

Abstract
Ras proteins are small GTPases that regulate cellular growth and differentiation. Components of the Ras signaling pathway have been shown to be important during embryonic vasculogenesis and angiogenesis. Here, we report that Rasip1, which encodes a novel Ras-interacting protein, is strongly expressed in vascular endothelial cells throughout development, in both mouse and frog. Similar to the well-characterized vascular markers VEGFR2 and PECAM, Rasip1 is specifically expressed in angioblasts prior to vessel formation, in the initial embryonic vascular plexus, in the growing blood vessels during angiogenesis and in the endothelium of mature blood vessels into the postnatal period. Rasip1 expression is undetectable in VEGFR2 null embryos, which lack endothelial cells, suggesting that Rasip1 is endothelial-specific. siRNA-mediated reduction of Rasip1 severely impairs angiogenesis and motility in endothelial cell cultures, and morpholino knockdown experiments in frog embryos demonstrate that Rasip1 is required for embryonic vessel formation in vivo. Together, these data identify Rasip1 as a novel endothelial factor that plays an essential role in vascular development.
Keywords: vasculogenesis, angiogenesis, blood vessel, endothelium, migration, proliferation, Ras, Rasip1, VEGFR2, Flk-1
Introduction
The cardiovascular system, which includes the entire network of blood vessels and the heart, is the first functional organ system to form in the embryo. Defects in the structure and/or function of the cardiovascular system inevitably lead to early embryonic lethality (Cleaver and Krieg, 1999). Initially, the vasculature emerges from aggregation of angioblasts. Angioblasts are endothelial precursors that arise from mesodermal cells that differentiate either within blood islands, structures composed of hematopoietic cells (blood cell precursors) surrounded by a mantle of angioblasts, or within embryonic tissues as scattered cells. Vessels subsequently form via vasculogenesis, or the coalescence of individual angioblasts “in situ” to form primitive vascular ‘cords’, which then undergo tubulogenesis (Risau and Flamme, 1995). The first vessels consist of a relatively simple and homogeneous endothelial cell (EC) network of vessels, often termed a ‘plexus’. Subsequently, the complexity of the vasculature increases dramatically as new vessels sprout and extend from pre-existing vessels, via a process called angiogenesis (Risau, 1997). Angiogenic remodeling of blood vessels then transforms the initially simple, net-like, primary plexus, into a complex hierarchical network of large and small vessels, which includes specialized ECs, such as arteries and veins. As these vessels mature and stabilize, they become ensheathed by smooth muscle cells and pericytes. However, they continue to grow coordinately with organs and tissues, providing the tissues they perfuse with the nutrients and oxygen required for viability.
Most of the molecular mechanisms responsible for blood vessel formation are not yet well understood. For decades, much attention was given to the role of vascular endothelial growth factor (VEGF) and its influence on EC migration and proliferation (Ferrara et al., 2003; Yancopoulos et al., 2000). Recently, however, discovery of a host of endothelial ‘guidance’ cues, which either attract or repel ECs and shape individual blood vessels, has broadened our understanding of how cell-cell signaling influences the morphogenesis of individual vessels and the vascular network as a whole. These signaling molecules include the Eph-ephrins (Kuijper et al., 2007), bone morphogenetic proteins (BMPs) (Lebrin et al., 2005; Moser and Patterson, 2005; Park et al., 2006), transforming growth factors (TGFβs) (Lebrin et al., 2005), Notch and Notch ligands (Roca and Adams, 2007) and many others. In addition, a number of cell-autonomous factors have also recently been shown to be critical for proper EC behavior and blood vessel formation. Many of these factors, such as small GTPases Ras, Rho, Rac, Cdc42, Pak and their many effectors/modulators (Fryer and Field, 2005; Garnaas et al., 2008; Gitler et al., 2003; Kranenburg et al., 2004; Merajver and Usmani, 2005; Tan et al., 2008) are already known to drive basic cell processes such as cell migration, cell proliferation and establishment of cell polarity. Despite recent advances, the molecular mechanisms underlying much of blood vessel formation in vivo remain unclear, and elucidation of both extracellular signaling events and cell autonomous regulatory signaling cascades will advance our understanding of vascular specification and patterning, in both normal and pathological conditions (Coultas et al., 2005).
Many Ras family members and their regulators have been implicated in vascular development (Gitler et al., 2003; Henkemeyer et al., 1995; Tan et al., 2008), including EC migration (Sosnowski et al., 1993; Tan et al., 2008), capillary tube assembly (Connolly et al., 2002), angiogenesis (Aitsebaomo et al., 2004; Fryer and Field, 2005; Kranenburg et al., 2004; Merajver and Usmani, 2005), blood vessel homeostasis (Komatsu and Ruoslahti, 2005) and vascular permeability (Serban et al., 2008). Ras molecules are small GTPases widely shown to function as molecular switches coordinating multiple cellular behaviors like growth, proliferation, migration and differentiation. Ras GTPases cycle between the GTP-bound (active) and GDP-bound (inactive) states, under the influence of GAPs (GTPase Activating Proteins), and GEFs (GTPase Exchange Factors). Ras family proteins have been shown to activate signaling cascades downstream of VEGF (Cross et al., 2003; Kranenburg et al., 2004; Roberts et al., 2004). VEGF stimulation of ECs increases the amount of activated Ras, while dominant negative Ras constructs inhibit VEGF-induced endothelial proliferation, migration and assembly (Meadows et al., 2001). However, although the Ras pathway proteins have been implicated in vascular development, their exact role is not well understood.
