

Abstract
To evaluate potential antioxidant characteristics of organic selenium (Se), double knock-in transgenic mice expressing human mutations for the amyloid precursor protein (APP) and human presenilin-1 (PS-1) were provided a Se-deficient diet, a Se-enriched (Sel-Plex), or a control diet from 4 to 9 months of age followed by a control diet until 12 months of age. Levels of DNA, RNA, and protein oxidation as well as lipid peroxidation markers were determined in all mice and amyloid beta peptide (Aβ) plaques were quantified. APP/PS1 mice provided Sel-Plex showed significantly (P < 0.05) lower levels of Aβ plaque deposition and significantly decreased levels of DNA and RNA oxidation. Sel-Plex-treated mice showed no significant differences in levels of lipid peroxidation or protein oxidation compared to APP/PS1 mice on a control diet. To determine if diminished oxidative damage was associated with increased antioxidant enzyme activities, brain glutathione peroxidase (GSH-Px), glutathione reductase, and glutathione transferase activites were measured. Sel-Plex-treated mice showed a modest but significant increase in GSH-Px activity compared to mice on a normal diet (P< 0.5). Overall, these data suggest that organic Se can reduce Aβ burden and minimize DNA and RNA oxidation and support a role for it as a potential therapeutic agent in neurologic disorders with increased oxidative stress.
Keywords: Alzheimer’s disease, DNA oxidation, RNA oxidation, organo-selenium, antioxidants
Introduction
Oxidative damage to a variety of biomolecules has been implicated in the pathogenesis of several neurologic disorders including Alzheimer’s disease (AD) (reviewed [1]). Free radicals generated by amyloid beta peptide (Aβ), a 40 or 42 amino acid peptide generated by gamma and beta secretase cleavage of the amyloid precursor protein (APP) may play a critical role in the oxidative damage associated with AD [2-4]. Because of its high oxygen consumption rate, high lipid content, and relatively limited antioxidant capacity compared to other organs, the brain is particularly susceptible to oxidative damage. Several studies show significantly increased oxidative damage to lipids, protein, DNA, and RNA in AD (Reviewed [1]). More recent studies suggest that oxidative damage associated with AD may begin during earlier stages of the disease including during anmestic mild cognitive impairment, the earliest clinical manifestation of AD [5]. To protect against oxidative damage, the brain employs a variety of small molecule antioxidants (vitamins E and C); enzymatic antioxidants including glutathione peroxidase (GSH-Px), a tetrameric cytosolic selenoprotein that catalyzes the reduction of organic hydroperoxides as well as H2O2 [6]; and thioredoxin reductase, a multifunctional selenoprotein that functions in the reduction of protein disulfides [7] and as a scavenger of reactive oxygen species [8].
Because selenium (Se) is an essential component of GSH-Px and thioredoxin reductase, there is considerable interest in the potential ability of Se and other dietary antioxidant supplements to modulate oxidative stress. Selenium, an essential trace element at low concentrations, has been shown to be a protective anti-carcinogenic [9]. In contrast, high concentrations of Se, particularly inorganic Se, may be genotoxic or carcinogenic [9], likely mediated through reactive oxygen species generation [10-13]. In addition, recent studies suggest that speciation of Se is also critical in determining whether it is toxic or protective [14]. Both organic (selenomethionine, selenocysteine, Se-methylselenocysteine) and inorganic (sodium selinite, sodium selenate) forms of Se are utilized by humans with similar efficiencies ultimately leading to production of selenoproteins [15]. Previous studies of sodium selenite show pretreatment of HT1000 cells with low concentrations of inorganic Se for 12 h decreased H2O2-induced apoptosis through activation of anti-apoptotic P13/Akt pathways and inhibition of the ASK1/JNK pathways [16]. Selenomethionine (SeMet), the major component of dietary Se, represents an organic form of Se that may be protective and provide enhanced antioxidant capacities [17]. Previous studies from our laboratory showed SeMet is protective against Aβ and iron/ hydrogen peroxide-mediated neuron death and is associated with increased GSH-Px activities [18]. In addition to numerous studies of Se in cancer prevention, a large NIH funded long term study of the efficacy of Se in the prevention of AD is currently underway [19].
To determine if the in vitro effects of organic Se are observed in an in vivo model of Aβ deposition, we provided APP/presenilin 1 (PS1) mice a Se-deficient, a Se-supplemented (Sel-Plex, 1.07 ± 0.04 μg/g Se), or a normal control diet (0.3 μg/g) from ages 4 to 9 months of age, a period during which Aβ production has been shown to occur but prior to extensive amyloid plaque deposition. The mice were then switched back to a normal control diet and maintained until 12 months of age. Quantification of amyloid burden was carried out using immunohistochemistry and Image J analysis. Levels of protein oxidation and lipid peroxidation were quantified using dot-blot immunochemistry. Oxidative modification of DNA and RNA was assayed using immunohistochemistry and confocal microscopy for 8-hydroxyguanine. To determine if Se supplementation increased antioxidant capacity, GSH-Px, glutathione reductase (GSSG-R) and glutathione transferase (GST) activities were analyzed.
Materials and methods
Animals
Double homozygous knock-in mice expressing a Swedish familial APP mutation and a humanized Aβ sequence and the 246L PS-1 mutation (APP/PS1) from Cephalon Inc. were used for these experiments. These animals develop amyloid plaques by 6 months of age [20] and show impairment of hippocampal long term potentiation [21]. In addition, deposition of Aβ in blood vessels is observed by 12 months of age and gradually increases. Alterations in Aβ solubility occur in APP/PS1 mice that closely resemble what is observed in AD. Thus, this model recapitulates many of the features of Aβ pathology in AD, including the deposition of diffuse amyloid plaques early and subsequent neuritic amyloid plaque formation and vascular deposits.
