KO mice (~27 hours) compared to WT mice (~46 hours). Reduced platelet production did not appear to contribute to the thrombocytopenia, because in SR-BI KO mice there was no decrease in either the number of young platelets in circulation or the number of splenic and bone marrow megakaryocytes. Indeed, there was an increase in splenic megakaryocytes (see Supplemental material). These results suggest that thrombocytopenia was likely due to abnormally rapid platelet clearance.
Mechanism underlying the reduced lifespan of circulating platelets in SR-BI KO mice
Next we determined if the reduced lifespan of platelets in SR-BI KO mice were a platelet cell autonomous property or if it were primarily dependent on the external environment of the platelets (i.e., plasma conditions). We isolated platelets from either WT or SR-BI KO donor mice, labeled them ex vivo with biotin, infused the labeled platelets into recipient WT or SR-BI KO mice and then determined clearance rates. Figure 2 shows the differences in the survival times of the platelets were primarily dependent on the genotype of the recipients, not the donors. Platelets from either WT or SR-BI KO mice exhibited almost identical, short (~20 h) lifetimes when infused into SR-BI KO recipients (squares), whereas the lifetimes for platelets from either donor were similar and longer (~43 h) in WT recipients (diamonds). For the WT recipients, there was a small, but reproducibly, longer lifetime observed for platelets from SR-BI KO donors than from WT donors. It is not clear if the somewhat larger fraction of ‘younger’ platelets from SR-BI KO mice (Supplemental Figure IC) contributed to this somewhat longer survival. Control experiments (Supplemental Materials) showed that severe thrombocytopenia does not per se reduce the half-life of infused platelets. Taken together these data indicate that the short lifespan of platelets in SR-BI KO mice, and their associated thrombocytopenia, appears to be a consequence primarily of the extracellular environment of the platelets rather than a platelet intrinsic phenomenon.
Figure 2. Survival of ex vivo biotinylated platelets after infusion into WT and KO recipients.
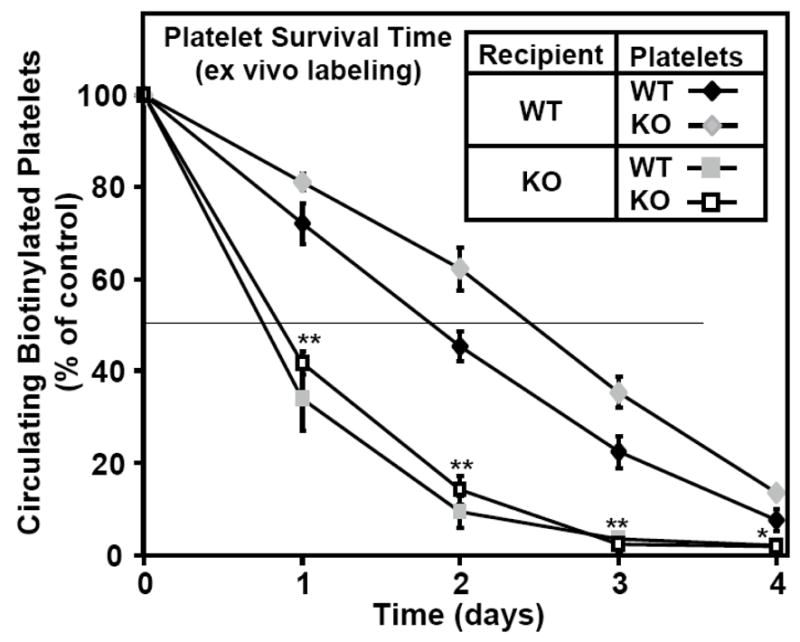
Platelets from wild type (WT, black diamonds or gray squares) or SR-BI KO (white squares or gray diamonds) mice were isolated, washed, biotinylated and intravenously infused into recipient mice of the indicated phenotypes (WT, diamonds; SR-BI KO, squares), and the percentage of labeled platelets in the circulation relative to day 0 immediately after infusion (100% of control) was determined as in Figure 1B. Results represent the average of two independent experiments (n=6–7). Values for donors of either genotype infused into SR-BI KO recipients were significantly lower than for WT recipients (*P<0.05 and **P<0.001).
