

Abstract
The process of HIV assembly requires extensive homomultimerization of the Gag polyprotein on cellular membranes to generate the nascent particle bud. Here we generated a full-length, monomeric Gag polyprotein bearing mutations that eliminated multimerization in living cells as indicated by fluorescence resonance energy transfer (FRET). Monomeric Gag resembled non-myristoylated Gag in its weak membrane binding characteristics and lack of association with detergent-resistant membranes (DRMs or lipid rafts). Monomeric Gag failed to assemble virus-like particles, but was inefficiently rescued into particles by wildtype Gag through the influence of the matrix domain. The subcellular distribution of monomeric Gag was remarkably different than either non-myristoylated Gag or wildtype Gag. Monomeric Gag was found on intracellular membranes and at the plasma membrane, where it induced the formation of plasma membrane extensions and ruffles. This study indicates that monomeric Gag can traffic to assembly sites in the cell, where it interacts weakly with membranes.
Keywords: HIV Assembly, multimerization, myristoylation, Gag, protein trafficking
INTRODUCTION
HIV-1 Pr55Gag (Gag) is a 55 kilodalton, myristoylated precursor polyprotein that forms the structural core of the developing viral particle. Gag is translated on cytoplasmic ribosomes, and subsequently must translocate to the plasma membrane of cells for productive assembly. Assembling particle buds appear by electron microscopy as dense patches on the plasma membrane (Gelderblom, 1991), representing a focal site of homomultimerization of Gag on the cytoplasmic face of the membrane. Plasma membrane curvature is elicited as multimerization progresses, until a complete (or apparently complete) spherical immature core has been formed. During or immediately following particle budding, the viral protease cleaves Gag and core maturation occurs, resulting in the formation of a conical viral core. Three major Gag cleavage products are formed in this maturation process; MA, CA, and NC, along two smaller peptides, SP1 and SP2. Within the mature viral particle, the myristoylated, N-terminal MA domain remains associated with the lipid envelope, while CA forms the shell of the mature capsid and NC remains associated with the viral RNA within the capsid structure.
Cryo-electron microscopic analysis of immature retroviral particles has shown that Gag molecules are arranged in a radial fashion, with the membrane binding MA domain in contact with the lipid envelope and the C-terminal ends pointing toward the center of the particle (Wilk et al., 2001). The Gag proteins of HIV, Rous sarcoma virus (RSV), and murine leukemia virus (MLV) have each been shown to form a spherical lattice composed of hexameric Gag rings (Briggs et al., 2006; Briggs et al., 2004; Fuller et al., 1997; Wilk et al., 2001; Yeager et al., 1998). Lateral Gag-Gag interactions within this lattice are critical to the formation and stability of the immature virion core. Several crucial contact areas between uncleaved Gag molecules have been identified. Biochemical and mutagenesis studies support the presence of critical interactions between Gag molecules in the C-terminal domain (CTD) of CA (Accola, Strack, and Gottlinger, 2000; Borsetti, Ohagen, and Gottlinger, 1998; von Schwedler et al., 1998). Mutations in key residues within the dimer interface of the CTD present in mature capsids disrupt the production of immature particles, suggesting that this interface is important in both structures (Datta et al., 2007a; von Schwedler et al., 2003). N-terminal domain (NTD) interactions have also been suggested to play a role within the immature lattice (Lanman et al., 2003; von Schwedler et al., 2003). Functional studies that preceded detailed structural studies revealed an important contribution of NC to multimerization and assembly, a contribution termed the “I” or interaction domain (Derdowski, Ding, and Spearman, 2004; Parent et al., 1995; Sandefur et al., 2000; Sandefur, Varthakavi, and Spearman, 1998; Weldon and Wills, 1993). The functional contribution of NC to assembly is likely due to its ability to bind nucleic acids, acting as a tether or nucleation point for Gag-Gag multimerization (Campbell and Rein, 1999; Campbell and Vogt, 1995). However, electron cryotomography has revealed that the NC region is not highly ordered in immature particles, while the CA NTD, CA CTD, and SP1 demonstrate clear hexagonal order (Wright et al., 2007). This study demonstrated that the CA and SP1 regions form a tightly-packed lattice of cup-shaped hexamers (Wright et al., 2007). A model for Gag-Gag interactions in immature particles is therefore becoming clearer, although defining the precise contacts between Gag monomers in the hexameric lattice of the immature particle will require additional, higher resolution studies.
Multimerization of Gag may begin at intracellular sites distant from the particle budding site. Distinct structural intermediates of Gag have been described (Dooher and Lingappa, 2004; Lee and Yu, 1998; Morikawa, Goto, and Momose, 2004; Morikawa et al., 1998) but remain to be firmly established. Oligomers of Gag may exhibit distinct characteristics contributing to particle assembly, such as distinct intracellular trafficking. To begin to approach this question, it would be useful to fully characterize the Gag monomer. We recently reported the use of a fluorescence resonance energy transfer (FRET)-based assay in living cells to define critical interfaces involved in assembly of the immature HIV particle (Li et al., 2007). Surprisingly, myristoylation was required for Gag-Gag interactions in this study, suggesting that membrane interactions conferred by the myristoylated N-terminus of MA are important for multimerization in cells. By combining amino acid substitutions within the NTD and CTD of CA with extensive substitution of basic residues in NC, we generated a myristoylated, monomeric Gag molecule. Here we characterize a full-length, myristoylated, monomeric Gag molecule expressed in mammalian cells. The protein is unable to form particles, but can be rescued in an inefficient manner into particles in a manner dependent upon the MA domain. Monomeric Gag is able to traffic to the plasma membrane, interacts weakly with cellular membranes, and exhibits a subcellular distribution pattern that is markedly distinct from that of wildtype Gag.
RESULTS
Characterization of a full-length monomeric Gag-CFP/YFP fusion protein
We previously used FRET to define domains and specific contacts contributing to Gag-Gag multimerization (Li et al., 2007). Surprisingly, multimerization was completely absent when paired Gag-CFP/YFP constructs were expressed lacking N-terminal myristic acid. In the context of myristoylated Gag-CFP/YFP, individual targeted substitutions within the NTD (M39A) or CTD (W184A/M185A) diminished but did not completely disrupt interaction. However, a combination of substitutions in the NTD (M39A), CTD (W184A/185A), and NC (NC15A, consisting of 15 substitutions of alanine for basic residues) resulted in complete loss of FRET. In our previous study, the substitutions were performed in the absence of MA and p6. In order to determine if these regions contributed to Gag-Gag interactions, we introduced these same substitutions into a full-length Gag protein construct and analyzed its ability to multimerize along with a number of controls.
The myristoylated Gag truncation construct SrcCANC demonstrated a substantial amount of FRET, indicated by a strong peak emission at 533 nm, while Myr(−) SrcCANC produced only a CFP emission curve (Fig. 1A, compare open circles with open triangles). Substitution of residues at the dimer interface alone (W184A/M185A) in the context of SrcCANC resulted in diminished FRET, but did not completely abolish Gag-Gag interactions (Fig. 1A open diamonds). In contrast, introduction of additional NTD and NC substitutions [SrcCA(M39A/W184A/M185A)NC15A] completely abolished FRET (Fig. 1A, “X”). These results recapitulated our previous findings (Li et al., 2007), and this construct will be referred to in this work as DMADP6 monomeric Gag. We next evaluated these same substitutions in the context of a full length Gag-CFP/Gag-YFP pair of constructs. The full-length construct [MACA(M39A/W184A/M185A)NC15AP6] similarly showed CFP fluorescence but lacked a discernable FRET peak at 533 nm (Fig 1B, “X”). These results indicated that MA and p6 did not restore multimerization to the monomeric truncated protein. The full-length, multiply-substituted construct will be referred to as full-length monomeric Gag in this report. Note that in each case the YFP fluorescence of the YFP binding partner was similar in level of expression as indicated by excitation at 513 and emission at 533 (data not shown).
