

Abstract
The treatment of severe lung disease often requires the use of high concentrations of oxygen coupled with the need for assisted ventilation, potentially exposing the pulmonary epithelium to both reactive oxygen species and nonphysiological cyclic stretch. Whereas prolonged hyperoxia is known to cause increased cell injury, cyclic stretch may result in either cell proliferation or injury depending on the pattern and degree of exposure to mechanical deformation. How hyperoxia and cyclic stretch interact to affect the pulmonary epithelium in vitro has not been previously investigated. This study was performed using human alveolar epithelial A549 cells to explore the combined effects of cyclic stretch and hyperoxia on cell proliferation and viability. Under room air conditions, cyclic stretch did not alter cell viability at any time point and increased cell number after 48 h compared with unstretched controls. After exposure to prolonged hyperoxia, cell number and [3H]thymidine incorporation markedly decreased, whereas evidence of oxidative stress and nonapoptotic cell death increased. The combination of cyclic stretch with hyperoxia significantly mitigated the negative effects of prolonged hyperoxia alone on measures of cell proliferation and viability. In addition, cyclic stretch resulted in decreased levels of oxidative stress over time in hyperoxia-exposed cells. Our results suggest that cyclic stretch, as applied in this study, can minimize the detrimental effects of hyperoxia on alveolar epithelial A549 cells.
Keywords: pulmonary epithelial cells, cell death
Mechanical Forces have been increasingly recognized to play an important role in the regulation of cell function, including the modification of cell growth, life span, surface receptor expression, cytoskeletal infrastructure, signal transduction, and gene expression (6, 25, 33). Physiologically relevant mechanical forces are known to occur in a variety of organs, including the skin, skeletal, vascular, and pulmonary systems (46). In both the fetal and mature lung, such forces have been shown to promote epithelial cell proliferation, differentiation, calcium mobilization, surfactant secretion, and prostacyclin production (27, 36, 39, 51). Alternatively, excessive mechanical stretch has been shown to be a contributing factor in the pathogenesis of ventilator-induced lung injury (5, 45). Repetitive cyclic stretch of pulmonary epithelial cells at nonphysiological amplitudes results in progressive cell injury, proinflammatory cytokine release, and necrotic and apoptotic cell death (12, 13, 33, 37).
Stretch-induced mechanotransduction is thought to result after activation of “mechanosensors” that transmit messages to the cell interior via the complex interaction of cell membrane proteins, the cytoskeletal network, and a number of intracellular signaling cascades (5, 33, 37). The response of pulmonary epithelial cells to mechanical stretch in vitro has been shown to vary considerably depending on the cell type, culture conditions, pattern, and duration of stretch exposure (4, 24–26). Because of the complexity of local distension patterns within the lung parenchyma that occur with ventilation, it is difficult to quantify the degree of stretch that actually occurs in vivo; however, a number of in vitro models have been developed to study the effects of stretch on pulmonary epithelial cells (10, 35, 43). In general, stretch amplitudes corresponding to changes in surface area (ΔSA) of <25–30% have been shown to be noninjurious and reflective of physiological stretch that occurs at less than total lung capacity (2, 43). Little is currently understood about how mechanical stretch within this physiological range alters cellular responses. It has been suggested that physical strain may actually prime cells to differentially respond in either a protective or synergistic fashion when exposed to additional harmful stimuli (10, 33, 37).
In addition to mechanical ventilation, the treatment of severe pulmonary disease often requires the use of high concentrations of oxygen (hyperoxia), which can result in the generation of increased reactive oxygen species (ROS) within the lung (8, 9). The production of ROS from oxidative stress causes enzyme inhibition, lipid peroxidation, and DNA damage and adversely alters gene regulation (1, 14, 18, 34, 44). In pulmonary epithelial cells, prolonged exposure to hyperoxia is known to cause cellular injury and/or death (9, 23, 32, 40). Previous studies have shown that hyperoxia kills human alveolar epithelial A549 cells by necrosis, with extensive cell swelling and death seen within a few days of exposure (19, 20, 21, 31).
Despite the clinical relevance of mechanical stretch and hyperoxic exposure to the lung, the combined effects of these factors applied in vitro to distal pulmonary epithelial cells have not been explored. In this study, we sought to determine the effects of physiological stretch in conjunction with prolonged hyperoxia on measures of cell proliferation, viability, and oxidative stress. Our data demonstrate that cyclic stretch, as applied in this study, attenuates the detrimental effects of hyperoxia on proliferation and nonapoptotic death in A549 pulmonary epithelial cells. In addition, cyclic stretch decreases cellular superoxide levels, which may be a contributing factor to these findings.
METHODS
Cell Culture
Human lung adenocarcinoma A549 cells (American Type Culture Collection CCL 185) were grown in Ham’s F-12 medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Sigma). Cells were maintained in a humidified chamber at 37°C in either room air (21% O2 and 5% CO2) or hyperoxic conditions (95% O2 and 5% CO2) both with and without cyclic stretch. Cells were seeded onto six-well collagen-coated, flexible-bottom, silicone elastomer BioFlex plates (Flexcell International, Hillsborough, NC) and allowed to grow to 60–70% confluence for assays looking at proliferation, viability, apoptosis, and cell morphology. At each time point, cell number was determined by manual counting using a hemacytometer.
Cyclic Stretch
A549 cells were subjected to mechanical stretch using the commercially available computer-driven FX-3000 Flexercell strain unit (Flexcell International, McKeesport, PA). This device uses a controlled vacuum to deform the monolayer of cells grown on top of the membrane. The vacuum produced a 16% elongation on the flexible bottom elastomer membranes at a frequency of 30 cycles/min (0.5 Hz). Cells were harvested after different durations of cyclic exposure. Control cells also were plated on elastomer plates to avoid variations based on attachment stratum.
Hyperoxia Exposure
A computer-driven Biospherix Oxycycler system (Reming Bioin-struments, Redfield, NY) was used to tightly control oxygen levels at 95% with 5% CO2 in the hyperoxic exposure cell groups. The Oxycycler chamber was custom designed to contain the FX-3000 Flexercell strain unit, allowing simultaneous regulation of both stretch and hyperoxic conditions.
Cell Proliferation Studies
Cell counts
Cell number was determined by manually counting cells from duplicate chambers with a hemacytometer.