Animal maintenance
For treatment, 4-month -old male APP/PS1 mice were randomized and placed on a Se-deficient diet (0.1 ± 0.1 μg/g Se) (n = 4), a Se-supplemented diet (1.0 ± 0.1 μg/g Se) (n = 5) (kindly provided by Alltech, Nicholasville, KY), or a normal control diet (0.3 μg/g Se) (n = 3) and maintained until 9 months of age. Because APP/PS1 mice begin to deposit amyloid plaques at 6 months of age we hypothesized that if Se can interrupt Aβ processing, it should be provided before the age at which definable amyloid plaques are deposited. The diet was continued until 9 months of age, a point at which amyoid deposition becomes more prominent. The ages of treatment were chosen to mimic the period during which changes in Aβ solubility may begin to occur in human AD subjects. The Se supplement was incorporated into the diet as a yeast extract prepared by growing yeast in Se-supplemented medium. The mice were maintained 2 per cage under 12 h light/dark cycles and provided food and water ad libitum. Animal care and treatment were in accordance with University of Kentucky Institutional Animal Care and Use Committee guidelines. At 9 months of age, the animals on Se-deficient or Se-supplemented diets were switched back to a normal control diet and maintained until 12 months of age. All animals were euthanatized by Halothane overdose using procedures consistent with the Panel on Euthanasia of the American Veterinarian Association followed by cervical dislocation at 12 months of age. The brains were quickly removed and dissected into the two hemispheres. One hemisphere was placed in 4% phosphate buffered paraformaldehyde for immunohistochemical studies and the other was immediately frozen in liquid nitrogen for biochemical assays.
Quantification of amyloid burden
After paraformaldehyde fixation for 7 days, the left hemisphere was dissected into 3 segments and embedded in paraffin. Sections (10 μm) were cut using a Shandon Finesse microtome,placed on Plus slides, and immunostained for Aβ using a monoclonal antibody raised against Aβ17-34 (Vector Laboratories, Burlingame, CA) and standard protocols. Aβ quantification was carried out in a blinded fashion by taking micrographs of 3 sections of the whole half hemisphere per brain using a 4X objective on a Nikon Eclipse E60 microscope. If sections were larger than the field, multiple micrographs with well defined margins were taken to cover the entire area of the section. The micrographs were converted to gray scale and percent total section area covered by Aβ deposits quantified using Image J (NIH) software. For analysis, threshold staining was set to the same intensity for all sections and areas calculated. Slides were analyzed without knowledge of animal treatment. Results were computed as mean ± SEM % total area occupied by amyloid plaques.
RNA and DNA oxidation
Levels of 8-hydroxyguanine (8-OHG), the predominant oxidative base adduct in DNA and RNA, were quantified using immunohistochemistry and confocal microscopy as previously described [22]. To visualize the cellular distribution of RNA oxidation, sections (10 μm) of paraffin-embedded cortex from APP/PS1 mice provided a control, Se-deficient, or Sel-Plex diet were cut using a Shandon Finesse microtome (Thermo Fisher Scientific, Waltham, MA), placed on Plus-slides, and rehydrated through xylene, descending alcohols, and water. Following rehydration sections were digested with 10 μg/ml proteinase K (Boehringer Mannheim, Indianapolis, IN) in phosphate buffered saline (PBS, pH 7.4) for 40 min at 37° C rinsed 3 times with Tris buffered saline (TBS; 150 mM tris-HCl, 150 mM NaCl, pH 7.6) and incubated with RNase free DNase-I (10 U/μl in PBS, Roche, Mannheim, Germany) for 2 hr at 37 C. The sections were rinsed 3 times in TBS and blocked in 10% normal goat serum in TBS at room temperature for 2 h and incubated overnight in a 1:100 dilution of anti-8-OHG antibody (QED Biosciences, San Diego, CA) in 1.5% goat serum/TBS. Following thorough rinsing in TBS, sections were incubated in a 1:1000 dilution of Alexa-488 conjugated IgG/1.5% goat serum/TBS for 2 h at room temperature. After 5 rinses in TBS and distilled/deionized water, sections were coverslipped using fluorescent anti-fade (Molecular Probes) and imaged using a Leica DM IRBE confocal microscope equipped with argon, krypton and HeNe lasers and a 40X oil objective. Confocal images were captured from a single z plane without optical sectioning and were the average of 3 scans. Images were captured from 10 fields/section with an average of 10 to 20 cells per field without knowledge of animal treatment. Fluorescence intensities of all cells in each field were quantified using Leica image analysis software. Mean fluorescence intensities were calculated for 8-OHG for each slide.
To visualize DNA oxidation subsequent sections were digested with proteinase K as described above followed by digestion of RNA with DNase free RNase (10U/ml in PBS, Roche, Mannheim, Germany) for 2 h at 37° C. The sections were then labeled for 8-OHG and imaged as described above.
Tissue processing for enzyme analysis
Tissue samples for enzyme assays, lipid peroxidation, and protein carbonyl assays were homogenized using a chilled Dounce homogenizer on ice in 1 mL HEPES buffer (pH 7.4) containing 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, 0.6 mM MgSO4, pepstatin A (1 μM), leupeptin (1.2 μM), aprotinin (0.1 μM) and PMSF (0.23 mM). Samples were centrifuged at 100,000 × g for 1 h at 4°C and the supernatant used for analyses. Total protein concentrations were determined using the Pierce BCA method (Sigma, St. Louis, MO).