Fig. 1. Emission scans representing FRET assay for the indicated pairs of Gag-CFP/YFP constructs.

SrcCANC (open circles) serves as a positive FRET control in both panels, Myr(−) SrcCANC (triangles) as a negative control that illustrates a CFP curve. Total YFP emission values are not shown in this figure but were measured separately, and were of approximately equal intensity for each comparison. (A) SrcCANC(M39A/W184A/M185A)NC15A pair fails to generate FRET (“X”). (B) Matrix and p6 regions do not restore FRET to monomeric Gag (curve marked by “X”). Data were obtained on a PTI scanning cuvette fluorometer following excitation at 433nm.
Myristic acid labelling of Gag constructs
Because lack of myristoylation had a dramatic effect in our FRET-based multimerization assay, we wished to confirm that the monomeric Gag proteins were myristoylated. We labeled these and control constructs with 3H-myristic acid and examined myristoylation after immunoprecipitation of Gag from cell lysates. Fig. 2A shows that wildtype Gag, monomeric Gag, and DMADP6 monomeric Gag were each myristoylated. The labeling was specific, as myristoylation-deficient constructs failed to take up the label but were present on Western blot (Fig. 1A and 1B). We conclude that the monomeric forms of Gag described here were appropriately myristoylated,
Fig. 2. Myristoylation of monomeric Gag constructs.

(A) 3H-myristic acid labeling of wildtype and monomeric Gag. Gag proteins were labeled for 8 hours, then harvested and immunoprecipitated with HIV patient sera and analyzed by SDS-PAGE and autoradiography. (B) A fraction of the lysates were analyzed by Western blotting using CA183 monoclonal antibody to demonstrate that all Gag proteins were produced in this experiment.
Monomeric Gag is deficient in formation of virus-like particles (VLPs)
Because Gag must generate a lattice made of homomultimers in the developing particle, it should not be possible for a monomeric Gag molecule to form VLPs. In order to evaluate our candidate monomeric Gag for particle formation, we compared the cellular and supernatant p24 content after transfection of 293T cells with wildtype or monomeric Gag. At 36 hours after transfection, cells and supernatants were harvested and assessed for p24 antigen content. A significant amount of particle release was apparent from wildtype but not monomeric Gag (Fig. 3A, S lanes). The intracellular levels of Gag were higher for monomeric Gag, consistent with a defect in particle production. A very small amount of Gag was released into the supernatant by cells expressing monomeric Gag, constituting just over 2% of the total Gag produced (Fig. 1A, right panel). In order to determine if this inefficiently released Gag was present in the form of VLPs, we analyzed the supernatants by pelleting particulate material through a 20% sucrose cushion, followed by equilibrium centrifugation on linear sucrose gradients. Wildtype Gag attained a peak density of 1.155 grams/ml (Fig. 3B). When performed in parallel and plotted on the same scale, no monomeric Gag was apparent at particle densities (Fig. 3B, left panel). However, a small amount of light Gag was detected at a density of 1.138 grams/ml (Fig. 3B, right panel, note differences in scale on y-axis). The nature of the inefficiently released, light Gag in this experiment is unclear. This density is not dissimilar from microvesicle density, although the sharpness of the peak would not be characteristic of microvesicles. We cannot rule out the possibility that very inefficient formation of light particles may occur through remaining weak Gag-Gag interactions not detected by FRET. However, it seems more likely to us that a fraction of the myristoylated monomer is exiting the cell in a membrane-bound form other than a true VLP, perhaps through release of some of the plasma membrane extensions induced upon expression of monomeric Gag (discussed below).
Fig. 3. Comparison of particle production by wildtype and monomeric Gag protein.
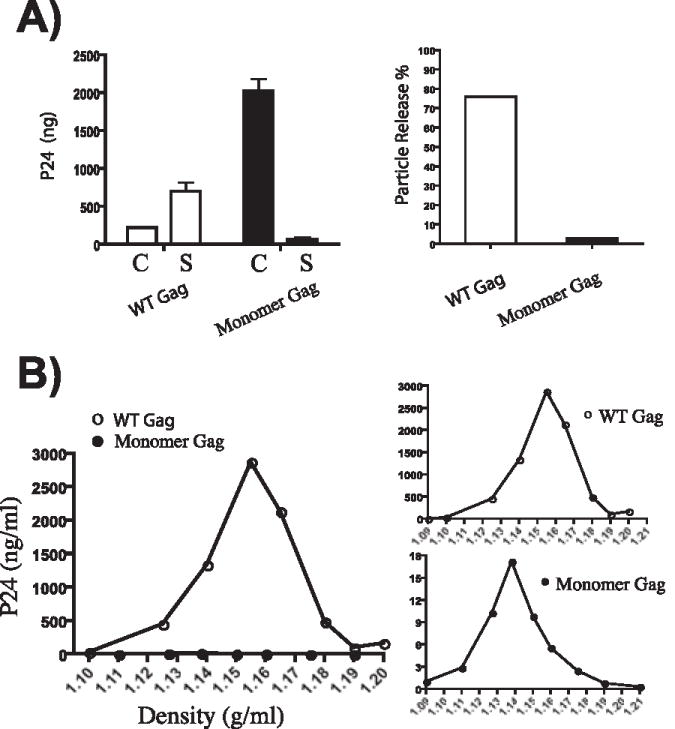
(A) Wildtype (open bars) and monomeric Gag (closed bars) were expressed in 293T cells. Cells and supernatants were harvested at 24 hours post-transfection, and the p24 antigen present in cells (C) or supernatants (S) was measured by antigen capture ELISA. Results are presented as total p24 (left) and as percentage of particle release (right). (B) Pelleted supernatants were analyzed after equilibrium density centrifugation on linear 20–60% sucrose gradients. Note that monomer Gag was not detectable on the same scale as wildtype Gag (left panel, closed vs. open circles). Using a different scale, a peak of p24 antigen was detected at a low particle density (1.138 g/ml, bottom gradient).
Analysis of particle formation by transmission electron microscopy
In order to directly address the potential formation of particles by monomeric Gag, we performed transmission electron microscopic analysis of cells expressing this molecule and compared them with those from wildtype Gag. HeLa cells were used in this analysis, and cells were fixed at 24 hours after transfection. Protein levels on a per-cell basis were similar in this experiment and in those reported for 293T cells, although transfection efficiency was slightly lower in HeLa (data not shown). At least two grids from each of three separate experiments were analyzed. Immature particle formation was easily detected in every case for wildtype Gag (Fig. 4A and 4B). Despite scanning the majority of cell sections present on each grid for particle formation, encompassing sections of several hundred total cells, none was seen in cells expressing monomeric Gag (representative cell shown in Fig. 4C and 4D). Notably, despite high cellular concentrations of protein, we were unable to detect intracellular particle formation by monomeric Gag. Some cells in the sections from monomeric Gag-expressing cells exhibited long cellular extensions (Fig. 4C), which may correlate with findings presented below from fluorescence microscopy.