DNA synthesis
Cell proliferation under the different experimental conditions was assessed using [3H]thymidine incorporation corrected for cell number. Cells pulsed for 12 h with 1 μCi/ml [3H]thymidine (Amersham, Arlington Heights, IL). On the morning after the pulse, monolayers were rinsed with phosphate-buffered saline and DNA was precipitated with 10% trichloroacetic acid for 24 h. Samples were then denatured in 0.5 M NaOH and analyzed using a Tri Carb 2500 TR liquid scintillation analyzer (Packard, Meriden, CT).
Assessment of Cell Viability and Death
Calcein acetoxymethyl ester assay
After the different experimental manipulations were completed, 100 μl of cell suspension were transferred to clear 96-well flat-bottomed microtiter plates (Costar, Corning, NY). Positive control wells were treated with 100 μl of methanol to induce maximal death. To each well, we added 25 μM calcein acetoxymethyl ester (AM) (Molecular Probes, Eugene, OR). The cell-permeant esterase substrate calcein AM is nonfluorescent until converted by enzymatic activity to highly fluorescent calcein, which is retained within live cells and imparts an intense green fluorescence (15). Cells were incubated in the dark for 20 min to avoid photodynamic effects. The fluorescence of the calcein generated within the cells was analyzed using the BIO-Tek spectrophotometer plate reader (Winooski, VT), with KC4 analysis software, at an excitation wavelength of ~485 nm and an emission wavelength of ~530 nm. This fluorometric microplate assay allowed quantification of green fluorescence, with the amount detected proportional to the number of viable cells. Samples were harvested after 24, 48 and 72 h of exposure.
LIVE/DEAD viability assay
The LIVE/DEAD viability/cytotoxicity kit (Molecular Probes) provides a two-color fluorescence-based cell viability assay that allows the simultaneous determination of live and dead cells, using calcein AM and ethidium homodimer-1 (EthD-1), respectively. Eth-D-1 undergoes a fluorescence enhancement upon binding nucleic acids and produces a bright red fluorescence. This dye is excluded from cells that have intact plasma membranes but is readily able to enter dead cells. Thus live cells fluoresce green, whereas dead cells fluoresce red.
Cell suspensions of 100 μl were transferred to 96-well clear, flat-bottomed microtiter assay plates (Costar). Positive control wells were treated with 100 μl of methanol to induce maximal death. EthD-1 (17 μM) and calcein AM (10 μM) were added to the wells, and the cells were incubated in the dark for 20 min to avoid photodynamic effects. Cells were analyzed using fluorescence microscopy with a Nikon Optiphot-2 microscope (Nikon Instruments, Melville, NY) equipped with a mercury lamp and Nikon UV filters. Images were taken using a Nikon DXM-1200F high-resolution digital camera with ACT-1 imaging software (Nikon Instruments). Cell counts were made based on the number of live cells (green) vs. dead cells (red) that were seen in random, noncontiguous fields until >250 cells total were counted.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity was measured using a cytotoxicity detection kit (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s protocol. From each well, 100 μl of medium were removed and the remaining cells were lysed by adding 100 μl of 1% Triton X-100 solution. The samples were incubated in the dark for 30 min with buffer containing NAD+, lactate, and tetrazolium. LDH converts lactate to pyruvate, generating NADH. The NADH then reduces tetrazolium (yellow) to formazan (red), which was detected by fluorescence (490 nm) using a BIO-Tek spectrofluorometer plate reader with KC4 analysis software. LDH release was expressed as a percentage of the LDH in the medium relative to the total LDH lysate.
Apoptosis Assays
Caspase-3 activity
Harvested cells and supernatants were collected and fixed in 0.5% paraformaldehyde for 10 min at 37°C. They were permeabilized in 90% methanol for 30 min at 4°C and then incubated with cleaved caspase-3 (Asp 175), a rabbit monoclonal antibody (Cell Signaling, Beverly, MA), for 30 min at room temperature, followed by staining with a secondary antibody, FITC-conjugated donkey anti-Rabbit IgG (Jackson ImmunoResearch, West Grove, PA) for 30 min at room temperature. Samples were run on a BD FACSCalibur flow cytometer and analyzed using BD Cell Quest software (BD BioSciences, San Jose CA). Cells exposed to 50 μM hydrogen peroxide for 6 h served as positive apoptotic controls. Cells exposed to room air alone were used to define the basal level of apoptotic cells. Samples were harvested after 24, 48 and 72 h of exposure.
Annexin V-APC staining
Annexin V-APC was used to quantitatively determine the percentage of cells within each exposure group undergoing early apoptosis. The standard flow cytometry viability probe 7-aminoactinomycin (7-ADD) was used to distinguish viable from nonviable cells within each exposure group. Cells were harvested and then stained with APC-conjugated annexin V-APC and 7-ADD according to the annexin V-APC staining protocol (BD Biosciences, Rockville, MD). Unstained cells, cells stained with annexin V-APC alone (no 7-AAD), and cells stained with 7-ADD alone (no annexin V-APC) were used as controls to set up compensation and quadrants. Cells were run on a BD FACSCalibur flow cytometer and analyzed using BC Cell Quest software. Cells exposed to room air alone were used to define the basal level of apoptotic and dead cells. Cells exposed to 50 μM hydrogen peroxide for 6 h served as positive apoptotic controls. Samples were harvested after 24, 48, and 72 h of exposure.
Cell Morphology
BioFlex plate (Flexcell, Hillsborough, NC) collagen-coated, flexible-well bottoms were removed with a scalpel and placed on a standard microscope slide. Cells were fixed with 4% paraformaldehyde and stained with hematoxylin and eosin. Images of random, noncontiguous fields of A549 cells were photographed with an Olympus Vanox AHB53 microscope (Olympus America, Melville, NY) at ×60 magnification using a SPOT RT camera with SPOT software version 3.5 (Diagnostic Instruments, Sterling Heights, MI). Five fields per slide were obtained from at least three separate experiments for each treatment. One observer under blinded conditions viewed samples.
