

Abstract

A de novo transformation of N-allyl-N-sulfonyl ynamides to amidines is described featuring a palladium-catalyzed N-to-C allyl transfer via ynamido-palladium-π-allyl complexes.
Our involvement in the studies of Huisgen’s azide-[3 + 2]1–3 cycloadditions employing ynamides4–7 led us to an exciting possibility. As shown in Scheme 1, under copper(I)-catalyzed conditions,8 while triazolyl copper intermediates 1 could be trapped with electrophiles other than proton to afford more substituted triazoles 2,9,10 when R2 = Ts, it could also readily lose N2 in a retro-[3 + 2] manner to give ynamido-copper complexes 3a in equilibrium with ketenimine-copper complexes 3b. A series of elegant studies have since appeared reporting nucleophilic trappings of 3 in both inter- and intramolecular fashion, leading to amidines and amidates.11–14 The potential of harvesting new reactivities from ynamido-metal complexes captured our attention. Consequently, we examined a different pathway that can provide general access to ynamido-metal π-allyl complexes 5a and 5b from N-allyl-N-sulfonyl ynamides 4. We report here a de novo synthesis of pharmacologically useful amidines15–18 from ynamides featuring a palladium-catalyzed N-to-C allyl transfer through ynamido-π-allyl complexes.
Scheme 1.

Generating Ynamido-Metal Complexes.
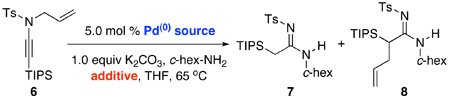
While identifying a suitable palladium catalyst for our intended reaction pathway was not difficult, we found two amidine products. As shown in Table 1, when treating N-allyl-N-sulfonyl ynamide 6 with 5 mol% of Pd(PPh3)2Cl2 in the presence of c-hex-NH2 in THF at 65 °C, both amidines 7 and 8 were observed.19 Intriguingly, the ratio of 7 and 8 depended upon the amount of c-hex-NH2 that was used. A greater amount of c-hex-NH2 [3–5 equiv] predominantly led to the formation of 7 in which the allyl group is lost [entries 1 and 2], while 1.0 equiv of c-hex-NH2 and/or addition with the use of syringe pump began to favor the formation of 8 in which the allyl group had undergone an N-to-C transfer [entries 3 and 4].
Table 1.
Effect of Equivalents of Amines and Pd(0) Sources.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry |
 [mol %] [mol %] |
amine equiv | time [h] | yield [%]a | 7 | 8 | |
| 1 | Pd(PPh3)2Cl2 | -- | 5.0 | 2 | 92 | 5 | |
| 2 | -- | 3.0 | 2 | 63 | 24 | ||
| 3 | -- | 1.0 | 2 | 44 | 45 | ||
| 4 | -- | 1.0: syringe pump addition | 2 | 11 | 73 | ||
| 5 | Pd(PPh3)4 | -- | 3.0 | 3 | 0 | ≥95 | |
| 6 | Pd(dppe)Cl2 | -- | 3.0 | 48 | 30 | <5 | |
| 7 | Pd(dppf)Cl2 | -- | 3.0 | 24 | 95 | 0 | |
| 8 | Pd2(dba)3 |
 [10.0] [10.0] |
3.0 | 24 | 40 | 59 | |
| 9 | Pd2(dba)3 |
 [10.0] [10.0] |
3.0 | 3 | 0 | ≥95 | |
| 10 | Pd2(dba)3 |
 [10.0] [10.0] |
3.0 | 24 | <5 | 95 | |
 | |||||||
All are isolated yields.
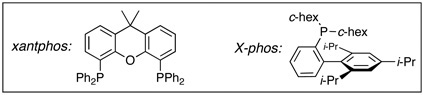
Moreover, a quick screening of palladium sources revealed that the allyl transfer is catalyst dependent [Table 1]. At 3.0 equiv of c-hex-NH2 in comparison with Pd(PPh3)2Cl2 [see entry 1], Pd(PPh3)4 gave exclusively allyl transferred amidine 8 [entry 5], while P d(dppe)Cl2 and Pd(dppf)Cl2 [entries 6 and 7] reverted back to favor amidine 7 with Pd(dppf)Cl2 giving a better yield [entry 7]. Sensing that these contrasts could be due to the differences either in the initial oxidation state of the palladium metal, or more likely, their respective ligands, we examined Pd2(dba)3 along with 10 mol% of various phosphine ligands. While BINAP was not useful [entry 8, potential ee was not analyzed], we found that both xantphos20 and X-phos21 [entries 9 and 10] represent excellent ligand systems for promoting the allyl transfer, with the former phosphine ligand [see entry 9] providing a much faster reaction.