Fig. 4. Transmission electron microscopic analysis of particle production.

(A) Wildtype Gag particles were easily detected in all fields; shown is one representative image at low power (A, bar = 2.4 mm) and higher magnification from the boxed region is shown below (B, bar = 305 nm). (C) No virus-like particles were seen in cells expressing monomeric Gag. Shown is a representative cell, with prominent cell ruffles and filamentous extensions of the plasma membrane. Bar = 3.2 mm in C, 750 nm in D.
Membrane binding is inefficient, and detergent-resistant complexes are not formed by monomeric Gag
Membrane binding of myristoylated monomeric Gag might be expected to resemble binding of MA, which has low intrinsic affinity for negatively-charged membranes as a monomer (Dalton et al., 2007). To analyze the membrane binding of monomeric Gag, we performed equilibrium flotation centrifugation of cytosolic and membrane components of lysed cells expressing wildtype Gag, Myr(−) Gag, monomeric Gag, and DMADP6 monomeric Gag. In this experiment, proteins were expressed with a C-terminal YFP tag and Gag was quantified in each fraction by fluorescence intensity. Gradients were 50%/40%/10% step gradients of iodixanol as performed previously (Ding et al., 2003). Wildtype Gag associated with membranes efficiently, as evidenced by peak fluorescence at the 40%/10% interface (fraction 4, Fig. 5A). Myr(−) Gag also behaved as expected, with predominantly cytosolic signal and a small amount of membrane-bound protein. Both monomeric Gag proteins showed an intermediate phenotype. The peak of Gag in the membrane layer (fraction 4) was stronger than that for myr(−), but much weaker than wildtype Gag. We interpret these findings to mean that a lack of multimerization does not allow cooperative effects that are necessary for the efficient membrane binding seen with the wildtype. Next, we asked if monomeric forms of Gag can form detergent-resistant complexes (DRCs). This assay involves treatment of cell lysates with 1% Triton X-100 at 4 °C, and is identical to that used by some investigators to indicate lipid raft or “barge” association of Gag (Lindwasser and Resh, 2001; Ono and Freed, 2001). Alternatively, Ono and colleagues reported that iodixanol gradients (but not sucrose gradients) allow the flotation of detergent-resistant Gag multimers that are not membrane-associated as indicated by resistance to octylglucoside extraction (Ono et al., 2005). Wildtype Gag was present in the DRC fraction, while myr(−) and both monomeric forms of Gag were absent (Fig. 5B). We conclude that in this way monomeric Gag resembles myr(−) Gag; it weakly associates with membranes and cannot form DRCs. Note that the inability to form DRCs may indicate the lack of formation of a “floatable” Gag multimer, consistent with the findings of Ono and colleagues for iodixanol flotation gradients, or in some interpretations would indicate the inability of monomeric Gag to associate with lipid rafts or detergent-resistant membranes.
Fig. 5. Membrane flotation and detergent-resistant membrane formation.

(A) Membrane flotation in the absence of detergent. Cell lysates were layered onto the bottom of a 50%–40%–10% iodixanol gradient, and equilibrium flotation centrifugation was carried out to separate membranes (40%–10% interface, fraction 4) from cytosol (fraction 10–12), using YFP fluorescence as a marker of total Gag protein in the membrane or cytosol. (B) Flotation following treatment of cell lysates with 1% TX-100 on ice for 30 minutes. Fraction 4 represents Gag-YFP on detergent-resistant membranes or putative rafts. Top of gradients is to the left.
Monomeric Gag is not rescued into complexes when co-expressed with wildtype Gag
Myr(−) forms of Gag do not themselves form particles, but can be rescued into particles when co-expressed with wildtype, myristoylated Gag. Thus, the ability to rescue mutated Gag into complexes or into VLPs can be used as a means of mapping Gag-Gag interactions (Burniston et al., 1999). To determine if sufficient capacity for interaction remained in the monomeric Gag construct to support rescue, we co-expressed wildtype Gag-CFP with a myr(−) version of monomeric Gag-YFP and assessed for rescue of complexes by FRET. Fig. 6A shows that wildtype Gag-CFP efficiently rescues myr(−) Gag-YFP, as indicated by the FRET peak at 533 nm (open triangles). Myr(−) Gag-YFP proteins bearing substitutions that had diminished but not negated FRET in the context of a homologous pairing demonstrated an intermediate amount of rescue (normalizing for CFP fluorescence). These included Gag proteins bearing substitutions in CA but an intact NC [myr(−)CA(M39A/W184A/M185A)NC and myr(−)CA(W184A/M185A)NC]. Myr(−) Monomeric Gag (open diamonds) and Myr(−) DMADP6 monomeric Gag were not rescued into functional FRET pairs when co-expressed with wildtype Gag-CFP. These results support the conclusion that CA-CA interactions and NC-RNA interactions were sufficiently disrupted by the substitutions in both monomeric constructs to prevent interaction and rescue by the wildtype molecule.
Fig. 6. FRET rescue experiments.

The ability of wildtype Gag-CFP to interact with (rescue) a series of test constructs expressed as YFP fusions was assessed by scanning cuvette fluorometry as described in Materials and Methods. Wildtype Gag-YFP (triangles) serves as positive control. (A) Myr(−) full-length monomeric Gag-YFP and the indicated constructs were assessed for interaction with Gag-CFP by FRET. (B) Myr(+) full-length monomeric Gag-YFP and other indicated constructs were assessed for interaction with wildtype Gag-CFP by FRET.
We then asked if the Myr(+) Monomeric Gag construct could be rescued into a FRET pair by Myr(−) wildtype Gag. Fig. 6B includes Myr(−) WT Gag as a positive control as before (open triangles). Gag-CFP again failed to rescue Myr(−) monomeric Gag-YFP into a FRET pair (open diamonds). Similarly, myristoylated monomeric Gag was not rescued by wildtype Gag-CFP (closed circles). Thus the substitutions introduced in CA and NC were sufficient to disrupt Gag-Gag interactions even in the context of the myristoylated pairs.
Monomeric Gag is inefficiently incorporated into wildtype particles; role of the MA domain
Another assay for rescue of assembly-deficient Gag molecules is represented by the appearance of particle-deficient forms of Gag in the supernatant upon expression of the wildtype protein. We used wildtype Gag bearing a C-terminal FLAG epitope as the particle-competent rescue vehicle, and expressed particle-deficient forms of Gag bearing a C-terminal HA epitope. The upper blot in Fig. 7 was probed with anti-FLAG antibody. Note that for each lane where Gag-FLAG was expressed in the cell, a corresponding Gag band was detected in the pelleted supernatant fraction (Fig. 7, top). Proteins to be rescued are shown by anti-HA blotting in Fig. 7. Test constructs were approximately equally expressed in cells (Cell, lanes 1–7). Myr(−) wildtype Gag, full-length monomeric Gag, and myr(−) monomeric Gag each failed to form viral particles when expressed alone (Fig. 6B, supernatant lanes 9, 10, and 11, respectively). Expression of wildtype Gag with myr(−) Gag efficiently rescued this protein into particles (lane 12).