Mechanism by which the extracellular environment controls murine platelet survival time
The most obvious difference in the environments of the platelets in WT and SR-BI KO mice that might influence platelet survival is the characteristic dyslipidemia in SR-BI KO mice (e.g., high TC and UC:TC ratio)5–7 Figure 3A shows that filipin (fluorescent cholesterol binding dye10, 19) staining of platelets from SR-BI KO mice was 1.8-fold greater than that of platelets from WT mice (1.83±0.09 vs 1.00±0.02, P<0.001), indicating a significantly greater level of unesterified cholesterol in SR-BI KO platelets.
Figure 3. Cholesterol content and ultrastructure of platelets.

A. Platelets in blood from WT (black bar) and SR-BI KO (white bar) mice were stained with anti-GPIIbIIIa, then incubated with 50 μg/ml filipin (15 minutes, RT) to label unesterified cholesterol, and filipin staining was determined by flow cytometry (n=6–7, **P<0.001). B–D. Washed platelets from WT (B) and SR-BI KO (C, D) mice were visualized using standard transmission electron microscopy. Multi-lamellar, membrane-like structures often seen in SR-BI KO platelets are indicated by arrows. Scale bar = 500 nm.
Transmission electron microscopic analysis of platelets from WT (Figure 3B) and SR-BI KO (Figure 3C and D) mice showed that, while there were similar amounts of intracellular granules (dense granules or alpha granules), mitochondria and endoplasmic reticulum, there were two striking morphologic differences. First, SR-BI KO platelets were ~1.4-fold larger than WT platelets (relative sizes: 1.39±0.04 vs 1.00±0.03, respectively, P<0.001). This difference, determined by quantitative analysis of the micrographs, was also observed when the platelets were subjected to forward light scattering analysis (Supplemental Figure III). Second, many of the platelets from SR-BI KO, but not WT, mice contained unusual multi-lamellar structures that resemble multi-lamellar phospholipid membranes (arrows in Figure 3C and D). It seems possible that these multilamellar structures are generated to accommodate the excess unesterified cholesterol in the SR-BI KO platelets.
If excess cholesterol in platelets results in rapid clearance, then platelet unesterified cholesterol levels (filipin staining) and survival times should correlate. Therefore, we infused ex vivo biotinylated platelets from either WT or SR-BI KO donors into WT or SR-BI KO recipients and one day later isolated platelets and compared the levels of filipin staining of endogenous ‘resident’ platelets (not biotinylated) with those of the biotinylated infused platelets. Figure 4 shows that, in all of the recipient mice regardless of genotype, the filipin staining of the infused platelets (white bars) was virtually identical to that of the resident platelets (gray bars). There was relatively low staining in WT recipients (Figure 4, left) and high staining in SR-BI KO recipients (Figure 4, right), regardless of the genotype of the donor animals. Thus, in the circulation platelets relatively rapidly acquire from or release to lipoproteins and/or cells, unesterified cholesterol. The extent of filipin staining was inversely correlated with the survival time (compare Figures 2 and 4), suggesting that accumulation of excess unesterified cholesterol in the platelets may have contributed to their more rapid clearance from the circulation.
Figure 4. Cholesterol content of ex vivo biotinylated platelets after infusion into WT or SR-BI KO recipients.
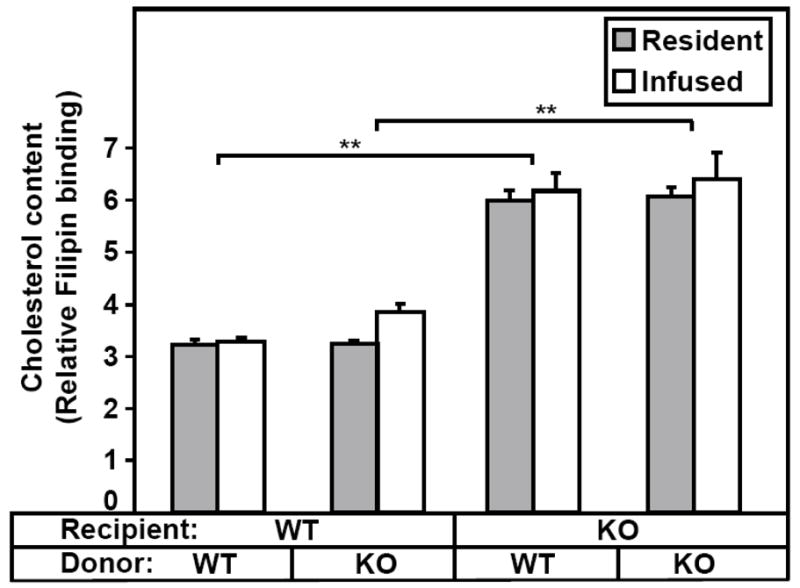
Platelets from WT or SR-BI KO mice were labeled with biotin and infused into WT or SR-BI KO recipients. Next day the recipient mice were bled, the platelets stained with GPIIbIIIa-APC, PE-SA and filipin, and the cholesterol content (filipin staining) of resident (non-biotinylated) and infused platelets was determined by flow cytometry. (n=3–4). The data are from one of two similar independent experiments (**P<0.001).