Recently, Mitin and colleagues reported the identification of a novel Ras-interacting protein, Rasip1/Rain, which displays the characteristics of an endomembrane Ras effector (Mitin et al., 2004). Their experiments showed that Rasip1 possesses a Ras-associating domain (RA), homologous to the RA domains of other Ras effectors, and that Rasip1 preferentially binds to the GTP-loaded form of Ras, both in vitro and in vivo. In addition, they demonstrated that transfected Rasip1 localizes to a perinuclear, juxta-Golgi region in intact COS cells and is recruited to the Golgi by active Ras. Its enrichment in adult lung and high expression in transformed EC lines suggested the possibility that Rasip1 is expressed by ECs (Mitin et al., 2006).
In an effort to discover unknown regulators of blood vessel development, we performed a microarray screen that transcriptionally profiled embryonic aortal ECs (Xu and Cleaver, unpublished). Among numerous EC-enriched transcripts, we identified Rasip1. Here, we show that expression of Rasip1 is strikingly restricted to the endothelium of the developing vasculature, in both frog and mouse, and we demonstrate that Rasip1 is essential for proper endothelial cell angiogenic assembly and migration, both in vivo and in vitro. We propose that Rasip1 plays important roles during vasculogenesis and angiogenesis, possibly regulating the function of Ras proteins in ECs.
Materials and Methods
Isolation of Rasip1 sequence
A pYX plasmid containing mouse Rasip1 cDNA piece (1047–3170bp, spanning exons 4 through 7) was obtained from OpenBiosystems (BC072584). For making longer in situ probes, the full-length coding region (2886bp) of Rasip1 was amplified from E8.5 mouse cDNA by RT-PCR, using 5’ primer ATGCTATCTGGTGAACGAAAG and 3’ primer TCAAGGTGTCGAAGCCACCG. PCR fragments were inserted into pGEM-T-Easy Vector (Promega) by TA cloning. Xenopus tropicalis Rasip1 partial coding region sequence (1151bp, exon2-exon7) was cloned by RT-PCR using primers: 5’ primer ATTAAGGGAAAGAGAAGAAAGCATCT and 3’ primer GCATACAGTGTCTTGGTCAGATAATATAC. The amplified fragment was subcloned into pGEM-T-Easy Vector (Promega) by TA cloning.
Embryos and Histology
CD1 embryos were collected from pregnant females (E7.5 through E15.5) after dissection in ice-cold PBS buffer and fixed in 4% paraformaldehyde in PBS solution overnight at 4°C with gentle rocking. The amnion was removed during dissection for better probe penetration. Embryos were washed three times in PBS for 5 min, and dehydrated using a series of ethanol washes. Embryos were then stored in 75% ethanol at −20°C. Postnatal tissue was collected and fixed in a similar manner.
For wax sectioning of embryos following in situ hybridization, the embryos were fixed and dehydrated as described above. Embryos were rinsed twice in 100% ethanol for 5 min, twice in xylene at room temperature (RT) for 10 min, then a mixture of 1:1 paraplast:xylene at 60°C for 10 min, then a series of 100% paraplast at 60°C (McCormick Scientific). The embryos were then embedded and sectioned with a Biocut 2030 microtome. For examination, the sections were placed on glass slides, deparaffinized in xylene twice for 5 min each and mounted on SuperfrostPlus glass slides (Fisher) using Permount (Fisher).
Digoxigenin-labeled RNA probes
Rasip1 (in pYX-plasmid) was linearized using EcoRI and an antisense Digoxigenin(Dig)UTP-labeled RNA probe was synthesized using T3 polymerase. VEGFR2 coding region was amplified from mouse E8.5 cDNA with primers 5’-GACGGAGAAGGAGTCTGTGC and 3’-GGGACAGGACCACTTCCAT and cloned into pGEM-T-Easy vector. This VEGFR2 clone was linearized using SpeI and an antisense Dig-labeled RNA probe was synthesized using T7 polymerase. A PECAM clone, containing 950bp of 3’UTR (and kindly provided by D. Melton), was linearized using XhoI and an antisense Dig-labeled RNA probe was synthesized using T3 polymerase. Probe synthesis was carried out at 37°C for 2 hrs: 1μg linearized plasmid, 2.0μl DIG-RNA labeling mix (Roche), 2.0μl 10X transcription buffer (Roche), 1.5μl Placental ribonuclease inhibitor (Promega), 1.0μl T3/T7 RNA polymerase (Roche), RNase free water to a final volume of 20μl. DNA template was removed using 2μl RQ1 DNase I (Promega), at 37°C for 15 min. The probes were then purified with Micro Biospin columns (Bio-RAD). 10x hybridization stock solution was prepared at a concentration of 10μg/ml in ‘prehyb’ solution: 50% Formamide (Fisher), 5×SSC (pH 4.5), 50μg/ml Ribonucleic acid from Torula yeast, Type VI (Sigma), 1% SDS, 50μg/ml Heparin (Sigma). Stock solution is stored at −80°C.