Fig. 7. Particle rescue experiments.
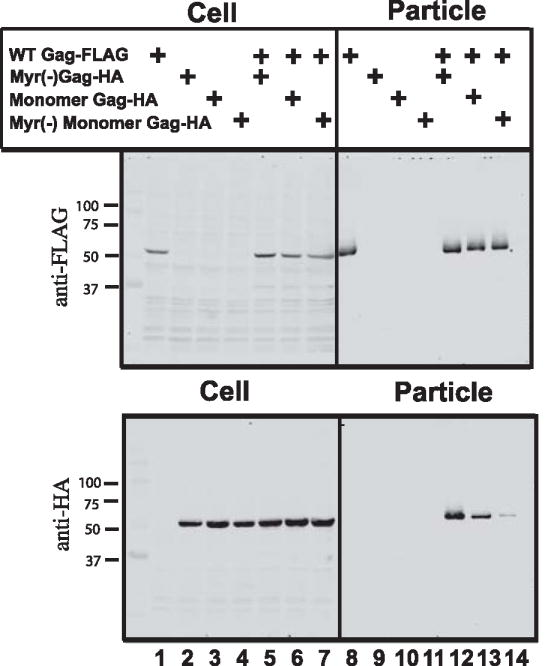
A FLAG-tagged wildtype Gag protein was co-expressed with HA-tagged Gag test constructs. Cell lysates and particles (pelleted through sucrose) were evaluated by Western blotting with anti-FLAG or anti-HA antibody. Top panel shows the wildtype Gag in cell lysates (left) and particles (right). Bottom panel shows protein production in cell lysates (left); the right panel here represents rescue of test constructs into particles by wildtype Gag. Blot shown is representative of three independent experiments.
Myristoylated, monomer Gag was also rescued into particles, albeit with reduced efficiency (lane 13). Myr(−) monomeric Gag was found in particles minimally (Fig. 7, see faint band in lane 14). The difference between rescue of myr(−) wildtype Gag and myr(−) monomeric Gag supports the idea that monomeric Gag is indeed unable to multimerize and be rescued (compare lanes 12 and 14). However, it is less clear what the partial rescue of the myristoylated monomer indicates. We speculate that the monomer is able to reach a common site of assembly at the plasma membrane through its myristoylated N-terminus, where it may be passively incorporated into the developing particle bud formed by the wildtype. This contention will be discussed in the section describing the subcellular localization of monomeric Gag later in this report.
To further investigate the requirements for the partial rescue described above, we asked whether MA was required. MA contains essential membrane binding and targeting functions for Gag, so it would be a reasonable hypothesis that MA may be required for monomeric Gag to reach the particle assembly site. MA-MA interactions or MA-RNA interactions could also contribute, although these were clearly not sufficient for multimerization as measured by FRET. We therefore repeated the particle rescue experiment with FLAG-tagged wildtype Gag and assembly-incompetent HA-tagged mutants, and included in this experiment monomeric Gag lacking MA (DMA monomeric Gag, faster migrating band in lanes 4 and 7, Fig. 8). Note that the DMA construct includes the v-src myristoylation signal at the N-terminus. Myristoylated, monomeric Gag was again rescued inefficiently by wildtype Gag (Fig. 8, lane 13). No rescue of DMA monomeric Gag was seen (lane 14). These results suggest that the MA region in the full-length monomer Gag contributed to the incomplete rescue of monomeric Gag. This effect could be a result of intracellular targeting to the particle assembly site, or potentially through MA-MA or MA- nucleic acid interactions.
Fig. 8. Role of MA in particle rescue.
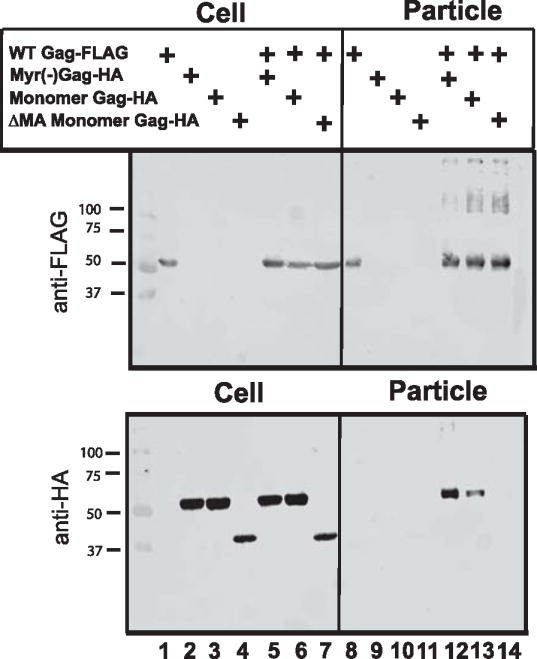
FLAG-tagged wildtype Gag was co-expressed with HA-tagged Gag test constructs as in Fig. 7. Inefficient but real rescue of the myristoylated, monomeric Gag is demonstrated in particle blot, while DMA monomeric Gag (faster migrating band, lanes 4 and 7) was not rescued (lane 14). Blot shown is representative of three independent experiments.
Monomeric Gag exhibits a unique intracellular distribution
One of the valuable features of studying a monomeric Gag protein is its potential to illuminate steps in the intracellular trafficking of Gag. Fluorescence confocal microscopy was performed to define the subcellular distribution of monomeric Gag vs. that of wildtype Gag. Monomeric Gag was first expressed in HeLa cells and stained with anti-MA polyclonal antisera or as an HA-tagged protein detected with anti-HA polyclonal sera. For comparison, wildtype Gag was examined in parallel using the same techniques, cells, and antisera. Monomeric Gag staining was found to be present throughout the cytoplasm, but was clearly accentuated on intracellular membranes, and lacked any bright puncta (Fig. 9A, 9G, and 9J). The intracellular pattern was membranous and somewhat reticular. The plasma membrane accentuation and reticular pattern contrasted significantly with non-myristoylated Gag, which was more diffusely distributed and did not mark the plasma membrane (Fig. 9B vs. 9A). The intracellular reticular pattern was somewhat suggestive of association with ER membranes to us; however we found little overlap when using a specific ER label (Fig. 9D, green is ER). Monomeric Gag tagged with HA (panel A) and untagged monomeric Gag (not shown) were essentially identical in distribution. In addition, the protein was clearly present in a thin layer underlying the plasma membrane. Remarkably, monomeric Gag but not wildtype Gag revealed long, thin extensions of the plasma membrane as well as prominent membrane ruffling (Fig. 9A, D, G, J). These extensions were consistently seen in cells that were not retracted, and were suggestive to us of the induction of true ruffles and cellular extensions rather than fixation artifacts. A careful analysis of wildtype Gag was performed in parallel, using identical fixation and staining techniques. Although this seemed to indicate that monomeric Gag induces membrane extensions, normal HeLa cells exhibited similar cellular extensions (data not shown), suggesting instead that wildtype Gag caused their loss. Live cell imaging confirmed the formation of long cellular extensions in the absence of any fixation (data not shown). The use of a palmitoylated membrane marker (ECFP-MEM) accentuated the colocalization of monomeric Gag and not wildtype Gag on membrane extensions (Fig. 9E vs. 9F). In contrast to monomeric Gag, bright puncta were apparent in the majority of cells expressing wildtype Gag (Fig. 9C). While puncta were easily discerned inside of cells, small puncta on the plasma membrane and surrounding cells, corresponding to particles seen by electron microscopy in Fig. 4, were also noted. However, the distinction in subcellular distribution between monomeric Gag and wildtype Gag may be better appreciated when both are expressed together with distinct tags. To compare wildtype and monomeric Gag in the same cell, we cotransfected HA-tagged monomeric Gag and FLAG-tagged wildtype Gag, and stained specifically with antibodies directed against each tag. Fig. 9G–I and Fig. 9J–L demonstrate the distinct patterns of the two proteins, with wildtype in red and monomeric Gag in green. Notably, expression of wildtype Gag revealed much more intracellular, punctate staining. Wildtype Gag did not appear to redistribute monomeric Gag extensively into Gag puncta, as the pattern of intracellular and plasma membrane staining was quite similar for monomeric Gag in the absence (Fig. 9A and B) or presence (Fig. 9D and G) of wildtype Gag. However, there were a few intracellular puncta that were brightly stained for wildtype and less intense but present for monomeric Gag, indicating specific colocalization/concentration at these intracellular sites. These are illustrated by the puncta near the asterisks in Fig. 9G and H. These colocalized puncta may represent the formation of intracellular vesicles where the molecules are co-recruited. An alternative explanation is that wildtype Gag particles are endocytosed from the plasma membrane as reported (Jouvenet et al., 2006), carrying along plasma membrane-associated monomeric Gag on the other (inner) face of the plasma membrane to common intracellular punctate sites. Together, these studies confirmed the association of monomeric Gag molecules with intracellular membranes and with the plasma membrane, demonstrated a propensity of monomeric Gag to induce the formation of plasma membrane extensions and ruffles, and showed that monomeric Gag itself is unable to form distinct plasma membrane or intracellular puncta.