Role of total and unesterified plasma lipoprotein cholesterol on platelet composition and metabolism
Hypercholesterolemia per se does not appear to be responsible for platelet abnormalities in SR-BI KO mice. For example, the platelet counts for wild-type control and apoE knockout (apoE−/−) mice, both on a pure C57Bl/6 background, did not differ ((6.89±0.31)×108 vs (6.46±0.42)×108 platelets/ml, respectively, P>0.05), despite sever hypercholesterolemia in apoE−/− mice (450±11 mg/dL vs 230±9 mg/dL for SR-BI KO, also see 5, 6, 17, 18). The UC:TC ratio in apoE−/− mice is essentially identical to that of WT mice (about half that in SR-BI KO mice)6, raising the possibility that a high UC:TC ratio may alter platelet structure and metabolism.
To explore this possibility, we examined platelets from three additional mouse strains with varying UC:TC ratios (Figure 5A): KO-Tg mice [normocholesterolemic, UC:TC=0.21 (similar to the ratio of 0.23 for WT, also see12)]; PDZK1−/− mice [hypercholesterolemic, UC:TC=0.254]; and SR-BI/apoE dKO mice [extreme hypercholesterolemia (9–10-fold greater than WT), UC:TC=~0.86]. Figure 5B shows that the relative levels of cholesterol (filipin staining) in the platelets of KO-Tg and PDZK1−/− mice (with essentially normal UC:TC) were not different than that in WT mice (P>0.05 for each comparison), whereas that for SR-BI/apoE dKO mice was 2.56- and 1.39-fold greater than that of WT and SR-BI single KO mice, respectively (for both, P<0.001). Figure 5C shows that the platelet counts for KO-Tg, PDZK1−/− and WT mice (all on mixed C57BL/6:129 genetic backgrounds) were not different (P>0.05). Similarly, there was no significant difference for PDZK1−/− and matched wild-type control mice on a pure Sv 129 genetic background [(1.17±0.08) ×109 and (1.35±0.07)×109 respectively, n=3, P>0.05)]. In contrast, the count for SR-BI/apoE dKO mice [(1.09±0.10)×108 platelets/ml] was 3.2-fold less than that of SR-BI single KO mice (Figure 5C, P<0.001). The platelet survival curves for KO-Tg and PDZK1−/− mice were similar to that of WT mice (see Supplemental Figure IV). These data show a strong correlation of platelet counts, platelet unesterified cholesterol levels and platelet survival times with the plasma lipoprotein UC:TC ratios and are consistent with the hypothesis that the abnormal UC:TC ratio in SR-BI KO mice is responsible, at least in part, for their thrombocytopenia.
Figure 5. Effects of altering SR-BI, PDZK1 and apoE expression on plasma UC:TC ratio, platelet cholesterol content and platelet count.