4-Hydroxynonenal and acrolein assay
Dot-blot analyses of 4-hydroxynonenal (HNE) and acrolein modified proteins were carried out using a Schleicher & Schuell dot-blot apparatus and the method of Saiki et al [23] with modification. Briefly, 20 μg aliquots of 100,000 × g supernatant protein were loaded in triplicate onto nitrocellulose in a 40-50 μL volume. Vacuum was applied until the solution was evacuated from the wells. After air drying, blots were incubated overnight in 5% dry milk in 0.05% Tween-20/tris buffered saline (TTBS) to block nonspecific binding of the primary antibody. Blots were probed with a 1:2000 dilution of anti-HNE (Alpha Diagnostic, San Antonio, TX) or anti-acrolein (United States Biological, Swampscott, MA) polyclonal antibodies for 1 h at room temperature, washed in TTBS 3 × 10 minutes and then incubated with a 1:3000 dilution of horseradish peroxidase-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) for 1 h [24]. Detection of bound antibodies was by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ) and exposure of the membrane to Hyperfilm (Amersham). Dots were quantified using Scion Image (NIH). The average of triplicate dots indicates protein bound HNE or acrolein content in an individual sample. Our previous study showed a linear response of immunostaining for protein samples incubated with increasing HNE or acrolein concentrations [24]. Protein carbonyls were determined using an oxy-blot kit (Chemicon, Temecula, CA) per manufacturer’s instructions. Briefly, 5 μg protein samples were derivatized with 10 mM 2,4-dinitrophenylhydrazine (DNPH) in the presence of 5 μL of 12% sodium dodecyl sulfate for 20 min at room temperature. The samples were then neutralized with 7.5 μL of the neutralization solution (2 M Tris in 30% glycerol) and blotted onto a nitrocellulose membrane using a Scheicher and Schull dot blot assembly. The membrane was washed with 10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20 (wash buffer) and blocked by incubation with 5% bovine serum albumin/wash buffer, followed by incubation with rabbit polyclonal anti-DNPH antibody (1:100 dilution) for 1 h. The membranes were washed 3 to 5 times with wash buffer and incubated with horse radish peroxidase-conjugated goat anti-rabbit antibody/TTBS for 1 h. Blots were developed using enhanced chemiluninescence per manufacturer’s instruction and staining intensity quantified using Scion Image.
Enzyme activity assays
The method for assay of total superoxide dismutase (SOD) (Cu/Zn- and MnSOD) activity is that of Misra and Fridovich [25] as modified by Mizuno [26]. Briefly, 70-μl aliquots of the 100,000 × g supernatant were placed in UV-grade cuvettes with 1.46 mL 68mM NaH2PO4 containing 1.35 mM EDTA (pH 7.8), 100 μL 4 mM xanthine and 170 μL 3.53 mM epinephrine (pH 11.5). Following incubation at 30°C for 5 min, 10 μL xanthine oxidase (Calbiochem, San Diego, CA) was added to the solution and the absorbance followed at 320 nm (Genesys 10 UV, ThermoSpectronic, Waltham, MA) for 3 minutes. The blank for the assay contained 70 μL tissue supernatant boiled in water for 5 minutes. For MnSOD, the same protocol was followed with the addition of 200 μL 20 mM KCN. Enzyme activities are expressed as units per microgram protein, where 1 unit reduces the absorbance change by 50%. Activities are expressed as mean ± SEM percent of control activity. Cu/Zn activity was obtained by subtracting MnSOD activity from total activity.
For GSH-Px activity, the method of Paglia and Valentine [27] as modified by Mizuno [26] was followed, which measures the rate of oxidation of reduced glutathione to oxidized glutathione by H2O2 as catalyzed by GSH-Px present in the tissue supernatant. For the assay, 100 μL aliquots of the 100,000 × g supernatant were added to 1.68 mL 68 mM KH2PO4 buffer (pH 7.0) containing 1mM EDTA, 100 μL 2 mM NADPH, 10 μL 66 U/mL GSSG reductase and 10 μL 200 mM sodium azide. The assay was initiated with 100 μL 15 mM H2O2. The decrease in absorbance was followed for 2 minutes at 340 nm. Quantification was based on a molar absorption coefficient of 6,270 M-1 cm-1 for NADPH. The blank for the enzyme assay consisted of 100 μL supernatant heated at 100 °C for 5 min, and its value was subtracted from the sample value to correct for nonenzymatic oxidation of glutathione. Activity of the enzyme is expressed in units/μg total protein, with 1 unit = 1 nmol NADPH oxidized per minute, and final data for activity is expressed as mean ± SD percentage of control.
GST activity was assayed using the method of Ricci et al [28], and measures the rate of conjugation of 7-chloro-4-nitrobenzo-2-oxa-1, 3-diazole (NBD-Cl) with glutathione catalyzed by glutathione transferase. The NBD-Cl complex is a stable, yellow compound that strongly absorbs at 419 nm with a molar absorption coefficient of 14.5 mM-1cm-1. For the assay, 1.6 mL of 0.1 M sodium acetate buffer (pH 5.0) was mixed with 200 μL 0.2 mM NBD-Cl and 100 μl 0.5 mM reduced glutathione. The reaction was initiated by addition of 100 μL aliquots of the 100,000 × g tissue supernatant. The absorbance was followed for 3 min. Enzyme activity was expressed in units/μg total protein, with 1 unit = 1 nmol NBD-Cl complex formed per minute. Results are expressed as mean ± SD percentage of control.