Whole mount in situ hybridization
Whole mount in situ hybridization in mouse embryos was carried out using a protocol adapted from D. Wilkinson’s Method (Wilkinson, 1999). Briefly, embryos stored in 75% ethanol at −20°C were rehydrated in stepwise fashion to PBST. Then, the embryos were treated with 10μg/ml proteinase K (time treated varied with age of tissue; 2min-30min), fixed in a 0.2% gluteraldehyde/4% paraformaldehyde (PFA) solution, and pre-hybridized at 65°C for 1 hour. The samples were transferred into hybridization mix, containing 1μg/ ml Dig-labeled probes described above. The in situ hybridization post-hybridization washes and antibody incubation were carried out using a Biolane HTI automated incubation liquid handler (Holle & Huttner). Color development was carried out using BM purple solution (Roche). Frog in situ hybridization was carried out using a similar standard in situ hybridization protocol (Costa et al., 2003).
In situ hybridization on sections
Paraffin sections (on glass slides) were washed 3×3min in PBS, followed by a 10min treatment with 15μg/ml proteinase K. Sections were then rinsed in PBS, fixed in 4% PFA for 5 min, and incubated for 10min in acetylation solution: mix of 2.66ml Triethanolamine, 350μg HCl, 750μg acetic anhydride and 200ml water. Prehybridization was carried out in plastic slide mailers (Fisher) containing hybridization buffer at RT for 1 hour. Slides were then transferred to a humidified chamber (humidified with 50% formamide/5×SSC) for probe hybridization (probe at 1μg/ml) with 100μl probe/slide (covered with glass coverslips) at 68°C overnight.
Slides were washed post-hybridization in 2×SSC at 72°C just long enough to allow coverslips to separate. Then slides were rinsed in 0.2×SSC at 72°C and RT for 1x1min, respectively, then MBST buffer at RT (100mM Maleic acid, 150mM NaCl, pH7.5, 0.1% Tween20). Slides were incubated in blocking solution (2% blocking reagent (Roche) and 5% heat-inactivated sheep serum in MBST) for 1 hour at RT. Anti-Dig alkaline phosphatase conjugated antibody was applied on slides in a chamber humidified with MBST (250μl of 1/4000 anti-Dig antibody (Roche)), covered with parafilm and incubated at 4°C overnight. Slides were washed for 3×30min in MBST after antibody incubation, and treated in NTMT (100mM NaCl, 100mM Tris, pH9.5, 50mM MgCl2, 0.1% Tween20) for 3×5min. Color reaction was carried out using BM purple as described above. For microscopic examination, slides were sealed and coverslipped using Permount (Fisher).
VEGFR2 null embryo generation and β-Galactosidase reaction
VEGFR2 null embryos were generated by mating Flk1(VEGFR2)-lacZ heterozygous males and females (kindly provided by Drs. Janet Rossant and Eli Keshet). Embryos were dissected manually in ice cold PBS. Embryos lacking blood vessels were identified visually, by the absence of yolk sac blood vessels, and genotypes (of either embryos or adults) were confirmed by PCR, using primers to lacZ; 5’primer GGTGGCGCTGGATGGTAAGC, 3’primer CGCCATTTGACCACTACC, which yield a 630bp PCR fragment. For the β-Galactosidase reaction Flk-1(VEGFR2)+/- and Flk-1(VEGFR2)-/- embryos (or isolated organs) were fixed in 5mM EGTA (pH 8.0), 0.2% gluteraldehyde, 2mM MgCl2 and PBS solution for 15 min on ice. After fixation, embryos were rinsed 3 times for 5 min in PBS. 50 mM Potassium Ferrocyanide (K4Fe(CN)6·3H2O) and Potassium Ferricyanide (K3Fe(CN)6) solutions, stored at RT in dark, were used to make lacZ staining solution: 20 mM K4Fe(CN)6·3H2O, 20 mM K3Fe(CN)6, 2 mM MgCl2, 0.02% NP-40, add water or 1×PBS to 500 μl. Staining solution was warmed to 37°C before adding X-Gal (Growcells) to avoid X-Gal precipitation. 4 μl of 100 mg/ml X-Gal stock (in dimethyl formamide) was then added to the lacZ staining solution. Embryos were placed in staining solution and color reaction was allowed to develop at 37°C overnight. When staining was evaluated to be optimal, embryos were washed with PBS 3 times for 5 min each, post-fixed in 4%PFA overnight, and transferred to 80% glycerol for viewing.