The generality of this allyl transfer could be established very quickly via three perspectives, leading to the synthesis of a diverse array of amidines. First, we employed a range of primary amines including allyl amine [entry 3 in Table 2], propargyl amine [entry 4], and anilines [entries 5–9]. Secondly, we examined a series of secondary amines including the use of p-Ns-substituted ynamide [see 20 in Figure 1], indoline [see 26], tetrahydroquinoline [see 27], and imidazole [see 28].
Table 2.
Amidine Synthesis Using Primary Amines.a
| entry | primary amines | 4-pentenyl-amidines | yield [%]b | |
|---|---|---|---|---|
| 1 |  |
 |
9 | ≥95 |
| 2 | 10 | 90 | ||
| 3 | 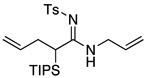 |
11 | 73 | |
| 4 | 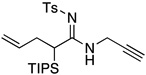 |
12 | 76 | |
| 5 |  |
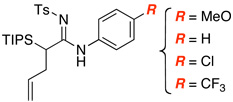 |
13 | 67 |
| 6 | 14 | 85c | ||
| 7 | 15 | 78d | ||
| 8 | 16 | 54 | ||
| 9 |  |
 |
17 | 52 |
All reactions utilized ynamide 6, 5.0 mol % Pd(PPh3)4, 1.0 equiv K2CO3, 3.0 equiv RNH2, THF [conc = 0.05 M], 65 °C, 5–8 h.
Isolated yields.
1.0 equiv of amine was used.
Reaction time was 24 h.
Figure 1.

Secondary Amines in the Amidine Synthesis.a,b
a. Reaction conditions: 5.0 mol % Pd2(dba)3, 10.0 mol % of xantphos, 1.0 equiv K2CO3, 3.0 equiv R2NH, THF [conc = 0.05 M], 65 °C, 1.5–6 h. b. Isolated yields. c. 10.0 mol % Pd2(dba)3, 20.0 mol% of xantphos, and 5.0 equiv R2NH were used. d. The only successful example in using 5.0 mol % Pd(PPh3)4.
Thirdly, we explored ynamides 29a–e with variations on the acetylenic substituent [Table 3]. It is noteworthy that while Pd(PPh3)4 was effective in promoting allyl transfer when using primary amines, it was not useful for secondary amines [with the exception of 22] and only the usage of Pd2(dba)3 and xantphos led to allyl transferred amidines.
Table 3.
Ynamide Substituent Effect.
 | |||||
|---|---|---|---|---|---|
| entry | ynamides | R1 = | amidines | NR2 = | isolated yield [%] |
| 1 | 29a | TBDPS | 30a | pyrrolidinyl | 95 |
| 2 | 29b | TBS | 30b | pyrrolidinyl | 94 |
| 3 | 29c | TES | 30c | pyrrolidinyl | 87 |
| 4 | 29d | (CH2)3OTBS | 30d | c-hex-NH | 41 |
| 5 | 29e | c-hex | 30e | pyrrolidinyl | 54 |
| 6 | 29e | c-hex | 30f | c-hex-NH | 69 |
A proposed model consistent with our observations is shown in Scheme 2. While all evidence points toward the presence of ynamido-P d-π-allyl complexes 5a in equilibrium with the ketenimine complex 5b through an oxidative addition,22,23 the pathway clearly diverged thereafter depending upon the concentration of the amine HNR2 and the nature of the ligand. We believe the first equivalent of HNR2 effectively gave the amidinyl Pd-π-allyl complexes 31a and 31b via nucleophilic addition to 5b. An ensuing reductive elimination of 31a and/or 31b would lead to respective allyl transferred amidines 32a and 32b, and 32a appears to tautomerize favorably to 32b.
Scheme 2.

A Proposed Mechanistic Model.
However, this reductive elimination step appears to be less favored when an excess of amine was used. Consequently, Pd-complexes 33 could be attained likely through a direct nucleophilic attack on the Pd-π-allyl motif, thereby leading to the loss of the respective allyl amines,24 and ultimately, the formation of the non-allyl transferred amidines 34 after reductive elimination. In addition, this de-allylative pathway is also consistent with the fact that when using the more nucleophilic secondary amines [relative to primary amines]25 and P d(PPh3)4, non-allyl transferred amine product predominated. Consequently, in all cases, either a controlled amount of HNR2, or a slow addition of HNR2, or more bulky ligands such as X-phos21 and/or bidentate ligands with unique bite angles such as xantphos20,26 that presumably promote reductive elimination could be employed to favor the formation of allyl transferred amidines 32b.