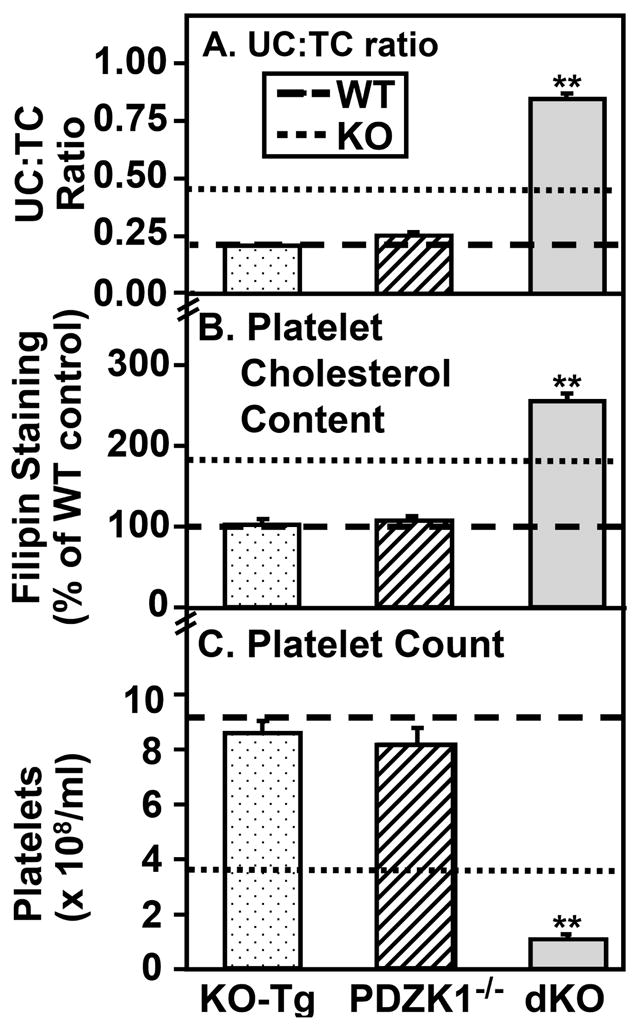
Blood was drawn from WT mice (dashed lines), SR-BI KO mice (dotted lines), SR-BI KO mice expressing a primarily liver specific SR-BI transgene (KO-Tg, stippled bars), PDZK1−/− mice (hatched bars), and SR-BI KO/apoE−/− double knockout mice (dKO, gray bars) and plasma unesterified-to-total cholesterol ratios (UC:TC) (panel A, n=7–18), platelet cholesterol contents (panel B, filipin staining, n=7) and platelet counts (panel C, n=7–13) were determined (**P<0.001).
Analysis of platelet function in vitro
The kinetics and extent of aggregation of platelet-rich plasma from WT and SR-BI KO mice were similar after stimulation by the thrombin receptor agonist PAR4 peptide (1 mM) (Figure 6A). However, abnormally low aggregation was reproducibly observed in SR-BI KO platelets stimulated with either 1 μM (Figure 6B) or 5 μM (Supplemental Figure V) of the weak agonist ADP, although the extent of reduction varied in different experiments (~30–100% reduction). Thus, SR-BI deficiency also led to a modest defect in in vitro platelet function.
Figure 6. Effects of platelet agonists PAR4 peptide and ADP on platelet aggregation.
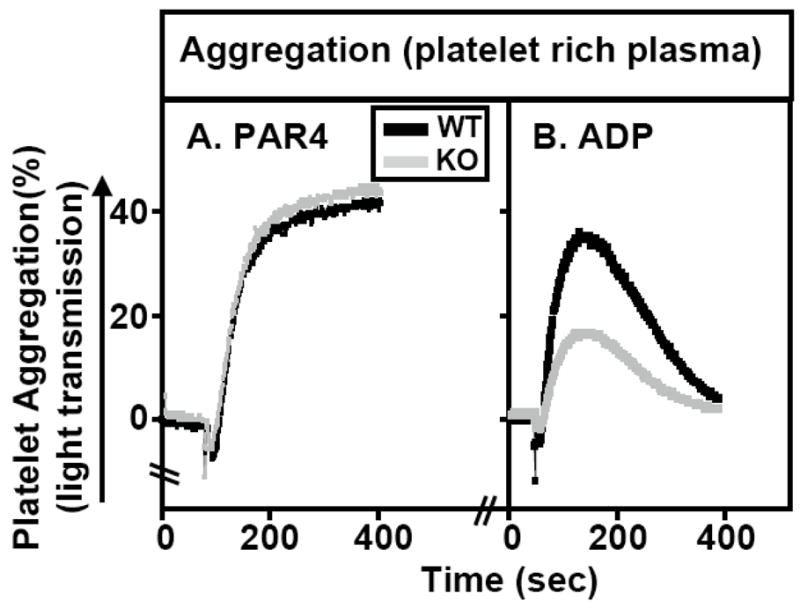
Platelet-rich plasma from WT (black lines) and SR-BI KO (gray lines) were isolated and platelet aggregation as a function of time after adding the agonists PAR4 peptide (1 mM, panel A) or ADP (1 μM, panel B) was measured as increased light transmission in an aggregometer as described in Methods. Aggregation traces from one of three independent experiments are shown.