siRNA transfection and endothelial cell assays
siRNAs were ordered from IDT-DNA as the TriFECTa Kit. Sequences: siHPRT: 5’-AAUUUCAAAUCCAACAAAGUCUGGCUU. siRasip1: 5’ -CCAUCUCUAGCACUUUCUCCUGUACAA. The transfection was carried out in the 24-well plate format. For each well, 1.25μl of 20μM dicer substrate siRNA was diluted in 50μl of Opti-MEM I Reduced Serum Medium (Invitrogen). 1 μl of Lipofectamine 2000 (Invitrogen) transfection reagent was diluted in another 50μl of Opti-MEM I Reduced Serum Medium. After 5 minutes incubation at RT, the diluted siRNA and the transfection reagent were combined together, and incubated for 20 minutes at RT. MS1 cells (ATCC) were plated on a 24-well plate with a density of 5 × 104 cells/well in 400μl DMEM containing 10% FBS and without penicillin/streptomycin. The pre-mixed 100μl transfection complexes were then added drop-wise on top of the cells. After gentle mixing by rocking the plate back and forth, the cells were incubated at 37°C in a 5% CO2 incubator prior to following assays.
For analyzing transcription of the targeted genes in these assays, cells were trypsinized 72 hours post transfection, and the total RNAs were isolated using an Rneasy Mini Kit (Qiagen). First strand cDNAs were made using M-MLV Reverse Transcriptase (Promega) based on manufacturer standard protocols. Rasip1 primers: 5’ primer GGAGCAGCTTACGGACTGAC, 3’ primer CCATCGTCTACCAACCCAAC. HPRT primers: idtDNA HPRT primer set. β-Actin primers: 5’ primer GTTGGTTGGAGCAAACATCC, 3’ primer AGGGAGACCAAAGCCTTCAT. The transcripts were amplified in a 30-cycle polymerase chain reaction.
‘Tube-formation assays’ were carried out in a 96-well plate. 50μl of Matrigel (BD Matrigel 354234) was thawed on ice and plated on the bottom of each well. ECs cultured in one well of a 24-well plate (90% confluency) were trypsinized, plated in one Matrigel coated well of a 96-well plate and cultured at 37°C. When using wild type cells, the angiogenic aggregation of ECs (or ‘tubes) starts to occur within a few hours. For better viewing, cells were stained with 1μM fluorescent dye Calcein-AM (Cell Biolabs) before microscopic examination. Quantification of angiogenic branchpoints was accomplished by counting observable branchpoints within 8 representative areas within each plated well. ‘Branchpoints’ are defined as the intersection point of two linear, vessel-like vascular structures, as previously defined by others (Hellstrom et al., 2007).
‘Wound-healing’ assays were carried out 72 hours post-transfection. Briefly, the cell monolayer is scratched using a sterile P200 pipet tip to create a ‘cell-free’ area (the wound, width of ~600μm). The cells were then immediately washed once with DPBS to remove detached cells from the wound area. Cells on the scratched plate are then allowed to recover and migrate into the ‘cell-free’ area. Images were acquired immediately after scratching and rinsing, and also after an overnight incubation at 37°C for comparison of wound width. Distance migrated was calculated as half of the total change in width.
Cell proliferation was analyzed in cultured ECs by Ki67 staining. 72 hours post-transfection of siRasip1, cells were washed 3 times in PBS, and then fixed in 4% PFA for 10 min at RT. For better antibody penetration, the cells were incubated in PBSN (0.1% NP-40 in PBS) for 15 min with gentle rocking. Cells were then incubated in blocking solution (5% donkey serum (sigma), 1% BSA (Fisher) in PBSN) for 30 min at RT. Primary rabbit anti-Ki67 antibody (Vector laboratories) and secondary antibodies (Alexa488 conjugated anti-rabbit, Invitrogen) were applied at 1:500 dilution in blocking solution at RT for 1 hour. Cells were rinsed 3×5 min in PBSN following each antibody incubation. Cells were then mounted in Vectashield mounting media (Vector laboratories) and examined using a Zeiss Axiovert fluorescent microscope.
Morpholino (MO) knockdown of Xenopus Rasip1
Xenopus tropicalis embryos were injected with 16ng Rasip1-MO (Gene-tools) into 1 cell at the 2-cell stage for assessment of vascular defects using in situ hybridization, or into both cells for assessment of transcript knockdown by RT-PCR. Embryos were allowed to develop to stage 32, then fixed in preparation for in situ hybridization. Morpholino-injected embryos were fixed in MEMFA (0.1M MOPS pH7.4, 2mM EGTA, 1mM MgCl2, 4% PFA), transferred to 100% ethanol and stored at -20°C. For evaluation of transcript knockdown efficiency, embryos were allowed to develop to either stage 25/26 or 29/30 and frozen directly on dry ice for RT-PCR.