Fig. 9. Subcellular distribution of monomeric Gag differs markedly from wildtype.

Cells expressing Gag constructs were fixed and stained with anti-Gag or with antibodies against epitope tags and examined by confocal fluorescence microscopy. (A) Monomeric Gag-HA stained with anti-HA antisera. (B) Non-myristoylated, untagged Gag stained with anti-MA antisera. (C) Wildtype Gag stained with anti-p24 monoclonal antibody. (D) Monomer Gag is shown in red in this panel, and endoplasmic reticulum staining in green. (E) pECFP-MEM is shown in blue, marking the plasma membrane, and monomer Gag in red. Areas of light blue/white signal indicate colocalization. (F) Wildtype Gag stained with anti-MA antibody is shown in red, and pECFP-MEM in blue. (G–I) Coexpression of monomeric Gag-HA and wildtype Gag-FLAG. Monomeric Gag-HA is shown in green (G and I). Wildtype Gag-FLAG is shown in red (H and I). (K–L) Coexpression of monomeric Gag-HA and wildtype Gag-FLAG, same technique as in (G–I). Size bars indicate 16 mm in each panel.
DISCUSSION
Oligomerization of Gag is a central feature of retrovirus assembly. In vitro models of assembly have provided a number of insights into the process of Gag-Gag oligomerization. Formation of particles from purified Gag protein in vitro can be stimulated by nucleic acids (Campbell and Rein, 1999; Campbell and Vogt, 1995), and oligonucleotides as short as 22 nucleotides promoted the assembly of Rous sarcoma virus (RSV) particles (Ma and Vogt, 2002). This distance represented twice the binding site size of a Gag monomer, suggesting that formation of the Gag dimer is the critical nucleation step leading to the polymerization of Gag. Recombinant, purified HIV Gag exists in monomer-dimer equilibrium in solution, as elegantly shown by sedimentation equilibrium measurements (Datta et al., 2007b). Remarkably, interaction with the inositol phosphate IP6 converted the equilibrium to monomer-trimer, suggesting that interaction between MA and IP6 creates a trimer that may be the critical building block for polymerization of Gag (Datta et al., 2007b). After the formation of a critical small oligomer, higher order oligomers must be generated through lateral interactions in order to generate the hexameric lattice that forms the developing shell of the immature capsid. Details of how and where this process occurs within the cell remain to be elucidated.
Studies of HIV assembly performed using recombinant protein in vitro such as those described above have yielded a number of critical insights into Gag-Gag interactions that are directly applicable to an understanding of particle assembly in mammalian cells. The importance of the dimer interface within the CA CTD, the role of the RNA-NC interaction in catalyzing assembly, the ability of dimerizing leucine zippers to replace the nucleic acid binding function of NC, among a number of other findings, hold true both in cells and in vitro (Accola, Strack, and Gottlinger, 2000; Campbell and Rein, 1999; Campbell and Vogt, 1995; Chang et al., 2008; Gross, Hohenberg, and Krausslich, 1997; Guo and Liang, 2005; Li et al., 2007; von Schwedler et al., 2003; Zhang et al., 1998). Examination of the multimerization of Gag expressed in mammalian cells may also reveal some differences, however. Gag expressed in cells is myristoylated, and is translated in the complex environment of the cellular cytoplasm. Cellular membranes are readily available for interactions with the myristoylated N-terminus. We found previously that myristoylation was crucial for Gag-Gag multimerization in cells, and correlated with the occurrence of FRET signal on cellular membranes (Li et al., 2007). While this requirement is not absolute, as indicated by the formation of intracellular particles by myr(−) Gag in some high-level expression systems (Gheysen et al., 1989), it may indicate a requirement for interaction at the N-terminus of the molecule to elicit efficient assembly under normal conditions. One possibility is that Gag simply concentrates on membranes through the membrane-binding motif in MA, and that upon concentration and nucleic acid tethering by NC, higher-order multimerization proceeds. Another intriguing possibility is that myristoylation results in formation of a trimeric assembly intermediate, as recently suggested by the Rein group (Datta et al., 2007b). They found that the interaction with inositol hexakisphosphate (IP6) in solution acts as a switch to induce extension of the folded Gag molecule, suggesting that this conformational change may be responsible for the change from monomer-dimer to monomer-trimer equilibrium. In cells, if the trimer is the fundamental unit from which polymerization of the Gag lattice proceeds, a myristoylated N-terminus may replace the in vitro requirement for IP6 binding and allow trimers to form.
By studying a monomeric Gag protein, we hope to gain insights into early events in assembly. A number of important questions might be asked using a monomeric Gag protein. For example, does Gag move as a monomer to the plasma membrane? Are domains involved in interactions with host proteins functional in the monomer, or do they require an oligomeric intermediate? This report describes our initial characterization of the monomeric form of Gag that we had derived from FRET-based studies in live cells. Monomeric Gag was unable to form virus-like particles, at least that we could detect by electron microscopy, and was released from the cell very inefficiently. Wildtype Gag could not rescue monomeric Gag into complexes as judged by FRET, and could not rescue the myr(−) monomer into particles. Interestingly, although the monomeric, myristoylated Gag was excluded from lipid rafts (using the biochemical fractionation criteria of flotation on detergent-resistant membranes (Ding et al., 2003; Lindwasser and Resh, 2001; Ono and Freed, 2001), it must have reached the same membrane microdomains from which wildtype particles were budding in order to gain incorporation into particles. The appearance of monomeric Gag at the plasma membrane suggests that a multimeric intermediate is not absolutely required for transit to the site of assembly. It remains possible, however, that an assembly intermediate forms within the cell for specific trafficking, and that the kinetics or path of transit will differ between monomeric Gag and wildtype Gag.