Statistical analysis
Two-way ANOVA and Dunnett’s post hoc test were used for differences in enzyme activity and oxidative stress markers using the commercially available ABSTAT (AndersonBell Corp, Arvada, CO) software. Significant differences were set at P < 0.05.
Results
Amyloid burden
Consistent with previous studies [21] immunostaining of sections of cortex for amyloid beta peptide showed the presence of numerous amyloid plaques throughout the cortex of animals maintained on a normal diet (Figure 1A, 2). In contrast, quantification of Aβ burden in the cortex of animals maintained on the Sel-Plex diet showed a significant (P < 0.05) decrease in the volume occupied by Aβ-positive amyloid plaques (Figures 1B, 2) compared to animals maintained on normal control or Se-deficient diets. Comparison of Se-deficient animals to those maintained on a normal control diet showed no significant differences in amyloid burden.
Fig. 1.

Representative micrographs of APP/PS1 mice provided a normal diet (A) or a Se-supplemented diet (Sel-Plex) (B) immunstained for amyloid beta peptide. Note a significant decrease in Aβ burden in APP/PS1 mice provided the Sel-Plex diet.
Fig. 2.
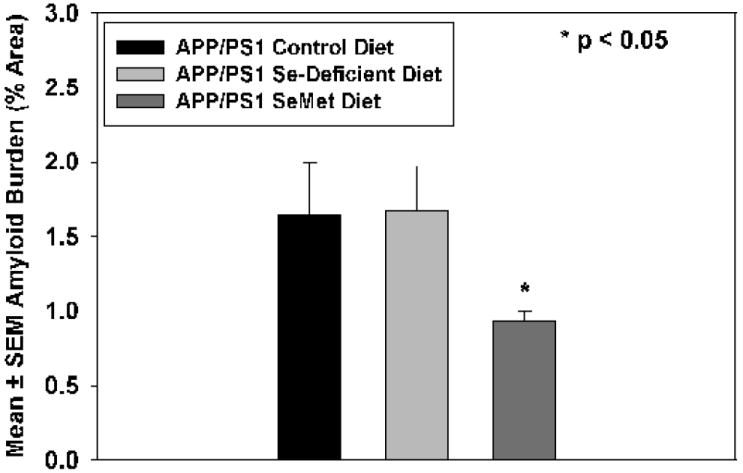
Mean ± SEM amyloid plaque area (% total area) in APP/PS1 mice provided a normal control diet, a Se-deficient diet ,or the Sel-Plex diet. The Aβ plaque area was significantly decreased(P < 0.05) in mice provided Sel-Plex.
Lipid peroxidation and protein oxidation
Quantification of levels of lipid peroxidation as demonstrated by protein bound HNE and acrolein showed no significant differences between any of the three treatment groups, although mice on a Se-deficient diet demonstrated a modest but statistically insignificant increase in HNE (Figure 3). Quantification of levels of oxidized protein showed a modest decrease in mice maintained on the Sel-Plex diet, although the difference was not statistically significant. In contrast, levels of oxidized protein were significantly (P < 0.05) elevated (~40%) in APP/PS1 mice maintained on a Se-deficient diet (Fig. 3).
Fig. 3.
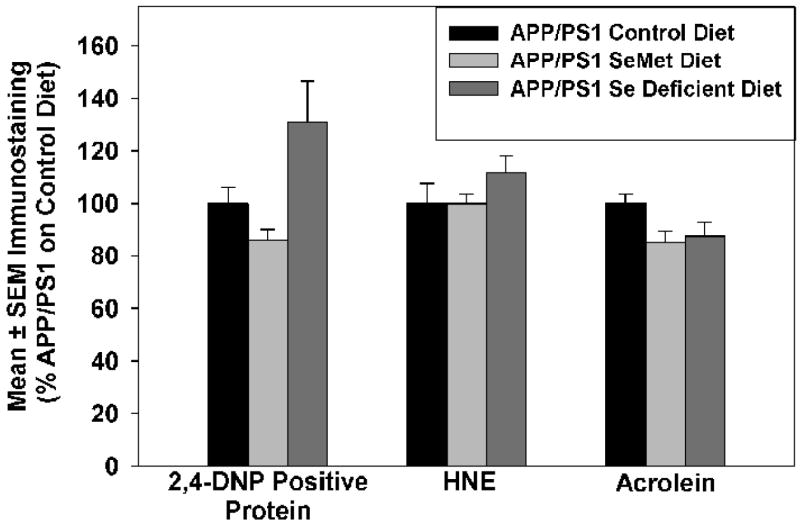
Mean ± SEM protein-bound HNE and acrolein and oxidized protein in mice provided a normal control, Se-deficient, or Sel-Plex diet. Levels of protein-bound HNE or acrolein were not significantly different between any of the three groups.
DNA and RNA oxidation
To quantify levels of RNA and DNA oxidation, sections of cortex were subjected to DNase free RNase treatment for DNA analysis or RNase free DNase for RNA analysis and immunostained for 8-OHG, the predominant oxidative adduct resulting from hydroxyl radical attack of DNA and RNA. Figure 4 shows representative confocal micrographs of 8-OHG immunostaining in DNA (A) or RNA (B). Quantification of immunostaining intensity shows animals maintained on the Sel-Plex diet had significantly less DNA oxidation (66.9 ± 6.2% APP/PS1 mice on a control diet) compared to APP/PS1 mice maintained on a control diet or Se-deficient diet (Fig. 5). Similarly, levels of 8-OHG in RNA were significantly lower (45.6 ± 10.6%) in animals provided the Sel-Plex enriched diet compared to animals on a normal control diet. There were no significant differences in 8-OHG levels in RNA of animals of a Se-deficient diet compared to animals provided a normal control diet.