Superoxide Measurement by Dihydroethidium Flow cytometry
Intracellular superoxide production was measured using the cell-permeable, dual-fluorescent probe dihydroethidium (DHE; Molecular Probes), which is very sensitive to superoxide but very insensitive to hydrogen peroxide (47, 53). DHE emits a blue fluorescent signal, but in the presence of superoxide, DHE is dehydrogenated to ethidium, which then intercalates with negatively charged DNA and emits a red fluorescent signal (11). Once formed, ethidium is extremely stable in the cell (53). Cells were detached by trypsinization and pelleted by centrifugation at 300 g for 5 min at 25°C. Cells were resuspended in 20 μM DHE and incubated for 30 min at 37°C in the dark. Cells were centrifuged to pellet form at 300 g for 5 min at 25°C and resuspended in 1% paraformaldehyde in HBSS to fix cells. Fluorescence intensity was measured using a BD FACSCalibur flow cytometer. For each analysis, 20,000 events were collected and analyzed using FL1 and FL2 channels. Data were analyzed using BC Cell Quest software. Percent gated cell count values were determined using an FL2 channel, with a right shift in the FL2 channel detecting orange-red fluorescence representing the presence of superoxide. Mean percent values were determined from three separate experiments.
Statistics
All data are presented as means ± SE. All experiments were performed at least three times unless otherwise stated. Paired Student’s t-test was used to assess differences between two groups. One-way analysis of variance (ANOVA) was performed when more than two groups were compared, followed by a multiple comparison test (Tukey or Holm Sidak post hoc methods). A P value <0.05 was considered significant. Statistical analyses were performed using SigmaStat 3.1 (Leesburg, VA).
RESULTS
Effects of Hyperoxia and Stretch on Cell Proliferation
Cyclic stretch under room air conditions increased cell number compared with unstretched controls after 48 h but not after 24 or 72 h (P = 0.002; Fig. 1A). Cell number markedly decreased after exposure to hyperoxia after all time points compared with both stretched and unstretched room air groups (P < 0.05). Compared with cells exposed to hyperoxia alone, cell number significantly increased during hyperoxic exposure when coupled with stretch after 48 and 72 h (P < 0.05). [3H]thymidine incorporation was also markedly decreased following exposure to hyperoxia after 48 and 72 h (P < 0.05; Fig. 1B) but again significantly increased when combined with cyclic stretch (P < 0.002).
Fig. 1.
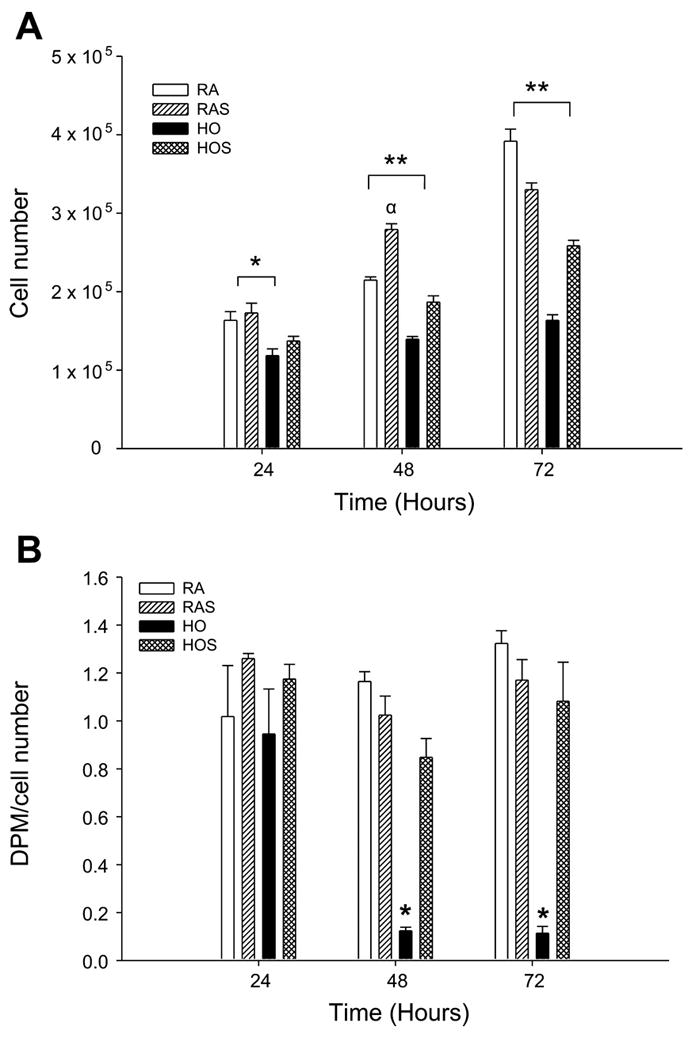
A: effects of hyperoxia and stretch on cell number. A549 cells were exposed to room air (RA), room air with stretch (RAS), hyperoxia (HO), and hyperoxia with stretch (HOS) for 24, 48, and 72 h. Cell number was determined by manually counting cells from duplicate chambers with a hemacytometer. Values are expressed as means ± SE, with n = 3 independent experiments. *P < 0.05, RA and RAS vs. HO. **P < 0.05, RA, RAS, and HOS vs. HO. αP < 0.05, RA vs. RAS. B: effects of hyperoxia and stretch on [3H]thymidine incorporation. A549 cells were exposed to RA, RAS, HO, and HOS for 24, 48, and 72 h. Cell proliferation was assessed as [3H]thymidine incorporation corrected for cell number. Cells were pulsed for 12 h with 1 μCi/ml [3H]thymidine and analyzed using a liquid scintillation analyzer. Values are expressed as means ± SE, with n = 3 independent experiments. *P < 0.05, RA, RAS, and HOS vs. HO. DPM, disintegrations per minute.
Effects of Hyperoxia and Stretch on Cell Viability
To determine whether stretch and hyperoxia affected cell viability, we used the calcein AM assay. As shown in Fig. 2, cells exposed to hyperoxia for 72 h displayed decreased green fluorescence, signifying decreased viability, compared with cells exposed to room air, room air with stretch, and hyperoxia with stretch (P < 0.05). There were no differences between groups after 24 and 48 h of exposure (data not shown).
Fig. 2.

Effects of hyperoxia and stretch on cell viability. A549 cells were exposed to RA, RAS, HO, and HOS for 24, 48, and 72 h. Cell suspensions were transferred to 96-well flat-bottomed microtiter plates, and 25 μM calcein AM was added to each well. Cells were incubated in the dark and analyzed using a spectrophotometer plate reader at an excitation wavelength of ~485 nm and an emission wavelength of ~530 nm. Methanol-exposed cells serve as positive controls for maximal cell death (lowest viability). Values are expressed as means ± SE, with n = 3 independent experiments. *P < 0.05, HO vs. RA, RAS, and HOS.