Rescue of the myristoylated, monomeric Gag into virus-like particles by wildtype Gag required a contribution from MA. The appearance of monomeric Gag at the plasma membrane by immunofluorescence analysis, together with the absence of FRET between wildtype and monomer, suggests that this rescue may represent passive incorporation at the particle assembly site, or that weak interactions between MA domains may recruit monomeric protein into particles but not allow direct interaction or the correct positioning of the C-terminal fluorescent proteins required to generate a FRET signal. There are yet other potential explanations for the contribution of MA to this rescue. A number of reports have established an RNA binding role for MA (Burniston et al., 1999; Chang et al., 2008; Cimarelli et al., 2000; Ott, Coren, and Gagliardi, 2005; Purohit et al., 2001), and some have suggested that MA can partially compensate for the loss of the RNA tethering role of NC (Burniston et al., 1999; Ott, Coren, and Gagliardi, 2005). It is possible therefore that the incorporation of monomeric Gag into wildtype particles was mediated by MA-RNA interactions. A monomeric Gag molecule targeted to the cytoplasmic face of the plasma membrane could interact with the RNA delivered there by wildtype Gag and subsequently be incorporated into the particle, without invoking direct protein-protein interaction. We favor the explanation that the colocalization at particle assembly sites with wildtype Gag allowed the monomeric protein to be incorporated through weak MA-MA interactions. Additional studies will be required to test this hypothesis.
The subcellular distribution of monomeric Gag was strikingly different than that of wildtype Gag. One major notable finding was the lack of any particle puncta or any bright intracellular puncta. This is of course not surprising for a monomeric Gag, as the bright puncta represent multimers of Gag, either virus-like particles, developing virus-like particles or multimers on intracellular membranes, or endocytosed virus-like particles (Jouvenet et al., 2006; Larson et al., 2005). The finding that monomeric Gag lines the plasma membrane, and is somewhat concentrated on ruffles and on plasma membrane extensions, suggests that some differential binding to the cytoplasmic face of the plasma membrane may occur. A significant fraction of monomeric Gag also associates with intracellular membranes based on imaging results. The interaction of monomeric Gag with membranes is likely to be of low affinity, even with myristic acid and the positively-charged surface of MA available for interaction with the membrane. Dalton and coworkers examined the contribution of myristic acid to the affinity of recombinant HIV MA for artificial membranes (Dalton et al., 2007), and found that membrane binding was increased by myristic acid but remained very weak. Dimerization greatly enhanced membrane interactions in this in vitro system and enhanced membrane binding in transfected cells (Dalton et al., 2007). Monomeric Gag may not generate optimal exposure of myristate, as Gag-Gag interactions provide conformational changes that trigger the myristyl switch (Tang et al., 2004). Interaction with PI(4,5)P2 can also trigger the myristyl switch (Saad et al., 2006), however, and we see no reason that the population of monomeric Gag associated with the cytoplasmic face of the plasma membrane should not be able to interact with PI(4,5)P2. Cooperative effects on binding would clearly be present for oligomers and absent in the monomer, and are likely to play a major role in stabilizing membrane interactions of oligomeric forms of Gag.
This report establishes the monomeric nature of Gag bearing M39A/W184A/M185A/NC15A mutations, and establishes its subcellular localization in transfected cells. This monomeric Gag will be a useful tool in further dissection of steps involved in HIV assembly. It will be of interest to define the kinetics of movement of monomeric Gag to the plasma membrane, and contrast this with movement of wildtype Gag or Gag puncta. Further mutagenesis should clarify the nature of the contribution of MA to incorporation of monomeric Gag into wildtype virus-like particles. Ultimately it will be desirable to trigger dimer or trimer linkages between monomers within live cells, which should facilitate studies of the kinetics of assembly and trafficking of specific Gag multimers.
MATERIALS AND METHODS
Plasmid construction
The Gag coding sequences for all constructs were derived from the codon-optimized HXB2 gag gene from plasmid pVRC3900 (Huang, Kong, and Nabel, 2001). Gag proteins were initially cloned for expression as fusion constructs with the cerulean variant of cyan fluorescent protein (CFP)(Rizzo et al., 2004) or the Venus variant of yellow fluorescent protein (YFP)(Nagai et al., 2002). CFP and YFP fusions were expressed at the C-termini of Gag proteins produced in plasmid vectors pcDNA 4/TO and pcDNA 5/TO (Invitrogen). Most of the YFP/CFP fusion constructs in this study were described in our earlier work (Li et al., 2007). MACA(M39A/W184A/M185A)NC15AP6-CFP was created from SrcCA(M39A/W184A/M185A)NC15A-CFP using PCR mutagenesis to create a full-length Gag-CFP with the indicated substitutions in vector pcDNA4/TO. A YFP-tagged construct was created in an identical fashion from SrcCA(M39A/W184A/M185A)NC15A-YFP. Untagged MACA(M39A/W184A/M185A)NC15AP6 (monomeric Gag) was created from MACA(M39A/W184A/M185A)NC15AP6-CFP by PCR cloning of the Gag region as a HindIII-BamH1 fragment with the wildtype stop codon introduced; this fragment was cloned into the same pcDNA4/TO vector. Untagged DMADP6 monomeric Gag was created from srcCA(M39A/W184A/M185A)NC15A-CFP by PCR cloning as a HindIII-BamHI fragment, introducing a stop codon following the final codon of NC and ligating into the HindIII and BamH1 sites of pcDNA5/TO. FLAG and HA fusions (WT Gag FLAG-tagged, Myr(−) WT Gag HA, monomer Gag HA, myr(−) monomer Gag HA and DMA monomer Gag HA) were generated by PCR using HindIII sites at the 5′ end and BamHI at the 3′ end, together with the coding sequence for the specific tag on the 3′ oligo, and ligated into pcDNA 3.1+ (Invitrogen). Myr(−) constructs included a G2A coding change within the 5′ oligo sequence. All constructs were sequenced throughout the gag gene. Oligonucleotide primers used for all PCR amplifications to generate constructs are available upon request. pECFP-MEM expresses a palmitoylated form of CFP and was obtained from Clontech.
Cells and transfections
293T cells were used for fluorometer-based studies and for particle production studies. HeLa cells were employed in EM and confocal microscopic analysis. Cell lines were maintained in Dulbecco’s modified Eagle medium with 10% fetal bovine serum supplemented with penicillin and streptomycin at 37°C in 5% CO2. Transfections were performed using the calcium phosphate transfection method or via lipofection with Lipofectamine 2000 (Invitrogen).