Fig. 4.
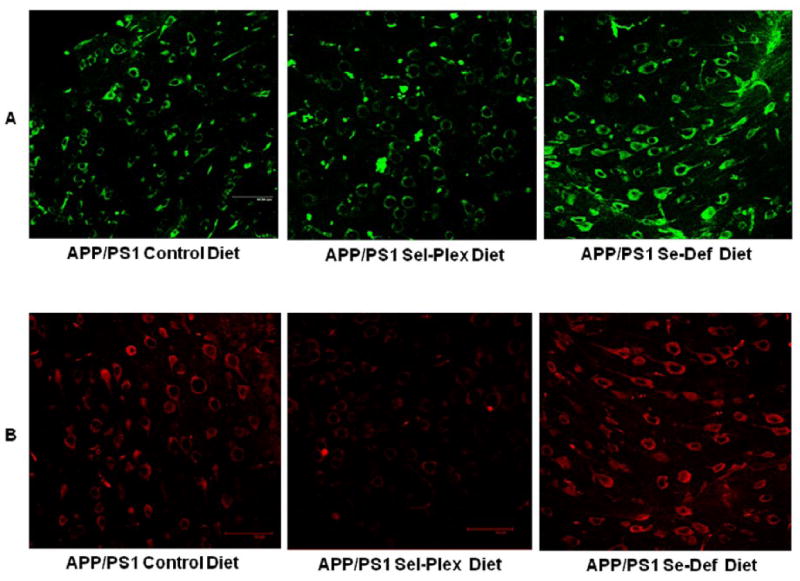
Representative confocal micrographs of APP/PS1 mice provided control, Sel-Plex supplemented, or Se-deficient diets immunostained for 8-hydroxyguanine in DNA (A) or RNA (B). Scale bar = 50 μm. Note the pronounced decrease of 8-OHG immunostaining in both RNA and DNA of mice provided Sel-Plex.
Fig. 5.

Mean ± SEM 8-OHG immunostaining (% APP/PS1 mice on control diet) in DNA and RNA. Note a significant decrease of 8-OHG immunostaining in DNA and RNA of mice provided Sel-Plex (P < 0.05).
Antioxidant enzymes
To determine if Se supplementation alters antioxidant enzyme capacity, activities of GSH-Px and activities of GSSG-R, were determined. Figure 6 shows Se supplementation did not significantly alter GSSG-R or GST activities. However, animals provided the Sel-Plex diet showed a modest but statistically significant increase in GSH-Px activity. In a surprising finding, GSH-Px activities were also significantly increased in animals provided a Se-deficient diet.
Fig. 6.

Mean ± SEM activities of glutathione peroxidase (GSH-Px), glutathione reductase (GSSG-R) and glutathione transferase (GST) of mice provided a control, Sel-Plex, or Se-deficient diet. GSH-Px activity in Sel-Plex treated mice was increased modestly yet significantly (P<0.05)
Discussion
Multiple studies show significant alterations of markers of oxidative stress including HNE and acrolein (markers of lipid peroxidation), protein oxidation, RNA and DNA oxidation in the brain in AD including subjects early in disease progression (MCI) (reviewed reviewed [1]). Oxidative stress, possibly mediated by Aβ, has been suggested to be central to the pathogenesis of AD [3, 29]. One of the goals of preventive or early treatment of AD is to decrease oxidative stress.
The present study used the APP-PS1 knock-in mouse as a model of amyloid plaque formation that has some similarities to those in AD. Our study shows that Sel-Plex-treated animals have significantly decreased RNA and DNA oxidative damage compared to mice on a normal diet or a Se-deficient diet. An increase in GSH-Px activity also was found in animals treated with Sel-Plex compared to mice on a normal diet. In addition, these oxidative and antioxidant changes were accompanied by a significant decrease in amyloid burden in the Sel-Plex- treated animals.
In addition to the effect on nucleic acid oxidation, animals fed Sel-Plex showed a 20% decrease in HNE and a 15% decrease in protein oxidation, although the differences were not statistically significant for either compared to animals on a normal diet. This suggests that Sel-Plex causes a generalized decrease in oxidative damage, which is most pronounced in DNA and RNA.
Our data showing pronounced levels of RNA and DNA oxidation in APP/PS1 mice are consistent with multiple studies of RNA and DNA oxidation in subjects with a variety of neurological disorders including late AD (LAD) (reviewed [30]. In studies of RNA oxidation, Nunomura et al. [31] showed significantly increased cytoplasmic staining of 8-OHG in early AD brain that decreased as amyloid beta peptide and neurofibrillary tangle burden increased. Increased RNA oxidation has also been observed in a presymptomatic subject who expressed a familial AD mutation [32] and Down syndrome subjects with early AD pathology [33] leading to the suggestion that RNA oxidation is an early event in the pathogenesis of AD. Studies of DNA oxidation in the progression of AD show similar findings with significantly increased levels of 8-OHG in nuclear and mitochondrial DNA from subjects with MCI and LAD (reviewed [1].