Discussion
A distinctive feature of the SR-BI KO mice is that they are hypercholesterolemic with abnormally large circulating HDL particles and an abnormally high unesterified-to-total plasma cholesterol ratio (UC:TC).5–7 In the current study we found that murine hypercholesterolemia per se (e.g., in apoE−/− mice) does not induce thrombocytopenia. However, our analysis of SR-BI KO and other related transgenic animals shows a correlation of an abnormally high plasma UC:TC ratio with abnormally high platelet cholesterol content, rapid platelet clearance and thrombocytopenia. We also found that one day after platelets from either SR-BI KO or WT donor mice were infused into recipient mice, the cholesterol content of the infused platelets, as well as the platelet clearance rates, mirror those of the resident endogenous platelets (either WT or SR-BI KO recipients), regardless of the genotype of the donor. We conclude that the high UC:TC ratio in the plasma lipoproteins of SR-BI KO mice is probably responsible for the rapid platelet clearance and thrombocytopenia. Although it is possible that the high plasma UC:TC ratio, independently of its effects on cholesterol accumulation in platelets, induces increased activity of the reticuloendothelial system responsible for platelet clearance, it seems likely that the accumulation of high unesterified cholesterol in the platelets themselves is the principle cause of their rapid clearance.
Platelets can acquire cholesterol from plasma through exchange with plasma lipoproteins.20, 21 Several dyslipidemic states, such as spur cell anemia,22, sitosterolaemia,23, 24 acanthocytosis,21 and Tangier disease25, 26 are associated with concurrent defects in platelets and RBCs. Hyperlipidemia and dyslipidemia have been linked with reduced platelet survival time and thrombocytopenia (e.g., inverse correlation of plasma total cholesterol and platelet survival time),23, 27, 28 although the mechanism by which circulating lipids induce thrombocytopenia remains uncertain. Additionally, several cholesterol-independent factors29–31, including shedding32 or clustering33 of cell surface GP1bα are known to influence platelet lifespan. Elevated shedding of GP1bα in SR-BI KO mice is unlikely to cause thrombocytopenia, because GP1bα levels on their platelets were ~1.44±0.04-fold higher than those from WT mice (see Supplemental Material).
There are striking similarities of the influence of SR-BI deficiency on platelets (this study) and RBCs10, 11 with respect to cellular composition and morphology (e.g., excess cholesterol), clearance rate (accelerated), and steady state levels in the blood (thrombocyotopenia and anemia). These similarities are particularly noteworthy given the substantial differences in the mechanisms governing the production and clearance of these two blood cell types.13, 29, 34–36 It is possible that neither RBCs or their immediate precursors nor platelets have adequate capacity to process the large amounts of intracellular unesterified cholesterol that accumulate in SR-BI KO mice. These cells might not be able either to effectively convert the excess cholesterol to cholesteryl esters for storage37 or to maintain sufficient levels of cholesteryl ester stores in stable cytoplasmic lipid droplets. Indeed, almost all of the cholesterol in platelets is normally unesterified cholesterol.38 As a consequence, these cells might be especially susceptible to toxic effects of excess unesterified cholesterol accumulation.