Scanning cuvette fluorometry for FRET
Cells cotransfected with Gag-CFP and Gag-YFP constructs were analyzed at 36 hours posttransfection. Cells were washed with phosphate-buffered saline (PBS) and then harvested in PBS for analysis by scanning fluorometry. Cells in suspension or gradient fractions were analyzed by scanning fluorometry in a PTI T-format scanning cuvette spectrofluorometer (Photon Technology International, Lawrenceville, NJ). For FRET analysis, samples were excited at 433 nm, and an emission scan range is from 460 nm to 550 nm. For the analysis of relative YFP Fluorescence intensity, samples were excited at 513 nm, with a resulting emission scan from 524 nm to 550 nm. Data were collected from at least three independent experiments for each construct, and the curves shown are representative of the three measures obtained. Photomultiplier gain was adjusted slightly to normalize the CFP peak intensity in each arm of the experiment in order to facilitate comparison of the relative amounts of YFP (FRET) signal relative to CFP.
Isolation of cytosolic and membrane fractions
Cells were harvested for analysis 36 hours following transfection. One 10 cm2 tissue culture dish of nearly confluent 293T cells was used in each experimental sample. Cells were washed in NTE buffer (100 mM NaCl, 10 mM Tris-Cl [pH 8.0], 1 mM EDTA [pH 8.0]) and then allowed to swell in hypotonic buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA [pH 8.0]) plus protease inhibitors for 20 minutes on ice. Cells were then broken by Dounce homogenization. After the buffer was adjusted to 0.1 M NaCl, the nuclei and unbroken cells were removed by centrifugation at 1000 × g for 10 min. The supernatants containing cytosolic and membrane components were then adjusted to 50% iodixanol from a stock solution of 60% iodixanol (Nycomed Pharma, Oslo, Norway), and 40% and 10% solutions of iodixanol were layered sequentially on top. The preparation was then centrifuged in a Beckman SW41 rotor at 41,000 rpm for 2 h at 4°C, after which 12 fractions were harvested from the top of the gradient (from fraction 1 to fraction 12); and then all the fractions were analyzed by cuvette fluorometry. For analysis of detergent-resistant membranes, postnuclear cellular fractions were treated wit 1% TX-100 on ice for 30 minutes prior to the fractionation procedure.
Sucrose Gradient Sedimentation
Supernatants were harvested 36 hours after transfection with the indicated Gag protein expression plasmids. Cells and debris in supernatants were removed by centrifugation at 1000 × g for 10 minutes, followed by filtration through 0.45 um syringe driven filter unit (Millipore). Particles were then concentrated and purified by pelleting through a 20% sucrose cushion in a Beckman SW32 rotor at 30,000 rpm for 2 h at 4 °C. Particle pellets were resuspended in phosphate-buffered saline (PBS), and were subjected to equilibrium gradient centrifugation on a 20–60% linear sucrose gradient in an SW-41 rotor at 35,000 rpm for 12–14 hours at 4 °C. 12 fractions were harvested from the top of the gradient (from fraction 1 to fraction 12) and then all the fractions were analyzed by cuvette fluorometry and refractive index, from which the density of each fraction was calculated.
Myristic Acid labeling
Myristic acid incorporation was examined using Gag expression in 293T cells. Complete culture medium was replaced by Dulbecco’s Modified Eagle Medium containing 5% delipidated fetal calf serum for 30 minutes, followed by incubation in the same media supplemented with 50 μCi of 3H-myristic acid per milliliter (55 Ci/mmol; Amersham). The cells were incubated for an additional 8 hours, and then Gag proteins were immunoprecipitated with HIV patient sera and analyzed by SDS-PAGE and autoradiography. Western blotting of a fraction of the same cells was performed to analyze the total Gag population.
Antibodies and immunoblotting
Transfected 293T cells or particle pellets were harvested 36 hours posttransfection. Cells were lysed in radioimmunoprecipitation assay buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate). Particles were pelleted through 20% sucrose as described above, and resuspended in SDS-PAGE load buffer. Cells and supernatants were examined by Western blotting using anti-green fluorescent protein rabbit serum (Invitrogen), rabbit anti-FLAG antibody (OctA-Probe) (Santa Cruz Biotechnology), mouse HA.11 Monoclonal Antibody (Covance) as the primary antibody and goat anti-rabbit (IRDye800; Li-Cor Biosciences) or goat anti-mouse (IRDye680; Li-Cor Biosciences) as the secondary antibody. Blots were imaged using infrared fluorescence detection using the Odyssey system (Li-Cor Biosciences).
Laser confocal fluorescence microscopy
HeLa cells were grown on glass coverslips and transfected with the indicated Gag or monomeric Gag expression constructs, or with Gag-FLAG and monomeric Gag-HA. Cells were washed and fixed after 18 hours posttransfection with 4% paraformaldehyde solution (Electron Microscopy Sciences) for 10 minutes. Primary antibodies for detection were rabbit anti-MA polyclonal (Varthakavi, Browning, and Spearman, 1999), anti-p24 monoclonal CA-183 (provided by Bruce Chesebro and Kathy Wehrly through the NIH AIDS Research and Reference Reagent Program), anti-FLAG F3165 (Sigma) or rabbit anti-HA H6908 (Sigma). ER staining was performed using the Alexa Fluor 488 endoplasmic reticulum staining kit from Invitrogen. Images were obtained using a spinning disk confocal microscope from Improvision (now part of Perkin-Elmer); each field was imaged using a 63X oil objective with an NA of 1.4. Images were analyzed using the Volocity software package (Improvision/Perkin-Elmer).
Electron Microscopy
Transfected 293T cells were fixed in 3% gluteraldehyde in cacodylate buffer. Dehydration, embedding, staining, and ultramicrotomy were performed for analysis by transmission EM as described before (Dong et al., 2005). Thin sections were analyzed on a Hitachi H-7500 transmission electron microscope.