To determine the ratio of viable to dead cells, we performed LIVE/DEAD analysis. There was an increase in cell death with hyperoxia exposure without stretch after 24, 48, and 72 h compared with the other study groups (P < 0.05; Fig. 3A). Hyperoxia combined with stretch did result in significantly less cell death at all time points compared with hyperoxia alone (P < 0.05), including a reduction from 27 to 10% at 72 h. Representative fluorescent microscopic images demonstrating this attenuation of cell death during hyperoxia with stretch at 72 h are shown in Fig. 3B. The number of dead cells with hyperoxia and stretch were equivalent to both room air groups after 48 h but were greater at both 24 and 72 h (Fig. 3A.). Cell death was not increased by cyclic stretch with room air at any of the time points.
Fig. 3.

A: effects of hyperoxia and stretch on cell viability and death. A549 cells were exposed to RA, RAS, HO, and HOS for 24, 48, and 72 h. With the use of a LIVE/DEAD assay, cell suspensions were transferred to 96-well microtiter assay plates. Positive control wells were treated with 100 μl of methanol to induce maximal death. Ethidium homodimer-1 (EthD-1; 17 μM) and calcein AM (10 μM) were added to the wells, and the cells were incubated in the dark and then analyzed using fluorescence microscopy. Cell counts were made on the basis of the number of live cells (green) vs. dead cells (red) that were seen in random, noncontiguous fields until ~250 cells total were counted. Values are expressed as means ± SE, with n = 3 independent experiments. *P < 0.05, HO vs. RA, RAS, and HOS. **P < 0.05, HOS vs. RA and RAS. B: effects of hyperoxia and stretch on cell viability and death. Fluorescent microscopy images represent the following groups of A549 cells after 72 h of exposure to RA (a), RAS (b), HO (c), and HOS (d). Cells were stained with calcein AM, which produces a bright green fluorescence in viable cells, and EthD-1, which produces a red fluorescence reflective of cell death. Scale bar, 100 μm. Images represent typical findings from 3 independent studies.
To further assess cell injury and necrotic death, we used an LDH assay. LDH release was significantly increased following hyperoxic exposure after 48 and 72 h. The addition of cyclic stretch to hyperoxia attenuated LDH release at both of these time points, resulting in measured levels equivalent to room air exposed cells (P < 0.01, Fig. 4). Cyclic stretch under room air conditions did not alter LDH release at any of the time points.
Fig. 4.

Lactate dehydrogenase (LDH) release assay. A549 cells were exposed to RA, RAS, HO, and HOS for 24, 48, and 72 h. Samples were assayed for LDH release, a marker of cell death, in culture medium. Values are expressed as means ± SE, with n = 3 independent experiments. *P < 0.01, HO vs. RA, RAS, and HOS.
Apoptosis Assays
Because the A549 cells exposed to hyperoxia demonstrated increased cell death, we investigated the mode of death. To detect evidence of apoptosis, we performed the caspase-3 activity and annexin V-APC flow cytometry assays. Caspase-3 activity, which detects early stages of apoptosis (48), was not increased in the stretch-exposed room air cells or the hyperoxia-exposed stretched and unstretched cells compared with cells exposed to room air alone in any of the groups at 24, 48, or 72 h (data not shown).
The annexin V-APC assay, which detects the loss of plasma membrane asymmetry seen in early apoptosis (48), also did not demonstrate evidence of apoptosis in the stretch-exposed room air cells or the hyperoxia-exposed stretched and unstretched cells compared with cells exposed to room air alone. A representative flow cytometry analysis plot of the different exposure groups at 72 h for annexin V-APC binding and 7-ADD staining is shown in Fig. 5.
Fig. 5.

Determination of apoptosis and necrosis. Cells were stretched, harvested at 72 h, stained with annexin V-APC and 7-aminoactinomycin (7-ADD) and analyzed using flow cytometry. Plots of flow cytometry analysis represent the following groups: RA (A), RAS (B), HO (C), and HOS (D). Intensity of 7-ADD staining (y-axis) is plotted vs. annexin V-APC intensity (x-axis). In all 4 plots, viable cells appear at bottom left (quadrant 1, annexin V negative/7-ADD negative), early apoptotic cells at bottom right (quadrant 2, annexin V positive/7-ADD negative), late apoptotic/necrotic cells at top right (quadrant 3, annexin V positive/7-ADD positive), and necrotic cells at top left (quadrant 4, annexin V negative/7-ADD positive).
Figure 6A demonstrates the changes in the percentage of necrotic (annexin V-APC negative and 7-ADD positive) cells at 48 and 72 h. The percentage of necrotic cells was significantly increased with hyperoxia compared with room air groups at both 48 (P < 0.01) and 72 h (P < 0.01). Although cells exposed to hyperoxia with stretch also had increased necrosis at 72 h compared with room air cells (P < 0.05), necrosis was significantly decreased compared with cells exposed to hyperoxia without stretch (P < 0.01). Figure 6B demonstrates the changes in the percentage of late-apoptotic/necrotic (annexin V-APC positive and 7-ADD positive) cell death. The effects of hyperoxia and stretch on cell death in this subgroup were similar to those shown in Fig. 6A, including the finding of less cell death with hyperoxia combined with stretch compared with hyperoxia alone (P < 0.05).
Fig. 6.