Because of the cholesterol accumulation and morphological abnormalities in the platelets in SR-BI KO mice, it was not surprising to find that these platelets exhibited a subtle, but reproducible, functional defect in vitro – reduced responsiveness to the agonist ADP in aggregation assays. It is noteworthy that Shattil and colleagues have shown that in vitro loading of platelets with unesterified cholesterol can increase intracellular cAMP levels.39 Although increased intracellular cAMP has been shown to reduce platelet sensitivity to ADP,40 Shattil et al and others reported increased sensitivity to ADP and epinephrine of platelets loaded with cholesterol in vitro,41 as well as platelets from hypercholesterolemic patients.41, 42 Furthermore, treatment of hypercholesterolemic patients with lipid lowering therapy has been reported to reduce response of platelets to ADP.27, 43 The effects of cholesterol loading of platelets, whether in vitro or in vivo, are clearly complex, possibly depending on the precise mechanism of loading (e.g., during or after platelet formation from megakaryocytes), and other features of lipoprotein metabolism (e.g., elevated plasma apoE, 5, 44, 45) also may impact platelets in SR-BI KO mice. The abnormal response of SR-BI KO platelet aggregation to ADP and normal response to the PAR4 peptide were observed in all experiments and clearly show that many key signaling and response pathways in these cells remained intact. Since the mice are on mixed genetic background, some of the observed variation in relative responses to ADP (30–100% reduction in induced aggregation) may have been due to the mixed genetic backgrounds. The mechanism underlying the reduced responsiveness to ADP and physiologic consequences, if any, remains to be determined.
The pathophysiologic consequences, if any, of the platelet abnormalities in SR-BI KO mice remain to be identified. We and others have shown that SR-BI is atheroprotective in a variety of murine models of atherosclerosis (e.g., enhanced atherosclerosis in mice with homozygous null mutations in the SR-BI gene8 (reviewed in2, 3). Platelets have been shown to contribute substantially to atherosclerosis46, 47 and unesterified cholesterol in RBCs (and possibly platelets) may contribute to cholesterol deposition in atherosclerosis.48 Thus, it is possible that the platelet abnormalities in SR-BI KO described here may contribute to the enhanced atherosclerosis observed in SR-BI-deficient mice.
Supplementary Material
Acknowledgments
We gratefully acknowledge Alpay Burak Seven, Michael S. Brown, Glenn Paradis, Maria Ericsson, Elizabeth J. Benecchi and Songwen Zhang for assistance, Olivier Kocher for providing PDZK1−/− mice, Stephen M. Cifuni, Tobias Goerge and Jerry Ware for GP1bα/IL-4R transgenic mice, and T.Y. Chang for helpful discussions. This work was supported by U.S. National Institutes of Health grants to MK and DW.
Footnotes
Conflict-of-interest disclosure: None
References
- 1.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 2.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 3.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 4.Kocher O, Yesilaltay A, Cirovic C, Pal R, Rigotti A, Krieger M. Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism. J Biol Chem. 2003;278:52820–52825. doi: 10.1074/jbc.M310482200. [DOI] [PubMed] [Google Scholar]
- 5.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Out R, Hoekstra M, Spijkers JA, Kruijt JK, van Eck M, Bos IS, Twisk J, Van Berkel TJ. Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res. 2004;45:2088–2095. doi: 10.1194/jlr.M400191-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miettinen HE, Rayburn H, Krieger M. Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J Clin Invest. 2001;108:1717–1722. doi: 10.1172/JCI13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, Andrews NC. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood. 2002;99:1817–1824. doi: 10.1182/blood.v99.5.1817. [DOI] [PubMed] [Google Scholar]