Finally, the efficacy of oxidative addition likely plays a role in the distribution between non-allyl and allyl transferred amidines because the choice of Pd(0) source appears to be critical. Specifically, a Pd(II) source could also serve as π-Lewis acid to activate the ynamide, leading to keteniminium Pd-complex 35 [Scheme 3]. After addition of the first equivalent of HNR2, de-allylation of the resulting N-allyl enamide 36 could take place with a second equivalent of HNR2. This process could be promoted by either Pd(0) or Pd(II), with the former initiating an oxidative addition while the latter again serving to activate the ketene-aminal motif. This assessment is consistent with the observation that non-allyl transferred amidines 34 were the major product when using Pd(II) sources.
Scheme 3.

Pd(II) Versus Pd(0) Source.
We have described here a de novo transformation of N-allyl-N-sulfonyl ynamides to a diverse array of amidines featuring a palladium-catalyzed N-to-C allyl transfer via ynamido-palladium-π-allyl complexes. Efforts in further developing synthetic methods involving these ynamido-palladium-π-allyl complexes are underway.
Supplementary Material
Experimental procedures as well as NMR spectra, and characterizations are available for all new compounds and free of charge via Internet http://pubs.acs.org.
Acknowledgement
We thank NIH [GM066055] for funding.
References
- 1.(a) Huisgen R. Angew Chem. 1963;75:604. [Google Scholar]; (b) Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Oxford: Pergamon Press; 1984. pp. 1–176. [Google Scholar]
- 2.For leading reviews, see: Meldal M, Tornoe CW. Chem. Rev. 2008;108:2952. doi: 10.1021/cr0783479. Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7. Bock VD, Hiemstra H, Van Maarseveen JH. Eur. J. Org. Chem. 2006:51. Katritzky AR, Zhang Y, Singh SK. Heterocycles. 2003;60:1225. Abu-Orabi ST. Molecule. 2002;7:302. Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.
- 3.For a review on organic azides, see: Bräse S, Gil C, Knepper K, Zimmermann v. Angew. Chem., Int. Ed. 2005;44:5188. doi: 10.1002/anie.200400657.
- 4.(a) Zhang X, Li H, You L, Tang Y, Hsung RP. Adv. Syn. Cat. 2006;348:2437. [Google Scholar]; (b) Zhang X, Hsung RP, You L. Org. Biomol. Chem. 2006;6:2679. doi: 10.1039/b606680a. [DOI] [PubMed] [Google Scholar]
- 5.For reviews on ynamides, see: Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei L-L. Tetrahedron. 2001;57:7575. Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379. Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455.
- 6.For chemistry of ynamides in the last two years, see: Couty S, Liegault B, Meyer C, Cossy J. Tetrahedron. 2009;65 ASAP. Deweerdt K, Birkedal H, Ruhland T, Skrydstrup T. Org. Lett. 2009;11:221. doi: 10.1021/ol802477d. Dooleweerdt K, Birkedal H, Ruhland T, Skrydstrup T. J. Org. Chem. 2008;73:9447. doi: 10.1021/jo801935b. Saito N, Katayama T, Sato Y. Org. Lett. 2008;10:3829. doi: 10.1021/ol801534e. Yasui H, Yorimitsu H, Oshima K. Bull. Chem. Soc. Jpn. 2008;81:373. Yasui H, Yorimitsu H, Oshima K. Chem. Lett. 2008;37:40. Istrate FM, Buzas AK, Jurberg ID, Odabachian Y, Gagosz F. Org. Lett. 2008;10:925. doi: 10.1021/ol703077g. Martínez-Esperón MF, Rodríguez D, Castedo L, Saá C. Tetrahedron. 2008;64:3674. Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. Yavari I, Sabbaghan M, Hosseini N, Hossaini Z. Synlett. 2007;20:3172. Hashimi ASK, Salathe R, Frey W. Synlett. 2007:1763. Rodríguez D, Martínez-Esperón MF, Castedo L, Saá C. Synlett. 2007:1963. Couty S, Meyer C, Cossy J. Synlett. 2007:2819. Movassaghi M, Hill MD, Ahmad OK. J. Am. Chem. Soc. 2007;129:10096. doi: 10.1021/ja073912a. Tanaka K, Takeishi K. Synthesis. 2007:2920. Kohnen AL, Dunetz JR, Danheiser RL. Organic Syn. 2007;84:88.