Results
Identification of Rasip1 expression in murine endothelial cells
To identify sequences enriched in the embryonic dorsal aortae, we carried out Affymetrix microarray screening of aortal ECs from E8.25 mouse embryos (Xu and Cleaver, unpublished). dChip (Li and Wong, 2001) and Genespring software analysis was used to compare array data (to non-vascular array sets) and extract endothelial-enriched sequences. We initially identified Rasip1 as an EST (AI853551) showing 50-fold enrichment in ECs over other tissues. A longer clone was acquired commercially (OpenBiosystems), allowing production of Dig-labeled antisense probes encompassing the region from exon 4 through exon 12 (~2000bp) of the Rasip1 transcript. The Rasip1 genomic structure has been previously described (Mitin et al., 2004), however no developmental expression or function has been reported.
Rasip1 is expressed in vascular endothelium during vascular plexus formation (E7.5-E10.0)
Using in situ hybridization, we characterized embryonic expression of Rasip1 in mouse embryos and found it to be principally expressed in vascular endothelium. At E7.0 Rasip1 is initially detected in the parietal yolk sac, in a punctate ring of cells (data not shown). Soon thereafter, at E7.5, expression expands to scattered cells of the extraembryonic yolk sac blood islands (Fig.1A). At E8.0, individual cells expressing Rasip1 within the extraembryonic mesoderm can be observed at increasingly ventrolateral locations, in regions previously described as containing angioblasts (Drake et al., 2000; Ferkowicz and Yoder, 2005). The punctate appearance of Rasip1 expression in extraembryonic tissues at this stage suggests that these cells are angioblasts (Fig.1B), as it closely resembles that of vascular endothelial growth factor receptor 2, VEGFR2 (or Flk1/KDR), and Tal1 both established markers for early angioblasts (Drake and Fleming, 2000). At E8.25-E8.5, Rasip1 is strongly expressed throughout the embryonic and extraembryonic endothelium in a pattern recognizable as the primary vascular plexus, including the endocardium, the forming dorsal aortae and the primordia of the cardinal veins (Fig.1C,D). During these stages, vasculogenesis of the principal embryonic blood vessels is occurring and major vessels are taking shape (i.e. parallel dorsal aortae in Fig.1D) (Walls et al., 2008).
Figure 1. Expression of Rasip1 in vascular endothelium during early embryogenesis.
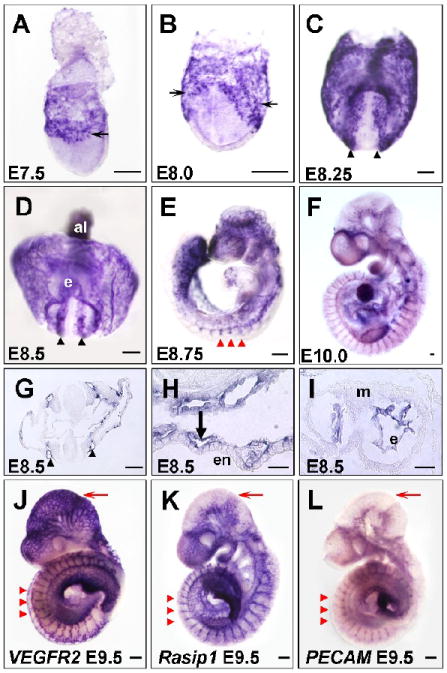
In situ hybridization showing expression of Rasip1 in embryonic vessels at stages indicated (A-I, K). A-F) Whole mount in situ hybridization showing whole stained embryos. Note expression in both scattered angioblasts (thin arrows), forming dorsal aortae (black arrowheads), intersomitic vessels (ISVs) (red arrowheads) and yolk sac vessels (thick arrows). G-I) Transverse sections of in situ hybridizations showing endothelial-specific expression of Rasip1 in G) dorsal aortae, H) yolk sac vessels, and I) heart endocardium. J-L) Comparison of Rasip1 expression with that of the vascular markers VEGFR2 and PECAM, in E9.5 embryos. Note overall similarity of expression, especially in the ISVs (red arrowheads) and trunk vessels. Note difference in intensity of vascular staining in distinct regions, such as the cephalic vessels (red arrows). al, allantois; e, endocardium; en, endoderm; m, myocardium. The scale bars represent 200μm in all panels except J-L, where they represent 50μm.
As embryogenesis continues, Rasip1 expression continues to be expressed in developing blood vessels. After embryonic turning, at E8.75, expression is evident in all large and small blood vessels, including the sprouting intersomitic/intersegmental vessels (ISVs) (Fig.1E). Expression of Rasip1 within ISVs of later embryos (Fig.1E,F,K) suggests a role not only during vasculogenesis, but also during extension of vascular sprouts, or angiogenesis. Transverse sections through E8.5 embryonic tissues reveal that expression is restricted to the endothelium in all tissues examined, including the dorsal aortae (Fig.1G) and yolk sac vessels (Fig.1H). In addition, Rasip1 is expressed within the endothelium of the endocardium, but not in myocardium (Fig.1I).