- 11.Meurs I, Hoekstra M, van Wanrooij EJ, Hildebrand RB, Kuiper J, Kuipers F, Hardeman MR, Van Berkel TJ, Van Eck M. HDL cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp Hematol. 2005;33:1309–1319. doi: 10.1016/j.exphem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Yesilaltay A, Morales MG, Amigo L, Zanlungo S, Rigotti A, Karackattu SL, Donahee MH, Kozarsky KF, Krieger M. Effects of hepatic expression of the high-density lipoprotein receptor SR-BI on lipoprotein metabolism and female fertility. Endocrinology. 2006;147:1577–1588. doi: 10.1210/en.2005-1286. [DOI] [PubMed] [Google Scholar]
- 13.Berger G, Hartwell DW, Wagner DD. P-Selectin and platelet clearance. Blood. 1998;92:4446–4452. [PubMed] [Google Scholar]
- 14.Kotze HF, Lotter MG, Badenhorst PN, Heyns AD. Kinetics of In-111-platelets in the baboon: II. In vitro distribution and sites of sequestration. Thromb Haemost. 1985;53:408–410. [PubMed] [Google Scholar]
- 15.Yesilaltay A, Kocher O, Pal R, Leiva A, Quinones V, Rigotti A, Krieger M. PDZK1 is required for maintaining hepatic scavenger receptor, class B, type I (SR-BI) steady state levels but not its surface localization or function. J Biol Chem. 2006;281:28975–28980. doi: 10.1074/jbc.M603802200. [DOI] [PubMed] [Google Scholar]
- 16.Karackattu SL, Picard MH, Krieger M. Lymphocytes are not required for the rapid onset of coronary heart disease in scavenger receptor class B type I/apolipoprotein E double knockout mice. Arterioscler Thromb Vasc Biol. 2005;25:803–808. doi: 10.1161/01.ATV.0000158310.64498.ac. [DOI] [PubMed] [Google Scholar]
- 17.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 18.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 19.Hayward RD, Cain RJ, McGhie EJ, Phillips N, Garner MJ, Koronakis V. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol Microbiol. 2005;56:590–603. doi: 10.1111/j.1365-2958.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- 20.Shattil SJ, Bennett JS, Colman RW, Cooper RA. Abnormalities of cholesterol-phospholipid composition in platelets and low-density lipoproteins of human hyperbetalipoproteinemia. J Lab Clin Med. 1977;89:341–353. [PubMed] [Google Scholar]
- 21.Ulibarrena C, Vecino A, Cesar JM. Red cell lipid abnormalities in acquired acanthocytosis are extended to platelets. Br J Haematol. 1994;87:614–616. doi: 10.1111/j.1365-2141.1994.tb08322.x. [DOI] [PubMed] [Google Scholar]
- 22.Doll DC. Surface morphology of platelets in spur-cell anemia. Hum Pathol. 1982;13:671–672. doi: 10.1016/s0046-8177(82)80013-0. [DOI] [PubMed] [Google Scholar]
- 23.Rees DC, Iolascon A, Carella M, O’Marcaigh AS, Kendra JR, Jowitt SN, Wales JK, Vora A, Makris M, Manning N, Nicolaou A, Fisher J, Mann A, Machin SJ, Clayton PT, Gasparini P, Stewart GW. Stomatocytic haemolysis and macrothrombocytopenia (Mediterranean stomatocytosis/macrothrombocytopenia) is the haematological presentation of phytosterolaemia. Br J Haematol. 2005;130:297–309. doi: 10.1111/j.1365-2141.2005.05599.x. [DOI] [PubMed] [Google Scholar]
- 24.Kruit JK, Drayer AL, Bloks VW, Blom N, Olthof SG, Sauer PJ, de Haan G, Kema IP, Vellenga E, Kuipers F. Plant sterols cause macrothrombocytopenia in a mouse model of sitosterolemia. J Biol Chem. 2007 doi: 10.1074/jbc.M706689200. [DOI] [PubMed] [Google Scholar]
- 25.Frohlich J, Godin DV. Erythrocyte membrane alterations and plasma lipids in patients with chylomicronemia and in Tangier disease. Clin Biochem. 1986;19:229–234. doi: 10.1016/s0009-9120(86)80032-7. [DOI] [PubMed] [Google Scholar]
- 26.Nofer JR, Herminghaus G, Brodde M, Morgenstern E, Rust S, Engel T, Seedorf U, Assmann G, Bluethmann H, Kehrel BE. Impaired platelet activation in familial high density lipoprotein deficiency (Tangier disease) J Biol Chem. 2004;279:34032–34037. doi: 10.1074/jbc.M405174200. [DOI] [PubMed] [Google Scholar]
- 27.Sinzinger H, Pirich C, Bednar J, O’Grady J. Ex-vivo and in-vivo platelet function in patients with severe hypercholesterolemia undergoing LDL-apheresis. Thromb Res. 1996;82:291–301. doi: 10.1016/0049-3848(96)00079-5. [DOI] [PubMed] [Google Scholar]