- 7.(a) Al-Rashid ZF, Johnson WL, Hsung RP, Wei Y, Yao P-Y, Liu R, Zhao K. J. Org. Chem. 2008;73:8780. doi: 10.1021/jo8015067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yao P-Y, Zhang Y, Hsung RP, Zhao K. Org. Lett. 2008;10:4275. doi: 10.1021/ol801711p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org. Lett. 2008;10:3477. doi: 10.1021/ol801257j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Al-Rashid ZF, Hsung RP. Org. Lett. 2008;10:661. doi: 10.1021/ol703083k. [DOI] [PubMed] [Google Scholar]; (e) Oppenheimer J, Hsung RP, Figueroa R, Johnson WL. Org. Lett. 2007;9:3969. doi: 10.1021/ol701692m. [DOI] [PubMed] [Google Scholar]; (f) You L, Al-Rashid ZF, Figueroa R, Ghosh SK, Li G, Lu T, Hsung RP. Synlett. 2007:1656. [Google Scholar]; (g) Li H, You L, Zhang X, Johnson WL, Figueroa R, Hsung RP. Heterocycles. 2007;74:553. [Google Scholar]; (h) Sagamanova IK, Kurtz KCM, Hsung RP. Organic Syn. 2007;84:359. [Google Scholar]; (i) Oppenheimer J, Johnson WL, Tracey MR, Hsung RP, Yao P-Y, Liu R, Zhao K. Org. Lett. 2007;9:2361. doi: 10.1021/ol0707362. [DOI] [PubMed] [Google Scholar]
- 8.Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JMJ, Sharpless KB, Fokin VV. Angew. Chem., Int. Ed. 2004;43:3928. doi: 10.1002/anie.200454078. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Hsung RP, Li H. Chem. Commun. 2007:2420. doi: 10.1039/b701040k. [DOI] [PubMed] [Google Scholar]
- 10.For related studies on these triazolyl copper intermediates, see: Gerard B, Ryan J, Beeler AB, Porco JA., Jr Tetrahedron. 2006;62:6405. Wu YM, Deng J, Li Y, Chen Q-Y. Synthesis. 2005:1314. Cassidy MP, Raushel J, Fokin VV. Angew. Chem., Int. Ed. 2006;45:3154. doi: 10.1002/anie.200503805. see footnote 11.
- 11.For intermolecular additions, see: Bae I, Han H, Chang S. J. Am. Soc. Chem. 2005;127:2038. doi: 10.1021/ja0432968. Cho SH, Yoo EJ, Bae I, Chang S. J. Am. Chem. Soc. 2005;127:16046. doi: 10.1021/ja056399e. Yoo EJ, Bae I, Cho SH, Han H, Chang S. Org. Lett. 2006;8:1347. doi: 10.1021/ol060056j. Kim SH, Jung DY, Chang S. J. Org. Chem. 2007;72:9769. doi: 10.1021/jo7016247. Cho SH, Chang S. Angew. Chem., Int. Ed. 2008;47:2836. doi: 10.1002/anie.200705940. Kim J, Lee SY, Lee J, Do Y, Chang S. J. Org. Chem. 2008;73:9454. doi: 10.1021/jo802014g. Yoo EJ, Ahlquist M, Bae I, Sharpless KB, Fokin VV, Chang S. J. Org. Chem. 2008;73:5520. doi: 10.1021/jo800733p.
- 12.For an intramolecular addition, see: Chang S, Lee M, Jung DY, Yoo EJ, Cho SH, Han SK. J. Am. Soc. Chem. 2006;128:12366. doi: 10.1021/ja064788i.
- 13.For a study using ynamides, see: Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745.
- 14.For other leading examples of trapping complexes such as 3, see: Cui S-L, Lin X-F, Wang Y-G. Org. Lett. 2006;8:4517. doi: 10.1021/ol061685w. Xu X, Cheng D, Li J, Guo H, Yan J. Org. Lett. 2007;9:1585. doi: 10.1021/ol070485x. Jin Y, Fu H, Yin Y, Jiang Y, Zhao Y. Synlett. 2007:901. Cui S-L, Wang J, Wang Y-G. Org. Lett. 2007;9:5023. doi: 10.1021/ol702241e. Cui S-L, Wang J, Wang Y-G. Org. Lett. 2008;10:1267. doi: 10.1021/ol800160q. For an earlier study on trapping of ynamido-lithium complexes, see: Fromont C, Masson S. Tetrahedron. 1999;55:5405.