We compared expression of Rasip1 with that of other known vascular markers, such as VEGFR2 and PECAM (Fig.1J-L and Suppl. Fig.1), and found that Rasip1 outlined almost identical vascular structures in the embryo. For instance, expression of all three markers was observed in aortae, ISVs, endocardium and vessels of the lateral plate and head mesoderm. Of note, different vascular beds appeared to express these three vascular markers with varying intensity. For instance, the head plexus of E9.5 embryos expressed VEGFR2 more robustly, while Rasip1 was more strongly expressed than either VEGFR2 or PECAM in the ISVs and endocardium (Fig.1J-L and Suppl. Fig.1). These differences reveal surprising endothelial heterogeneity at early stages of vascular development. Nonetheless, overall expression analysis suggests that Rasip1 is primarily restricted to vascular endothelium.
Rasip1 during late embryogenesis (E10.5-birth)
Analysis of Rasip1 transcripts later during development reveals their expression in established vessels. Expression could indeed be detected in the blood vessels of various organs throughout midgestion stages (Fig.2 and Suppl. Fig.2). Specifically, we found Rasip1 strongly expressed in the vessels of all embryonic organs and tissues examined, including heart (Fig.2B,E, Suppl. Fig.2B,C,E,H), lung (Fig.2B’,E’, Suppl. Fig.2C,D,G), head (Fig.2B”), limb bud (Fig.2E”), pancreas, spleen and stomach (Suppl. Fig.2J-L). When compared to the expression of VEGFR2 (Fig.2 columns A,D) and PECAM (Fig.2 columns C,F), we found that Rasip1 generally marked identical vascular beds, with only slight variations in expression intensity (Fig.2 columns B,E). Expression of Rasip1 in later embryonic vessels, after their formation via either vasculogenesis or angiogenesis, implies that it has a maintenance function in mature vessels. Indeed, Rasip1 continued to be expressed in the endothelium of vessels into postnatal stages (Suppl. Fig.3) and was detected in adult organs, particularly in the highly vascularized lung (Mitin et al., 2004).
Figure 2. Vascular expression of Rasip1 in embryonic organs and tissues.
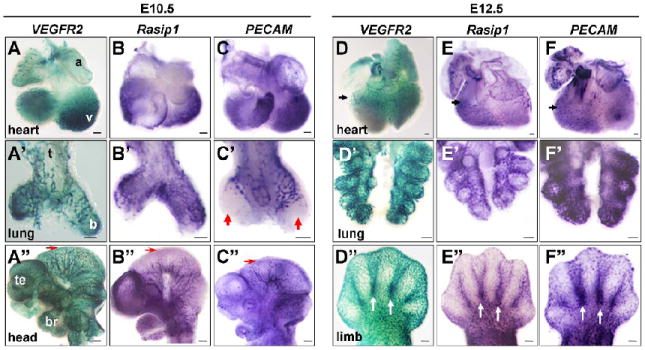
Flk1(VEGFR2)-lacZ whole mount β-galactosidase staining and whole mount in situ hybridization of vascular markers in isolated embryonic tissues, at stages indicated. A,D columns) Whole mount β-galactosidase staining using Flk1(VEGFR2)-lacZ embryos. Whole mount in situ hybridization of Rasip1 (B,E columns) and PECAM (C,F columns). A-F) Hearts. A’-F’) Lungs. A”-C”) Heads. D”-F”) Limb buds. Note similarity of expression of Rasip1 in most vessels, as marked by VEGFR2 and PECAM expression (black arrowheads). Expression of all three vascular markers can be observed in the endocardium of the ventricle trabeculae in the heart (A-C) and of the coronary vasculature (arrows, D-F). Expression of all three markers is evident in the proximal ECs of the early lung buds (A’-C’), although PECAM is not expressed in the ECs of the most distal tips of the buds at E10.5 (red arrowheads), while both VEGFR2 and Rasip1 are observed in this population. This heterogeneity is also observed in the cephalic vessels at E10.5, where VEGFR2 is robustly expressed in the most mediolateral/distal vessels of the mesencephalon, while Rasip1 and PECAM are expressed at lower levels (red arrows, A”-C”). Rasip1 is expressed in the vessels of the developing limb buds, including the interdigit vessels (white arrows, D”-F”). a, atria; b, bronchus; br, branchial arches; t, trachea; te, telencephalon; v, ventricle. The scale bars represent 100μm in A-F’ and 250μm in A”-F”.