Selenium, an essential trace element is a critical component in 25 selenoproteins [34] including iodothyronine deiodinases [35-38] selenoprotien P [39], seleno synthetases [40] and the antioxidant enzymes thioredoxin reductase [40-45] and glutathione peroxidases [6, 43, 46-50]. Dietary selenium is taken up from soil by plants and enters the human food chain as inorganic Se (sodium selenate or sodium selenite) that is converted to organoselenium (selenomethionine, selenocysteine and selenium methylselenocysteine) by animals [51]. Recommended daily allowances for Se are 50 – 70 μg/day for healthy adults [52, 53] with deficiencies occurring with intakes less than 11 μg/day and chemical selenosis occurring at intakes between 3200 and 5000 μg/day [54]. Selenomethinine and methinone are equally recognized during protein synthesis leading to incorporation of Se into proteins [15]. Selenomethionine may also be converted to selenocysteine by trans esterification ultimately leading to generation of H2Se, the key intermediate between reductive metabolism of Se and its methylation pathway and serves as a precursor for synthesis of selenocysteine [14, 55]. Selenomethionne may also be processed by methionine α,β lyase to generate methyl selenium [55].
Selenocysteine from dietary sources or converted from selenomethionine is converted to H2Se that may be reconverted to selenocysteine following conversion of H2Se to selenophosphate by selenophosphate sythetase [51]. Insetion of selenocysteine into proteins is initiated by the UGA condon in mRNA and forms the catalytic site of many selenoproteins [56].
Our previous study using cultured fetal rodent neurons showed that SeMet significantly decreased ROS generation against Aβ and iron/hydrogen peroxide and significantly increased GSH-Px activity. Others have shown that ebselen (2-phenyl-1, 2-benzisoselanazol-3(2H)-one), a Se-containing heterocylic compound, prevents nitric oxide-induced apoptosis of differentiated PC-12 cells by blocking activation of ASK1 and inhibition of p38 mitogen-activated protein kinase and JNK. Ebselen also appears to act as a GSH-Px mimetic. Studies of focal cerebral ischemia in rodents showed ebselen significantly decreases levels of HNE and levels of 8-OHG [57]. Another study showed that treatment of primary human keratinocytes with SeMet or sodium selenite protected against UV-induced oxidative damage [58]. These studies, our recent study, and the present study suggest that Se modulates oxidative stress in vivo and in vitro.
The most pronounced effect of Sel-Plex in our study was on DNA and RNA. Levels of 8-OHG were significantly decreased in both DNA and RNA in Sel-Plex fed animals compared to those on normal diets or Se-deficient diets. Although the reason for the decrease in DNA oxidation is not clear, it could be due to an increase in base excision repair as previously observed by Seo et al. in in-vitro studies [59]. We can only speculate that the decrease in RNA oxidation could relate to the general decrease of oxidative stress in the model.
Our finding of significantly decreased amyloid plaque in Sel-Plex treated animals compared to controls is of considerable interest. The role of Se in decreasing Aβ plaque formation is not clear. Previous studies show treatment of Chinese hamster ovary cells with Se decreased gamma secretase activity by ~50% [60]. In addition, Se alters levels of AP-1, a transcription factor associated with APP transcription, and induces prolonged stimulation of ERK leading to reduced Aβ secretion [61]. Although insufficient tissue was available in the current study to measure AP-1 or ERK levels, it is quite possible that a combination of these effects led to the decrease of Aβ plaque deposition that was observed.
Overall our data indicate that supplementation of low levels of organic Se leads to decreased oxidative damage to RNA and DNA in a transgenic mouse model of Aβ. This suggests that Se could be a neuroprotective agent possibly useful in prevent.
Acknowledgments
Supported by NIH grants 5-P01-AG05119 and 5-P30-AG028383 and by grants from the Abercrombie Foundation and the Healey Family Foundation. The authors thank Ms. Paula Thomason for technical and editorial assistance and Alltech Biotechnology for providing the Sel-Plex diet.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Markesbery WR, Lovell MA. Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol. 2007;64:954–956. doi: 10.1001/archneur.64.7.954. [DOI] [PubMed] [Google Scholar]
- 2.Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol Aging. 1995;16:661–674. doi: 10.1016/0197-4580(95)00066-n. [DOI] [PubMed] [Google Scholar]
- 3.Lovell MA, Xie C, Xiong S, Markesbery WR. Protection against amyloid beta peptide and iron/hydrogen peroxide toxicity by alpha lipoic acid. J Alzheimers Dis. 2003;5:229–239. doi: 10.3233/jad-2003-5306. [DOI] [PubMed] [Google Scholar]
- 4.Milton NG. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: implications for treatment. Drugs Aging. 2004;21:81–100. doi: 10.2165/00002512-200421020-00002. [DOI] [PubMed] [Google Scholar]
- 5.Petersen RC. Mild cognitive impairment: transition between aging and Alzheimer’s disease. Neurologia. 2000;15:93–101. [PubMed] [Google Scholar]