- 28.Steele P, Rainwater J. Effects of dietary and pharmacologic alteration of serum lipids on platelet survival time. Circulation. 1978;58:365–367. doi: 10.1161/01.cir.58.2.365. [DOI] [PubMed] [Google Scholar]
- 29.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 30.Bergmeier W, Burger PC, Piffath CL, Hoffmeister KM, Hartwig JH, Nieswandt B, Wagner DD. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003;102:4229–4235. doi: 10.1182/blood-2003-04-1305. [DOI] [PubMed] [Google Scholar]
- 31.McNeil HP, Chesterman CN, Krilis SA. Immunology and clinical importance of antiphospholipid antibodies. Adv Immunol. 1991;49:193–280. doi: 10.1016/s0065-2776(08)60777-4. [DOI] [PubMed] [Google Scholar]
- 32.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ Res. 2004;95:677–683. doi: 10.1161/01.RES.0000143899.73453.11. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmeister KM, Josefsson EC, Isaac NA, Clausen H, Hartwig JH, Stossel TP. Glycosylation restores survival of chilled blood platelets. Science. 2003;301:1531–1534. doi: 10.1126/science.1085322. [DOI] [PubMed] [Google Scholar]
- 34.Koury MJ, Sawyer ST, Brandt SJ. New insights into erythropoiesis. Curr Opin Hematol. 2002;9:93–100. doi: 10.1097/00062752-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Ferrant A, Cauwe F, Michaux JL, Beckers C, Verwilghen R, Sokal G. Assessment of the sites of red cell destruction using quantitative measurements of splenic and hepatic red cell destruction. Br J Haematol. 1982;50:591–598. doi: 10.1111/j.1365-2141.1982.tb01959.x. [DOI] [PubMed] [Google Scholar]
- 36.Broudy VC, Kaushansky K. Thrombopoietin, the c-mpl ligand, is a major regulator of platelet production. J Leukoc Biol. 1995;57:719–725. doi: 10.1002/jlb.57.5.719. [DOI] [PubMed] [Google Scholar]
- 37.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 38.Marcus AJ, Ullman HL, Safier LB. Lipid composition of subcellular particles of human blood platelets. J Lipid Res. 1969;10:108–114. [PubMed] [Google Scholar]
- 39.Sinha AK, Shattil SJ, Colman RW. Cyclic AMP metabolism in cholesterol-rich platelets. J Biol Chem. 1977;252:3310–3314. [PubMed] [Google Scholar]
- 40.Aszodi A, Pfeifer A, Ahmad M, Glauner M, Zhou XH, Ny L, Andersson KE, Kehrel B, Offermanns S, Fassler R. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. Embo J. 1999;18:37–48. doi: 10.1093/emboj/18.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shattil SJ, Anaya-Galindo R, Bennett J, Colman RW, Cooper RA. Platelet hypersensitivity induced by cholesterol incorporation. J Clin Invest. 1975;55:636–643. doi: 10.1172/JCI107971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldassarre D, Mores N, Colli S, Pazzucconi F, Sirtori CR, Tremoli E. Platelet alpha 2-adrenergic receptors in hypercholesterolemia: relationship between binding studies and epinephrine-induced platelet aggregation. Clin Pharmacol Ther. 1997;61:684–691. doi: 10.1016/S0009-9236(97)90104-1. [DOI] [PubMed] [Google Scholar]
- 43.Tekten T, Ceyhan C, Ercan E, Onbasili AO, Turkoglu C. The effect of atorvastatin on platelet function in patients with coronary artery disease. Acta Cardiol. 2004;59:311–315. doi: 10.2143/AC.59.3.2005187. [DOI] [PubMed] [Google Scholar]
- 44.Riddell DR, Owen JS. Inhibition of ADP-induced platelet aggregation by apoE is not mediated by membrane cholesterol depletion. Thromb Res. 1995;80:499–508. doi: 10.1016/0049-3848(95)00205-7. [DOI] [PubMed] [Google Scholar]
- 45.Higashihara M, Kinoshita M, Kume S, Teramoto T, Kurokawa K. Inhibition of platelet function by high-density lipoprotein from a patient with apolipoprotein E deficiency. Biochem Biophys Res Commun. 1991;181:1331–1336. doi: 10.1016/0006-291x(91)92084-w. [DOI] [PubMed] [Google Scholar]
- 46.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 47.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 48.Kolodgie FD, Burke AP, Nakazawa G, Cheng Q, Xu X, Virmani R. Free cholesterol in atherosclerotic plaques: where does it come from? Curr Opin Lipidol. 2007;18:500–507. doi: 10.1097/MOL.0b013e3282efa35b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.