Acknowledgments
This study was funded by AI40338 and AI067101. The work was supported in part by the Emory University Center for AIDS Research (P30 AI050409). We thank the Integrated Electron Microscopy Core at Emory University for assistance with transmission EM studies, and the James B. Pendleton Charitable Trust for a gift allowing the purchase of fluorescence imaging equipment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accola MA, Strack B, Gottlinger HG. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J Virol. 2000;74 (12):5395–402. doi: 10.1128/jvi.74.12.5395-5402.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsetti A, Ohagen A, Gottlinger HG. The C-terminal half of the human immunodeficiency virus type 1 Gag precursor is sufficient for efficient particle assembly. J Virol. 1998;72 (11):9313–7. doi: 10.1128/jvi.72.11.9313-9317.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Johnson MC, Simon MN, Fuller SD, Vogt VM. Cryo-electron microscopy reveals conserved and divergent features of gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J Mol Biol. 2006;355 (1):157–68. doi: 10.1016/j.jmb.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, Johnson MC. The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol. 2004;11 (7):672–5. doi: 10.1038/nsmb785. [DOI] [PubMed] [Google Scholar]
- Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J Virol. 1999;73 (10):8527–40. doi: 10.1128/jvi.73.10.8527-8540.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J Virol. 1999;73 (3):2270–9. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Vogt VM. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J Virol. 1995;69 (10):6487–97. doi: 10.1128/jvi.69.10.6487-6497.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Chang YF, Wang SM, Tseng YT, Huang KJ, Wang CT. HIV-1 matrix protein repositioning in nucleocapsid region fails to confer virus-like particle assembly. Virology. 2008;378 (1):97–104. doi: 10.1016/j.virol.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimarelli A, Sandin S, Hoglund S, Luban J. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J Virol. 2000;74 (7):3046–57. doi: 10.1128/jvi.74.7.3046-3057.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol. 2007;81 (12):6434–45. doi: 10.1128/JVI.02757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SA, Curtis JE, Ratcliff W, Clark PK, Crist RM, Lebowitz J, Krueger S, Rein A. Conformation of the HIV-1 Gag protein in solution. J Mol Biol. 2007a;365 (3):812–24. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SA, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, Kvaratskhelia M, Lebowitz J, Rein A. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J Mol Biol. 2007b;365 (3):799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdowski A, Ding L, Spearman P. A novel fluorescence resonance energy transfer assay demonstrates that the human immunodeficiency virus type 1 Pr55Gag I domain mediates Gag-Gag interactions. J Virol. 2004;78 (3):1230–42. doi: 10.1128/JVI.78.3.1230-1242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Derdowski A, Wang JJ, Spearman P. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J Virol. 2003;77 (3):1916–26. doi: 10.1128/JVI.77.3.1916-1926.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120 (5):663–74. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Dooher JE, Lingappa JR. Conservation of a stepwise, energy-sensitive pathway involving HP68 for assembly of primate lentivirus capsids in cells. J Virol. 2004;78 (4):1645–56. doi: 10.1128/JVI.78.4.1645-1656.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller SD, Wilk T, Gowen BE, Krausslich HG, Vogt VM. Cryo-electron microscopy reveals ordered domains in the immature HIV-1 particle. Curr Biol. 1997;7 (10):729–38. doi: 10.1016/s0960-9822(06)00331-9. [DOI] [PubMed] [Google Scholar]
- Gelderblom HR. Assembly and morphology of HIV: potential effect of structure on viral function. Aids. 1991;5 (6):617–37. [PubMed] [Google Scholar]
- Gheysen D, Jacobs E, de Foresta F, Thiriart C, Francotte M, Thines D, De Wilde M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell. 1989;59 (1):103–12. doi: 10.1016/0092-8674(89)90873-8. [DOI] [PubMed] [Google Scholar]
- Gross I, Hohenberg H, Krausslich HG. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur J Biochem. 1997;249(2):592–600. doi: 10.1111/j.1432-1033.1997.t01-1-00592.x. [DOI] [PubMed] [Google Scholar]
- Guo X, Liang C. Opposing effects of the M368A point mutation and deletion of the SP1 region on membrane binding of human immunodeficiency virus type 1 Gag. Virology. 2005;335 (2):232–41. doi: 10.1016/j.virol.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Huang Y, Kong WP, Nabel GJ. Human immunodeficiency virus type 1-specific immunity after genetic immunization is enhanced by modification of Gag and Pol expression. J Virol. 2001;75 (10):4947–51. doi: 10.1128/JVI.75.10.4947-4951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4 (12):e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman J, Lam TT, Barnes S, Sakalian M, Emmett MR, Marshall AG, Prevelige PE., Jr Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J Mol Biol. 2003;325 (4):759–72. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- Larson DR, Johnson MC, Webb WW, Vogt VM. Visualization of retrovirus budding with correlated light and electron microscopy. Proc Natl Acad Sci U S A. 2005;102 (43):15453–8. doi: 10.1073/pnas.0504812102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Yu XF. Identification and characterization of virus assembly intermediate complexes in HIV-1-infected CD4+ T cells. Virology. 1998;243 (1):78–93. doi: 10.1006/viro.1998.9064. [DOI] [PubMed] [Google Scholar]
- Li H, Dou J, Ding L, Spearman P. Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J Virol. 2007;81 (23):12899–910. doi: 10.1128/JVI.01280-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwasser OW, Resh MD. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J Virol. 2001;75 (17):7913–24. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YM, Vogt VM. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J Virol. 2002;76 (11):5452–62. doi: 10.1128/JVI.76.11.5452-5462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Goto T, Momose F. Human immunodeficiency virus type 1 Gag assembly through assembly intermediates. J Biol Chem. 2004;279 (30):31964–72. doi: 10.1074/jbc.M313432200. [DOI] [PubMed] [Google Scholar]
- Morikawa Y, Zhang WH, Hockley DJ, Nermut MV, Jones IM. Detection of a trimeric human immunodeficiency virus type 1 Gag intermediate is dependent on sequences in the matrix protein, p17. J Virol. 1998;72 (9):7659–63. doi: 10.1128/jvi.72.9.7659-7663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20 (1):87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc Natl Acad Sci U S A. 2001;98 (24):13925–30. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A, Waheed AA, Joshi A, Freed EO. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J Virol. 2005;79 (22):14131–40. doi: 10.1128/JVI.79.22.14131-14140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott DE, Coren LV, Gagliardi TD. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J Virol. 2005;79 (22):13839–47. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, Wills JW. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol. 1995;69 (9):5455–60. doi: 10.1128/jvi.69.9.5455-5460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Dupont S, Stevenson M, Green MR. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. Rna. 2001;7(4):576–84. doi: 10.1017/s1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22 (4):445–9. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103 (30):11364–9. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandefur S, Smith RM, Varthakavi V, Spearman P. Mapping and characterization of the N-terminal I domain of human immunodeficiency virus type 1 Pr55(Gag) J Virol. 2000;74 (16):7238–49. doi: 10.1128/jvi.74.16.7238-7249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandefur S, Varthakavi V, Spearman P. The I domain is required for efficient plasma membrane binding of human immunodeficiency virus type 1 Pr55Gag. J Virol. 1998;72 (4):2723–32. doi: 10.1128/jvi.72.4.2723-2732.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101 (2):517–22. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varthakavi V, Browning PJ, Spearman P. Human immunodeficiency virus replication in a primary effusion lymphoma cell line stimulates lytic-phase replication of Kaposi’s sarcoma-associated herpesvirus. J Virol. 1999;73 (12):10329–38. doi: 10.1128/jvi.73.12.10329-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stemmler TL, Klishko VY, Li S, Albertine KH, Davis DR, Sundquist WI. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. Embo J. 1998;17 (6):1555–68. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol. 2003;77 (9):5439–50. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon RA, Jr, Wills JW. Characterization of a small (25-kilodalton) derivative of the Rous sarcoma virus Gag protein competent for particle release. J Virol. 1993;67 (9):5550–61. doi: 10.1128/jvi.67.9.5550-5561.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk T, Gross I, Gowen BE, Rutten T, de Haas F, Welker R, Krausslich HG, Boulanger P, Fuller SD. Organization of immature human immunodeficiency virus type 1. J Virol. 2001;75 (2):759–71. doi: 10.1128/JVI.75.2.759-771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright ER, Schooler JB, Ding HJ, Kieffer C, Fillmore C, Sundquist WI, Jensen GJ. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. Embo J. 2007;26 (8):2218–26. doi: 10.1038/sj.emboj.7601664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Wilson-Kubalek EM, Weiner SG, Brown PO, Rein A. Supramolecular organization of immature and mature murine leukemia virus revealed by electron cryo-microscopy: implications for retroviral assembly mechanisms. Proc Natl Acad Sci U S A. 1998;95 (13):7299–304. doi: 10.1073/pnas.95.13.7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qian H, Love Z, Barklis E. Analysis of the assembly function of the human immunodeficiency virus type 1 gag protein nucleocapsid domain. J Virol. 1998;72 (3):1782–9. doi: 10.1128/jvi.72.3.1782-1789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]