- 6.Flohe L, Gunzler WA, Schock HH. Glutathione peroxidase: a selenoenzyme. FEBS Lett. 1973;32:132–134. doi: 10.1016/0014-5793(73)80755-0. [DOI] [PubMed] [Google Scholar]
- 7.Lippoldt A, Padilla CA, Gerst H, Andbjer B, Richter E, Holmgren A, Fuxe K. Localization of thioredoxin in the rat brain and functional implications. J Neurosci. 1995;15:6747–6756. doi: 10.1523/JNEUROSCI.15-10-06747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitsui A, Hirakawa T, Yodoi J. Reactive oxygen-reducing and protein-refolding activities of adult T cell leukemia-derived factor/human thioredoxin. Biochem Biophys Res Commun. 1992;186:1220–1226. doi: 10.1016/s0006-291x(05)81536-0. [DOI] [PubMed] [Google Scholar]
- 9.Spallholz JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med. 1994;17:45–64. doi: 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 10.Kramer GF, Ames BN. Mechanisms of mutagenicity and toxicity of sodium selenite (Na2SeO3) in Salmonella typhimurium. Mutat Res. 1988;201:169–180. doi: 10.1016/0027-5107(88)90123-6. [DOI] [PubMed] [Google Scholar]
- 11.Seko Y, Imura N. Active oxygen generation as a possible mechanism of selenium toxicity. Biomed Environ Sci. 1997;10:333–339. [PubMed] [Google Scholar]
- 12.Spallholz JE. Free radical generation by selenium compounds and their prooxidant toxicity. Biomed Environ Sci. 1997;10:260–270. [PubMed] [Google Scholar]
- 13.Terada A, Yoshida M, Seko Y, Kobayashi T, Yoshida K, Nakada M, Nakada K, Echizen H, Ogata H, Rikihisa T. Active oxygen species generation and cellular damage by additives of parenteral preparations: selenium and sulfhydryl compounds. Nutrition. 1999;15:651–655. doi: 10.1016/s0899-9007(99)00119-7. [DOI] [PubMed] [Google Scholar]
- 14.Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128:1845–1854. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 15.Shiobara Y, Yoshida T, Suzuki KT. Effects of dietary selenium species on Se concentrations in hair, blood, and urine. Toxicol Appl Pharmacol. 1998;152:309–314. doi: 10.1006/taap.1998.8537. [DOI] [PubMed] [Google Scholar]
- 16.Yoon SO, Kim MM, Park SJ, Kim D, Chung J, Chung AS. Selenite suppresses hydrogen peroxide-induced cell apoptosis through inhibition of ASK1/JNK and activation of PI3-K/Akt pathways. Faseb J. 2002;16:111–113. doi: 10.1096/fj.01-0398fje. [DOI] [PubMed] [Google Scholar]
- 17.Daniels LA. Selenium metabolism and bioavailability. Biol Trace Elem Res. 1996;54:185–199. doi: 10.1007/BF02784430. [DOI] [PubMed] [Google Scholar]
- 18.Xiong S, Markesbery WR, Shao C, Lovell MA. Seleno-L-methionine protects against beta-amyloid and iron/hydrogen peroxide-mediated neuron death. Antioxid Redox Signal. 2007;9:457–467. doi: 10.1089/ars.2006.1363. [DOI] [PubMed] [Google Scholar]
- 19.Kryscio RJ, Mendiondo MS, Schmitt FA, Markesbery WR. Designing a large prevention trial: statistical issues. Stat Med. 2004;23:285–296. doi: 10.1002/sim.1716. [DOI] [PubMed] [Google Scholar]
- 20.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- 21.Wu ZL, Ciallella JR, Flood DG, O’Kane TM, Bozyczko-Coyne D, Savage MJ. Comparative analysis of cortical gene expression in mouse models of Alzheimer’s disease. Neurobiol Aging. 2006;27:377–386. doi: 10.1016/j.neurobiolaging.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Lovell MA, Markesbery WR. Oxidatively modified RNA in mild cognitive impairment. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saiki I, Nayar R, Bucana C, Fidler IJ. A microassay for the rapid and selective binding of cells from solid tumors to mouse macrophages. Cancer Immunol Immunother. 1986;22:125–131. doi: 10.1007/BF00199126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 26.Mizuno Y. Changes in superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase activities and thiobarbituric acid-reactive products levels in early stages of development in dystrophic chickens. Exp Neurol. 1984;84:58–73. doi: 10.1016/0014-4886(84)90006-2. [DOI] [PubMed] [Google Scholar]
- 27.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–169. [PubMed] [Google Scholar]
- 28.Ricci G, Caccuri AM, Lo Bello M, Pastore A, Piemonte F, Federici G. Colorimetric and fluorometric assays of glutathione transferase based on 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole. Anal Biochem. 1994;218:463–465. doi: 10.1006/abio.1994.1209. [DOI] [PubMed] [Google Scholar]
- 29.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 30.Nunomura A, Honda K, Takeda A, Hirai K, Zhu X, Smith MA, Perry G. Oxidative damage to RNA in neurodegenerative diseases. J Biomed Biotechnol. 2006;2006:82323. doi: 10.1155/JBB/2006/82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 32.Nunomura A, Chiba S, Lippa CF, Cras P, Kalaria RN, Takeda A, Honda K, Smith MA, Perry G. Neuronal RNA oxidation is a prominent feature of familial Alzheimer’s disease. Neurobiol Dis. 2004;17:108–113. doi: 10.1016/j.nbd.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 33.Hofer T, Badouard C, Bajak E, Ravanat JL, Mattsson A, Cotgreave IA. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol Chem. 2005;386:333–337. doi: 10.1515/BC.2005.040. [DOI] [PubMed] [Google Scholar]
- 34.Stadtman TC. Discoveries of vitamin B12 and selenium enzymes. Annu Rev Biochem. 2002;71:1–16. doi: 10.1146/annurev.biochem.71.083101.134224. [DOI] [PubMed] [Google Scholar]