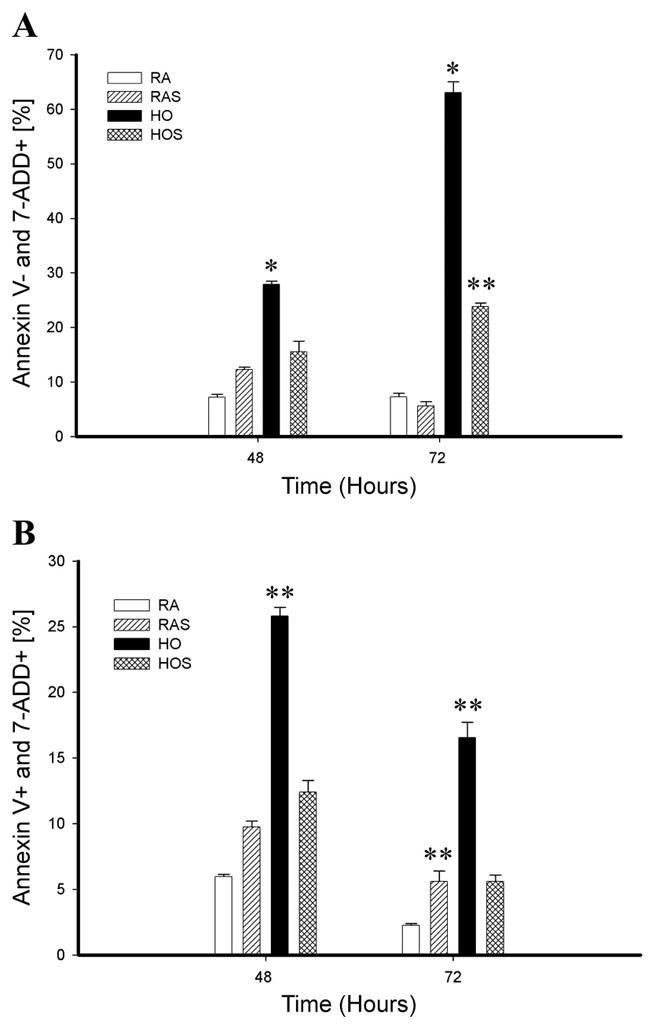
Annexin V-APC binding and 7-ADD staining. Flow cytometry data of A549 cells exposed to RA, RAS, HO, and HOS represent a comparison between groups at 48 and 72 h of exposure. A: the percentage of necrotic cells (annexin V-APC − and 7-ADD +). *P < 0.01, HO > RA, RAS, and HOS. **P < 0.05 HOS > RA and RAS. B: the percentage of late apoptotic/necrotic cells (annexin V-APC + and 7-ADD −). **P < 0.05, RAS > RA and HO > RA, RAS, and HOS.
The results of the annexin V-APC assay, in conjunction with the findings of the caspase-3 activity assay, suggest that cell death in A549 cells from hyperoxia occurs primarily via nonapoptotic mechanisms and that hyperoxia-induced cell death is attenuated with the addition of noninjurious cyclic stretch.
Effect of Hyperoxia and Stretch on Cell Morphology
Because hyperoxia may injure cells, we examined the morphological features of the different exposure groups by using light microscopy and hematoxylin and eosin staining. Features of apoptosis, such as cell shrinkage, prominent condensation of nuclear chromatin, and cell membrane blebbing, were not seen in any of the groups at any of the time points. As shown in Fig. 7, after 72 h of exposure, cells exposed to hyperoxia with stretch appeared less swollen and vacuolated compared with cells exposed to hyperoxia alone and more closely resembled the appearance of room air cells.
Fig. 7.
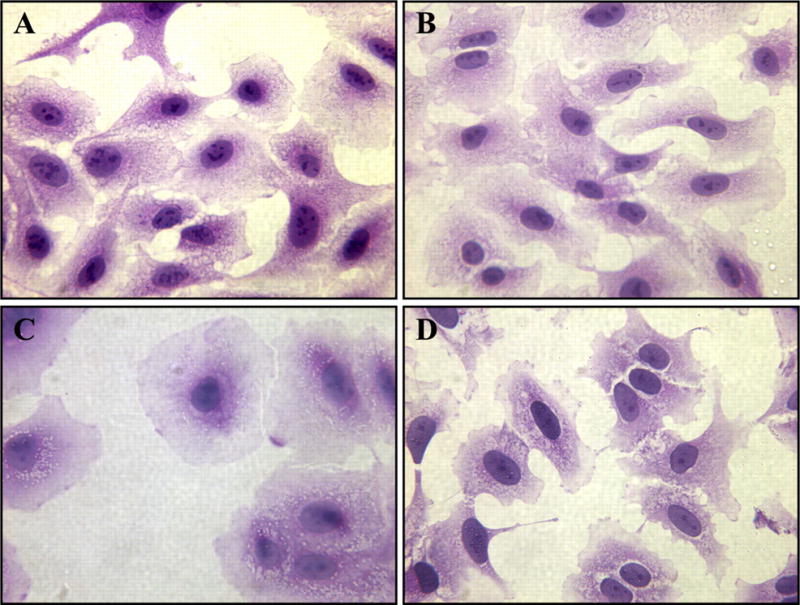
Effects of hyperoxia and stretch on A549 cell morphology. After 72 h of exposure to RA (A), RAS (B), HO (C), and HOS (D), A549 cells were stained with hematoxylin and eosin as described in METHODS. Scale bar, 50 μm.
Hyperoxia and Superoxide Production
To quantify superoxide levels in the different cells groups, we performed flow cytometry of DHE-stained cells. Representative flow cytometry histogram overlay plots of detected emission in the FL2 585/42 channel, measuring red fluorescence intensity of ethidium, are shown in Fig. 8. After 48 and 72 h, red fluorescence, indicating the presence of superoxide, was significantly increased in cells exposed to hyperoxia (27 and 42% FL2, respectively) compared with room air (5 and 6% FL2 positive, respectively; P < 0.05), room air with stretch (6% FL2 positive for both time points; P < 0.05), and hyperoxia with stretch (6 and 9% FL2 positive, respectively; P < 0.05). No differences in superoxide levels were noted between the room air groups and hyperoxia with stretch.
Fig. 8.

Superoxide detection with dihydroethidium (DHE) staining using flow cytometry. A549 cells were stained with DHE after 48 (A) and 72 h (B) of exposure to RA (thick shaded line), RAS (thin shaded line), HO (thick solid line), and HOS (thin solid line) to detect superoxide. Histogram overlay represents comparisons between groups after 24, 48, and 72 h of exposure. A right shift in the FL2 channel detecting orange-red fluorescence represents the presence of superoxide (nuclear binding of ethidium). P < 0.05, HO vs. RA, RAS, and HOS. P < 0.05, HOS vs. RA and RAS.
DISCUSSION
The treatment of respiratory failure in the developing and mature lung often necessitates prolonged exposure to both hyperoxia and mechanical stretch. The combined effects of oxygen toxicity and cyclic stretch on the distal pulmonary epithelium, however, have not been directly studied. With the use of an in vitro system simulating physiological distension, our data demonstrate that cyclic stretch attenuates the detrimental effects of hyperoxia on cell proliferation and nonapoptotic cell death in alveolar epithelial A549 cells. In addition, cyclic stretch coupled with prolonged hyperoxia resulted in decreased cellular superoxide levels, suggesting that stretch may be an important factor in reducing hyperoxia related injury.