Rasip1 expression is absent in vascularless embryos
To definitively test whether Rasip1 is restricted to vascular endothelium, we assessed its expression in VEGFR2 mutant embryos that lack vascular endothelium (Shalaby et al., 1995). First, we compared Rasip1 expression (Fig.3 column B) to that of Flk1(VEGFR2)-lacZ (Fig.3 column A) in VEGFR2+/- heterozygous mice, which display no detectable abnormalities, and found both outlined the developing vasculature as expected. However VEGFR2-/- homozygotes, which lack all blood vessels, exhibited no trace of Rasip1 expression by in situ hybridization (Fig.3 column C). Rasip1 and VEGFR2 are expressed in similar vascular domains in VEGFR2 heterozygotes, from E8.25 to E9.0, while in VEGFR2 null embryos we observed no Rasip1 expression in any embryonic region. These findings suggest that Rasip1 is expressed exclusively in VEGFR2-dependent cell types. Given that VEGFR2 is primarily expressed in and required for ECs (Shalaby et al., 1995; Yamaguchi et al., 1993), it is likely that Rasip1 is also expressed exclusively in those cell types, but not in other non-vascular mesodermal or mesenchymal cell populations. We propose that Rasip1 is a novel and largely specific marker of embryonic blood vessels throughout development.
Figure 3. Expression of Rasip1 is restricted to VEGFR2-dependent endothelium.
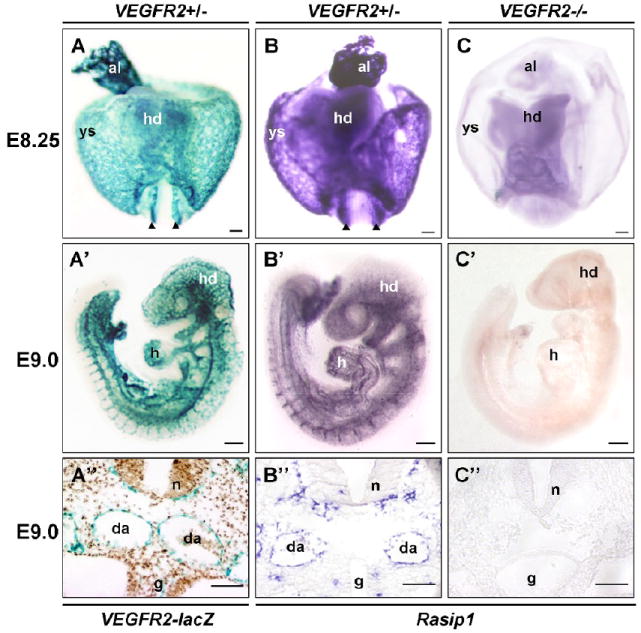
Whole mount in situ hybridization and β-galactosidase staining to detect expression of Rasip1 and VEGFR2. A,B,A’,B’) Comparison of Flk1(VEGFR2)-lacZ staining and Rasip1 expression. Note overall similarity of expression. Rasip1 expression closely resembles Flk1(VEGFR2)-lacZ expression at both E8.5 (A,B) and E9.0 (A’,B’). C,C’) VEGFR2-/- null embryos, lacking all endothelium. Embryos in C-C” have been stained by in situ hybridization for Rasip1 expression and allowed to develop same length of time as wildtype embryos in B-B”. Note complete lack of Rasip1 expression in these mutants. A”-C”) Sections through embryos in A’-C’ showing presence of aortae and perineural vascular plexus in wildtype embryos, while these vascular structures are missing in VEGFR2-/- embryos. al, allantois; g, gut tube; h, heart; hd, head; da or black arrowheads, dorsal aortae; n, neural tube; ys, yolk sac. The scale bars represent 200μm (A’-C’) and 50 μm (A-C, A”-C”).
Rasip1 is required for angiogenesis in cultured ECs
To identify the potential role of Rasip1 in ECs, we used an in vitro siRNA approach to knockdown endogenous Rasip1 expression in cultured mouse ECs (Fig.4A). To identify endothelial cell lines that expressed Rasip1, we screened a number of lines using RT-PCR. We found that MS1, bEnd.3 and SVEC endothelial cell lines (ATCC) all expressed Rasip1 to varying degrees, while non-vascular lines such as HEK293, Balb3 and NIH3T3 did not (data not shown), supporting the notion that Rasip1 is a marker of endothelium. MS1 cells expressed the highest levels of both Rasip1 and VEGFR2, therefore we chose this line for subsequent assays. When siRNAs targeting Rasip1 were transfected into MS1 cells, the cells displayed an abnormal ‘elongated’ morphology, compared to untreated (wildtype, WT) or positive (siHPRT) control cells within 5 days of transfection (Suppl. Fig.4). This suggested a specific effect, resulting from loss-of-function of Rasip1 in MS1 cells.
Figure 4. Rasip1 ablation in MS1 cells by transient siRNA transfection hinders endothelial tube formation and migration ability.