- 15.Greenhill JV, Lue P. Prog. Med. Chem. 1993;30:203. doi: 10.1016/s0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- 16.For a leading review on amidine derivatives serving as selective muscarinic agonists in the treatment of Alzheimer’s diseases, see: Messer WS, Jr, Dunbar PG. Muscarinic Agonists and the Treatment of Alzheimer's Disease. 1996:131–153.
- 17.Dunn PJ. In: Compreh. Org. Funct. Group Transform. II. Katritzky Alan R, Taylor Richard JK., editors. Vol. 5. Sandwich, UK: Amidines and N-Substituted Amidines. Pfizer Global Research and Development; 2005. pp. 655–699. [Google Scholar]
- 18.For recent examples of amidine synthesis, see: Yu RT, Rovis T. J. Am. Chem. Soc. 2008;130:3262. doi: 10.1021/ja710065h. Wang J, Xu F, Cai T, Shen Q. Org. Lett. 2008;10:445. doi: 10.1021/ol702739c. Malik H, Frederic B, Alexandre M, Jean-Jacques B. Org. Biom. Chem. 2006;4:3142. Katritzky AR, Cai C, Singh SK. J. Org. Chem. 2006;71:3375. doi: 10.1021/jo052443x. Kumagai N, Matsunaga S, Shibasaki M. Angew. Chem., Int. Ed. 2004;43:478. doi: 10.1002/anie.200352750.
- 19.See Supporting Information. Based on NOE experiments [see Supporting Information for details], these amidines adopt an E-geometry with respect to the C=N bond.
- 20.For a leading reference on xantphos, see: Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM. Organometallics. 1995;14:3081.
- 21.For leading references on X-phos, see: Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:6653. doi: 10.1021/ja035483w. Barder TE, Buchwald SL. J. Am. Chem. Soc. 2007;129:12003. doi: 10.1021/ja073747z.
-
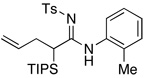
22.As shown below, a non-palladium involved pathway would entail an aza-Claisen type of rearrangement followed by trapping of the allylketenimine intermediate i with an external amine. However, while this pathway is indeed a possibility, it requires much higher temperature and longer reaction time. When carried out at 65 °C to 80 °C in THF, the reaction was sluggish and slow.
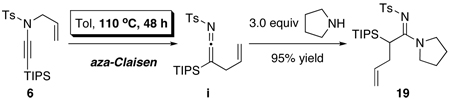 For a recent account on a related thermal transformation using ynol ethers, see:
Sosa JR, Tudjarian AA, Minehan TG. Org. Lett. 2008;10:5091. doi: 10.1021/ol802147h.
For a recent account on a related thermal transformation using ynol ethers, see:
Sosa JR, Tudjarian AA, Minehan TG. Org. Lett. 2008;10:5091. doi: 10.1021/ol802147h.
- 23.For leading reviews on Claisen rearrangements, see: Hill RK. In: Asymmetric Synthesis. Morrison JD, editor. New York: Academic Press; 1984. Wipf P. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Oxford: Pergamon Press; 1991. p. 827.
-
24.Because of their basicity, polarity, and/or volatility, the respective allyl amine byproducts [R2N-CH2CH=CH2] from de-allylation were difficult to isolate. However, we were able to isolate the following allylated amine ii when using piperizine.
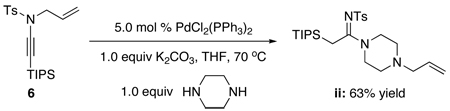
- 25.For a leading reference on relative nucleophilicity of amines, see: Brotzel F, Chu YC, Mayr H. J. Org. Chem. 2007;72:3679. doi: 10.1021/jo062586z.
- 26.Hartwig observed that in comparison with mono-dentate phosphine ligands, the usage of bidentate ligands such as xantphos leads to a much faster amidative cross-coupling. This is presumably due to the ability of amido-type carbonyl groups to engage in tight complexation with the palladium metal. As a result, when using xantphos, its unique bite angle promotes reductive elimination. See: Fujita K-I, Yamashita M, Puschmann F, Alvarez-Falcon MM, Incarvito CD, Hartwig JF. J. Am. Chem. Soc. 2006;128:9044. doi: 10.1021/ja062333n.


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures as well as NMR spectra, and characterizations are available for all new compounds and free of charge via Internet http://pubs.acs.org.