Tschumperlin et al. (41), using isolated whole rat lung, correlated changes in lung volume to epithelial basement membrane surface area, establishing a reference for applying physiologically relevant deformations to pulmonary epithelial cells in vitro. They found minimal ΔSA at low lung volumes but observed significant changes at the limits of lung inflation. Specifically, ΔSA of 12, 25, 37, and 50% stretch corresponded to ~60, 80, 100, and >100% of total lung capacity (41, 42). Subsequent experiments showed that the loss of alveolar epithelial barrier integrity occurred with a ΔSA of 37%, but not 12 or 25%, and that cellular injury is amplitude dependent, significantly increasing when stretch exceeds 37–50% ΔSA (2, 43). Additional studies have shown that repetitive cyclic stretch at amplitudes of 22–30% can induce both apoptotic and necrotic cell death in primary rat pulmonary epithelial cells (7, 12).
Consistent with the physiological stretch regimen chosen for our experiment, we did not find any evidence of increased cellular injury and/or death in stretched A549 cells in room air for up to 72 h. We observed only a minimal proliferative effect with 16% stretch at 0.5 Hz under room air conditions, noting increased cell counts after 48 h without corresponding significant changes in [3H]thymidine incorporation. Other investigators using different cell lines, culture conditions, and patterns of stretch have demonstrated a more pronounced proliferative response in stretched pulmonary epithelium. Chess et al. (4) elicited a proliferative response in H441 cells using 20% stretch at 60 cycles/min, and Liu et al. (26, 27) noted a stretch-induced mitogenic response in primary fetal rat lung cells in several experiments using a three-dimensional strain model.
A novel finding from our study was the significant effect of cyclic stretch on cell proliferation and viability under hyperoxic conditions. We found that stretch increased cell number and [3H]thymidine incorporation during prolonged hyperoxic exposure compared with hyperoxia alone. A549 cells exposed to hyperoxia without stretch appeared swollen and vacuolated and were killed by nonapoptotic cell death. The addition of stretch resulted in decreased cell injury and death from hyperoxia, as demonstrated by LDH, calcein AM viability, LIVE/DEAD viability, and annexin V-APC flow cytometry assays. Together, these findings suggest that noninjurious stretch, under certain conditions, can help attenuate the detrimental effects of prolonged hyperoxia on pulmonary epithelial cells.
There are a number of cellular responses known to occur after stretch in pulmonary epithelium that may have contributed, at least in part, to the observations seen in our study. Under room air conditions, mechanical stretch has been shown to induce the phosphorylation of both tyrosine and non-tyrosine kinase receptors, adenylate cyclase, guanylate cyclase, phospholipases A2 and C, and Ca2+ influx, resulting in a complex intracellular cascade of events (17, 18, 50). Mechanical stretch also has been shown to result in conformational changes of integrins leading to the activation of focal adhesion kinase and the mitogen-activated protein kinase (MAPK) pathway, which is involved in the control of cellular functions such as cell proliferation, inflammation, and programmed cell death (3, 49). In turn, the MAPK pathway signaling cascades may transduce the stretch-induced signal to the nucleus and stimulate gene transcription (30). How varying levels of stretch may alter intracellular signaling pathways in lung epithelium under hyperoxic and/or room air conditions, including effects on cell proliferation and viability, remains poorly understood and is the subject of ongoing investigation. Zhou et al. (52) recently have shown in intestinal epithelial cells that multiple signal transduction pathways are activated during oxidative stress-induced injury. Specifically, they found that phosphatidylinositol 3-kinase/Akt appears to serve as an important protective pathway during oxidative stress (52).
In an effort to characterize the level of oxygen toxicity induced in A549 cells after prolonged exposure to hyperoxia, we used DHE staining to measure superoxide levels. We were able to detect the presence of superoxide under hyperoxic, but not room air, conditions. Of interest, cyclic stretch significantly decreased the amount of superoxide present after 48 and 72 h of hyperoxia. It is possible that the potential beneficial effects of cyclic stretch observed in our study in A549 cells, including improved proliferation and viability during hyperoxia, may be related to a decreased presence of oxidative stress. Whether mechanical stretch can actually alter cellular superoxide levels, however, and if so by what mechanism, needs to be further explored.
Physiological levels of stretch may directly inhibit ROS generators or perhaps upregulate the expression of cellular antioxidant defenses, such as superoxide dismutase, glutathione, and/or catalase. Several recent studies have shown that the overexpression of antioxidant enzymes in pulmonary epithelial cells protects against oxidant injury (16, 22). Given that ROS have been shown to play a role in cell signaling through a variety of intracellular pathways, including proliferation and death, it is further possible that cyclic stretch may impact these pathways indirectly via alteration of ROS levels (20, 28). Regardless of its mechanism of action, the effect of mechanical stretch as previously discussed likely depends on the degree and duration of exposure, as well as the nature of the cellular conditions being studied.
In summary, we have shown that cyclic stretch, as applied under our experimental conditions, decreases the detrimental effects of hyperoxia on cell proliferation, nonapoptotic cell death, and oxidative stress in A549 cells. Further investigation is needed to elucidate the molecular mechanisms involved in stretch-induced mechanotransduction to determine how stretch modifies pulmonary epithelial responses under room air and hyperoxic conditions.
Acknowledgments
We thank Dr. Victoria Centonze Frohlich and Mark W. Blaylock for help with the images acquired in the Core Optical Imaging Facility, University of Texas Health Science Center, San Antonio, TX.
Footnotes
DISCLOSURES
The opinions expressed in this paper are solely those of the authors and do not represent the views of the United States Air Force, Department of Defense, or the United States Government.