- 35.Arthur JR, Nicol F, Beckett GJ. Hepatic iodothyronine 5’-deiodinase. The role of selenium. Biochem J. 1990;272:537–540. doi: 10.1042/bj2720537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Behne D, Kyriakopoulos A, Meinhold H, Kohrle J. Identification of type I iodothyronine 5’-deiodinase as a selenoenzyme. Biochem Biophys Res Commun. 1990;173:1143–1149. doi: 10.1016/s0006-291x(05)80905-2. [DOI] [PubMed] [Google Scholar]
- 37.Croteau W, Whittemore SL, Schneider MJ, St Germain DL. Cloning and expression of a cDNA for a mammalian type III iodothyronine deiodinase. J Biol Chem. 1995;270:16569–16575. doi: 10.1074/jbc.270.28.16569. [DOI] [PubMed] [Google Scholar]
- 38.Davey JC, Becker KB, Schneider MJ, St Germain DL, Galton VA. Cloning of a cDNA for the type II iodothyronine deiodinase. J Biol Chem. 1995;270:26786–26789. doi: 10.1074/jbc.270.45.26786. [DOI] [PubMed] [Google Scholar]
- 39.Motsenbocker MA, Tappel AL. Effect of dietary selenium on plasma selenoprotein P, selenoprotein P1 and glutathione peroxidase in the rat. J Nutr. 1984;114:279–285. doi: 10.1093/jn/114.2.279. [DOI] [PubMed] [Google Scholar]
- 40.Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346(Pt 1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SR, Kim JR, Kwon KS, Yoon HW, Levine RL, Ginsburg A, Rhee SG. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J Biol Chem. 1999;274:4722–4734. doi: 10.1074/jbc.274.8.4722. [DOI] [PubMed] [Google Scholar]
- 42.Lescure A, Gautheret D, Carbon P, Krol A. Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. J Biol Chem. 1999;274:38147–38154. doi: 10.1074/jbc.274.53.38147. [DOI] [PubMed] [Google Scholar]
- 43.Takahasi K, Sakurada T, Sakurada S, Kuwahara H, Yonezawa A, Ando R, Kisara K. Behavioural characterization of substance P-induced nociceptive response in mice. Neuropharmacology. 1987;26:1289–1293. doi: 10.1016/0028-3908(87)90089-x. [DOI] [PubMed] [Google Scholar]
- 44.Watabe S, Makino Y, Ogawa K, Hiroi T, Yamamoto Y, Takahashi SY. Mitochondrial thioredoxin reductase in bovine adrenal cortex its purification, properties, nucleotide/amino acid sequences, and identification of selenocysteine. Eur J Biochem. 1999;264:74–84. doi: 10.1046/j.1432-1327.1999.00578.x. [DOI] [PubMed] [Google Scholar]
- 45.Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase two modes of catalysis have evolved. Eur J Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- 46.Chu FF, Doroshow JH, Esworthy RS. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J Biol Chem. 1993;268:2571–2576. [PubMed] [Google Scholar]
- 47.Flohe LG, W A, Coschen G. The glutathione peroxidase reaction: A key to understand the selenium reqirement of mammls. Trace metals in health and disease. 1979:263–286. [Google Scholar]
- 48.Mills GC. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J Biol Chem. 1957;229:189–197. [PubMed] [Google Scholar]
- 49.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 50.Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- 51.Letavayova L, Vlckova V, Brozmanova J. Selenium: from cancer prevention to DNA damage. Toxicology. 2006;227:1–14. doi: 10.1016/j.tox.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 52.El-Bayoumy K. The protective role of selenium on genetic damage and on cancer. Mutat Res. 2001;475:123–139. doi: 10.1016/s0027-5107(01)00075-6. [DOI] [PubMed] [Google Scholar]
- 53.Whanger PD. Selenium and its relationship to cancer: an update. Br J Nutr. 2004;91:11–28. doi: 10.1079/bjn20031015. [DOI] [PubMed] [Google Scholar]
- 54.Reid ME, Stratton MS, Lillico AJ, Fakih M, Natarajan R, Clark LC, Marshall JR. A report of high-dose selenium supplementation: response and toxicities. J Trace Elem Med Biol. 2004;18:69–74. doi: 10.1016/j.jtemb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Meuillet E, Stratton S, Prasad Cherukuri D, Goulet AC, Kagey J, Porterfield B, Nelson MA. Chemoprevention of prostate cancer with selenium: an update on current clinical trials and preclinical findings. J Cell Biochem. 2004;91:443–458. doi: 10.1002/jcb.10728. [DOI] [PubMed] [Google Scholar]
- 56.Hatfield DL, Gladyshev VN. How selenium has altered our understanding of the genetic code. Mol Cell Biol. 2002;22:3565–3576. doi: 10.1128/MCB.22.11.3565-3576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imai H, Masayasu H, Dewar D, Graham DI, Macrae IM. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke. 2001;32:2149–2154. doi: 10.1161/hs0901.095725. [DOI] [PubMed] [Google Scholar]
- 58.Rafferty TS, Green MH, Lowe JE, Arlett C, Hunter JA, Beckett GJ, McKenzie RC. Effects of selenium compounds on induction of DNA damage by broadband ultraviolet radiation in human keratinocytes. Br J Dermatol. 2003;148:1001–1009. doi: 10.1046/j.1365-2133.2003.05267.x. [DOI] [PubMed] [Google Scholar]
- 59.Seo YR, Sweeney C, Smith ML. Selenomethionine induction of DNA repair response in human fibroblasts. Oncogene. 2002;21:3663–3669. doi: 10.1038/sj.onc.1205468. [DOI] [PubMed] [Google Scholar]
- 60.Zhu X, Raina AK, Perry G, Smith MA. Alzheimer’s disease: the two-hit hypothesis. Lancet Neurol. 2004;3:219–226. doi: 10.1016/S1474-4422(04)00707-0. [DOI] [PubMed] [Google Scholar]
- 61.Tung YT, Hsu WM, Wang BJ, Wu SY, Yen CT, Hu MK, Liao YF. Sodium selenite inhibits gamma-secretase activity through activation of ERK. Neurosci Lett. 2008;440:38–43. doi: 10.1016/j.neulet.2008.05.048. [DOI] [PubMed] [Google Scholar]