References
- 1.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 2.Cavanaugh KJ, Jr, Margulies SS. Measurement of stretch-induced loss of alveolar epithelial barrier integrity with a novel in vitro method. Am J Physiol Cell Physiol. 2002;283:C1801–C1808. doi: 10.1152/ajpcell.00341.2002. [DOI] [PubMed] [Google Scholar]
- 3.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 4.Chess PR, Toia L, Finkelstein JN. Mechanical strain-induced proliferation and signaling in pulmonary epithelial H441 cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L43–L51. doi: 10.1152/ajplung.2000.279.1.L43. [DOI] [PubMed] [Google Scholar]
- 5.Dos Santos CC, Slutsky AS. Mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol. 2000;89:1645–1655. doi: 10.1152/jappl.2000.89.4.1645. [DOI] [PubMed] [Google Scholar]
- 6.Edwards YS. Stretch stimulation: its effects on alveolar type II cell function in the lung. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:267–285. doi: 10.1016/s1095-6433(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 7.Edwards YS, Sutherland LM, Power JH, Nicholas TE, Murray AW. Cyclic stretch induces both apoptosis and secretion in rat alveolar type II cells. FEBS Lett. 1999;448:127–130. doi: 10.1016/s0014-5793(99)00357-9. [DOI] [PubMed] [Google Scholar]
- 8.Freeman BA, Crapo JD. Hyperoxia increases oxygen radical production in rat lungs and lung mitochondria. J Biol Chem. 1981;256:10986–10992. [PubMed] [Google Scholar]
- 9.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 10.Fujiwara K. Mechanical stresses keep endothelial cells healthy: beneficial effects of a physiological level of cyclic stretch on endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2003;285:L782–L784. doi: 10.1152/ajplung.00142.2003. [DOI] [PubMed] [Google Scholar]
- 11.Gertzberg N, Neumann P, Rizzo V, Johnson A. NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-α. Am J Physiol Lung Cell Mol Physiol. 2004;286:L37–L48. doi: 10.1152/ajplung.00116.2003. [DOI] [PubMed] [Google Scholar]
- 12.Hammerschmidt S, Kuhn H, Grasenack T, Gessner C, Wirtz H. Apoptosis and necrosis induced by cyclic mechanical stretching in alveolar type II cells. Am J Respir Cell Mol Biol. 2004;30:396–402. doi: 10.1165/rcmb.2003-0136OC. [DOI] [PubMed] [Google Scholar]
- 13.Hammerschmidt S, Kuhn H, Sack U, Schlenska A, Gessner C, Gillissen A, Wirtz H. Mechanical stretch alters alveolar type II cell mediator release toward a proinflammatory pattern. Am J Respir Cell Mol Biol. 2005;33:203–210. doi: 10.1165/rcmb.2005-0067OC. [DOI] [PubMed] [Google Scholar]
- 14.Hecht D, Zick Y. Selective inhibition of protein tyrosine phosphatase activities by H2O2 and vanadate in vitro. Biochem Biophys Res Commun. 1992;188:773–779. doi: 10.1016/0006-291x(92)91123-8. [DOI] [PubMed] [Google Scholar]
- 15.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 16.Ilizarov AM, Koo H, Kazzaz JA, Mantell LL, Yuchi L, Bhapat R, Pollack S, Horowitz S, Davis JM. Overexpression of manganese superoxide dismutase protects lung epithelial cells against oxidant injury. Am J Respir Cell Mol Biol. 2001;24:436–441. doi: 10.1165/ajrcmb.24.4.4240. [DOI] [PubMed] [Google Scholar]
- 17.Ingram AJ, Ly H, Thai K, Kang M, Scholey JW. Activation of mesangial cell signaling cascades in response to mechanical strain. Kidney Int. 1999;55:476–485. doi: 10.1046/j.1523-1755.1999.00276.x. [DOI] [PubMed] [Google Scholar]
- 18.Johnson GL, Vaillancourt RR. Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol. 1994;6:230–238. doi: 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 19.Jyonouchi H, Sun S, Abiru T, Chareancholvanich S, Ingbar D. The effects of hyperoxic injury and antioxidant vitamins on death and proliferation of human small airway epithelial cells. Am J Respir Cell Mol Biol. 1998;19:426–436. doi: 10.1165/ajrcmb.19.3.2862m. [DOI] [PubMed] [Google Scholar]
- 20.Kazzaz JA, Horowitz S, Li Y, Mantell LL. Hyperoxia in cell culture. A non-apoptotic programmed cell death. Ann NY Acad Sci. 1999;887:164–170. doi: 10.1111/j.1749-6632.1999.tb07930.x. [DOI] [PubMed] [Google Scholar]
- 21.Kazzaz JA, Xu J, Palaia TA, Mantell L, Fein AF, Horowitz S. Cellular oxygen toxicity-oxidant injury without apoptosis. J Biol Chem. 1996;271:15182–15186. doi: 10.1074/jbc.271.25.15182. [DOI] [PubMed] [Google Scholar]
- 22.Koo H, Davis JM, Li Y, Hatzis D, Opsimos H, Pollack S, Strayer MS, Ballard PL, Kazzaz JA. Effects of transgene expression of superoxide dismutase and glutathione peroxidase on pulmonary epithelial cell growth in hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2005;288:L718–L726. doi: 10.1152/ajplung.00456.2003. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Arita Y, Koo HC, Davis JM, Kazzaz JA. Inhibition of c-Jun N-terminal kinase pathway improves cell viability in response to oxidant injury. Am J Respir Cell Mol Biol. 2003;29:779–783. doi: 10.1165/rcmb.2003-0087RC. [DOI] [PubMed] [Google Scholar]
- 24.Liu M, Skinner SJ, Xu J, Han RN, Tanswell AK, Post M. Stimulation of fetal rat lung cell proliferation in vitro by mechanical stretch. Am J Physiol Lung Cell Mol Physiol. 1992;263:L376–L383. doi: 10.1152/ajplung.1992.263.3.L376. [DOI] [PubMed] [Google Scholar]
- 25.Liu M, Tanswell AK, Post M. Mechanical force-induced signal transduction in lung cells. Am J Physiol Lung Cell Mol Physiol. 1999;277:L667–L683. doi: 10.1152/ajplung.1999.277.4.L667. [DOI] [PubMed] [Google Scholar]
- 26.Liu M, Xu J, Souza P, Tanswell B, Tanswell AK, Post M. The effect of mechanical strain on fetal rat lung cell proliferation: comparison of two- and three-dimensional culture systems. In Vitro Cell Dev Biol Anim. 1995;31:858–866. doi: 10.1007/BF02634570. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Tanswell AK, Post M. Stretch-induced growth-promoting activities stimulate fetal rat lung epithelial cell proliferation. Exp Lung Res. 1993;19:505–517. doi: 10.3109/01902149309064360. [DOI] [PubMed] [Google Scholar]
- 28.Manasija-Radisavljevic Z, Gonzaliez-Flecha B. Signaling through Cdk2, importin-α, and NuMA is required for H2O2-induced mitosis in primary type II pneumocytes. Biochim Biophys Acta. 2003;1640:163–170. doi: 10.1016/s0167-4889(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 29.Mantell LL, Horowitz S, Davis JM, Kazzaz JA. Hyperoxia-induced cell death in the lung—the correlation of apoptosis, necrosis, and inflammation. Ann NY Acad Sci. 1999;887:171–180. doi: 10.1111/j.1749-6632.1999.tb07931.x. [DOI] [PubMed] [Google Scholar]
- 30.Oldenhof AD, Shynlova OP, Liu M, Langille BL, Lye SJ. Mitogen-activated protein kinases mediate stretch-induced c-fos mRNA expression in myometrial smooth muscle cells. Am J Physiol Cell Physiol. 2002;283:C1530–C1539. doi: 10.1152/ajpcell.00607.2001. [DOI] [PubMed] [Google Scholar]
- 31.Otterbein LE, Chin BY, Mantell LL, Stansberry L, Horowitz S, Choi AMK. Pulmonary apoptosis in aged and oxygen-tolerant rats exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 1998;275:L14–L20. doi: 10.1152/ajplung.1998.275.1.L14. [DOI] [PubMed] [Google Scholar]
- 32.Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann NY Acad Sci. 2003;1010:405–416. doi: 10.1196/annals.1299.074. [DOI] [PubMed] [Google Scholar]
- 33.Pugin J. Molecular mechanisms of lung cell activation induced by cyclic stretch. Crit Care Med. 2003;31:S200–S206. doi: 10.1097/01.CCM.0000057844.31307.ED. [DOI] [PubMed] [Google Scholar]
- 34.Roper JM, Mazzatti DJ, Watkins RH, Maniscalco WM, Keng PC, O’Reilly MA. In vivo exposure to hyperoxia induces DNA damage in a population of alveolar type II epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1045–L1054. doi: 10.1152/ajplung.00376.2003. [DOI] [PubMed] [Google Scholar]
- 35.Salva U, Olson LE, Waters CM. Mathematical modeling of airway epithelial wound closure during cyclic mechanical strain. J Appl Physiol. 2004;96:566–574. doi: 10.1152/japplphysiol.00510.2003. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Estaban J, Cicchiello LA, Wang Y, Tsai SW, Williams LK, Torday JS, Rubin LP. Mechanical stretch promotes alveolar epithelial type II cell differentiation. J Appl Physiol. 2001;91:589–595. doi: 10.1152/jappl.2001.91.2.589. [DOI] [PubMed] [Google Scholar]
- 37.Schumacker PT. Straining to understand mechanotransduction in the lung. Am J Physiol Lung Cell Mol Physiol. 2002;282:L881–L882. doi: 10.1152/ajplung.00043.2002. [DOI] [PubMed] [Google Scholar]
- 38.Shaikh AY, Xu J, Wu Y, He L, Hsu CY. Melatonin protects bovine cerebral endothelial cells from hyperoxia-induced DNA damage and death. Neurosci Lett. 1997;229:193–197. doi: 10.1016/s0304-3940(97)00307-8. [DOI] [PubMed] [Google Scholar]
- 39.Skinner SJ, Somervell CE, Olson DM. The effects of mechanical stretching on fetal rat lung cell prostacyclin production. Prostaglandins. 1992;43:413–433. doi: 10.1016/0090-6980(92)90125-d. [DOI] [PubMed] [Google Scholar]
- 40.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 41.Tschumperlin DJ, Margulies SS. Equibiaxial deformation-induced injury of alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol. 1998;275:L1173–L1183. doi: 10.1152/ajplung.1998.275.6.L1173. [DOI] [PubMed] [Google Scholar]
- 42.Tschumperlin DJ, Margulies SS. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J Appl Physiol. 1999;86:2026–2033. doi: 10.1152/jappl.1999.86.6.2026. [DOI] [PubMed] [Google Scholar]
- 43.Tschumperlin DJ, Oswari J, Margulies SS. Deformation-induced injury of alveolar epithelial cells. Effect of frequency, duration, and amplitude. Am J Respir Crit Care Med. 2000;162:357–362. doi: 10.1164/ajrccm.162.2.9807003. [DOI] [PubMed] [Google Scholar]
- 44.Ueda N, Shah SV. Endonuclease-induced DNA damage and cell death in oxidant injury to renal tubular epithelial cells. J Clin Invest. 1992;90:2593–2597. doi: 10.1172/JCI116154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uhlig S. Ventilation-induced lung injury and mechanotransduction: stretching it too far? Am J Physiol Lung Cell Mol Physiol. 2002;282:L892–L896. doi: 10.1152/ajplung.00124.2001. [DOI] [PubMed] [Google Scholar]
- 46.Vandenburgh HH. Mechanical forces and their second messengers in stimulating cell growth in vitro. Am J Physiol Regul Integr Comp Physiol. 1992;262:R350–R355. doi: 10.1152/ajpregu.1992.262.3.R350. [DOI] [PubMed] [Google Scholar]
- 47.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 48.Vermes I, Haanen C, Reutelingsperger C. Flow cytometry of apoptotic cell death. J Immunol Methods. 2000;243:167–190. doi: 10.1016/s0022-1759(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 49.Wang JG, Miyazu M, Matsushita E, Sokabe M, Naruse K. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem Biophys Res Commun. 2001;288:356–361. doi: 10.1006/bbrc.2001.5775. [DOI] [PubMed] [Google Scholar]
- 50.Wasilenko WJ. Phosphoinositides and cell growth. Adv Exp Med Biol. 1992;321:147–151. doi: 10.1007/978-1-4615-3448-8_16. [DOI] [PubMed] [Google Scholar]
- 51.Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 1990;250:1266–1269. doi: 10.1126/science.2173861. [DOI] [PubMed] [Google Scholar]
- 52.Zhou Y, Wang Q, Evers BM, Chung DH. Signal transduction pathways involved in oxidative stress-induced intestinal epithelial cell apoptosis. Pediatr Res. 2005;58:1192–1198. doi: 10.1203/01.pdr.0000185133.65966.4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuo L, Clanton TL. Detection of reactive oxygen and nitrogen species in tissues using redox-sensitive fluorescent probes. Methods Enzymol. 2002;352:307–325. doi: 10.1016/s0076-6879(02)52028-0. [DOI] [PubMed] [Google Scholar]