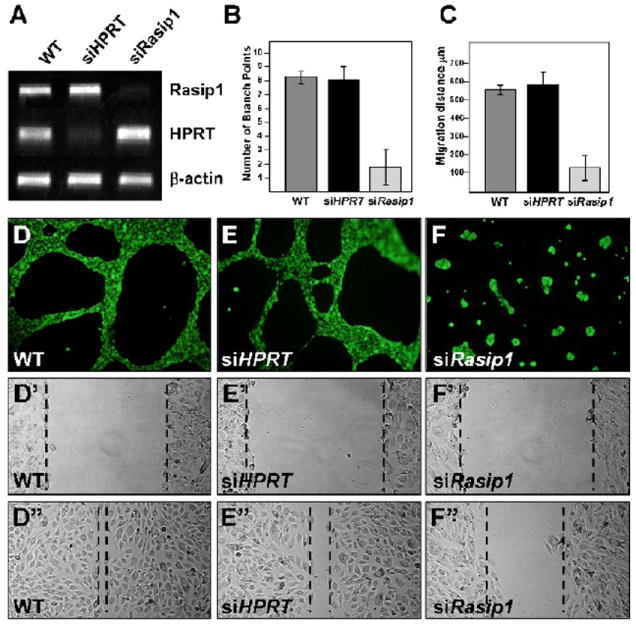
A) Semi-quantitative RT-PCR (30 cycles) shows the knockdown of Rasip1 at the mRNA level. HPRT control knockdown is also shown. B,D-F) siRasip1 treated MS1 cells fail to form “tubes” when plated on Matrigel. B) Quantification shows that formation of linear structures (tubes or cords) as measured by counting branching points in the vascular plexus, is decreased by approximately 85%. C,D’-F’, D”-F”) Knockdown of Rasip1 inhibits endothelial cell migration. siRasip1-treated cells show slow healing rate in “scratch assay”. While untreated cells migrate quickly into the cell free area (D’,D”), siRasip1-treated cells migrate less than half the distance during the same time period (F’,F”). C) Quantification of endothelial migration in the scratch assay was determined by difference in relative diameter of scratch, before and after healing (D’-F’, compared to D”-F”), measured in μm on y axis (as shown).
To examine whether reduction of Rasip1 levels might have a functional impact on MS1 cell behavior, we carried out both in vitro tube-formation and wound-healing assays. These assays allowed evaluation of the effect of Rasip1 knockdown on endothelial angiogenesis and cell motility, respectively. Strikingly, siRasip1-treated MS1 cells, in which Rasip1 transcript levels were significantly reduced (Fig.4A), almost completely lost the ability to form plexus-like vascular structures when cultured on Matrigel (Fig.4B,D-F). This observation supports the notion that Rasip1 is required for endothelial function. To evaluate the effect, we quantified the number of branch points created by the coalescence of ECs into cords/tubes, and found that these were reduced by over 85%.
In addition, ablation of Rasip1 function in MS1 cells also dramatically decreased EC migration ability. Using an in vitro scratch assay, the ‘healing rate’ of a scratch ‘wound’, across a monolayer of ECs, was significantly reduced. While unmanipulated or siHPRT transfected cells were able to heal the wound following overnight incubation (i.e. fill in the ~600μm cell-free wound area), siRasip1 transfected cells migrated only about 50% the distance over that same timeframe (Fig.4D’-F’,D”-F”). In sum, control cells could migrate approximately 300μm on each side to fill in the gap, while siRasip1 treated cells migrated less than 150μm. It is unlikely that this effect is indirect, as a result of decreased cell proliferation, since the timeframe of the healing study is too short to allow significant proliferation within the gap (wound healing assay is carried out overnight, and MS1 doubling time is approximately 24 hrs). In support, we detect no significant difference in endothelial proliferation by Ki67 staining in siRasip1-treated cells, following a short siRNA treatment of 3 days (Suppl. Fig.5). (Of note, we do detect a mild effect on EC proliferation over a longer treatment period of 6 days). Together, these results indicate that Rasip1 function is required in cultured ECs, for both angiogenic coalescence and cell motility, and is therefore likely to play important roles in blood vessel development.
Rasip1 is required for embryonic blood vessel formation
Bioinformatic comparison of Rasip1 sequences revealed that it was highly conserved across many different species, from human to lower vertebrates, including frog. The DILute domain of Rasip1, for instance, displayed almost 85% identity at the amino acid level between mouse and Xenopus tropicalis (Suppl. Fig.6). This high level of similarity suggested Rasip1 might also be expressed in the vessels of other species, such as frog, and might play a conserved role in vessel formation. Thus, to assay Rasip1 function during embryonic vessel formation in vivo, we examined its expression and function in Xenopus tropicalis embryos. This tractable model system provided us with an avenue for in vivo assays.
RT-PCR was used to amplify a fragment containing approximately 1900bp of the Rasip1 coding region from Xenopus tropicalis cDNA. Using this construct, we generated Dig-labeled probe for in situ hybridization and compared Rasip1 expression to known vascular markers (Fig.5A-H). We note that erg (Fig.5A,B), VE-Cadherin (Fig.5C), and msr (Fig.5D) expression patterns outline the early vasculature in Xenopus tropicalis, during both vasculogenesis and angiogenesis, as previously described in Xenopus laevis (Baltzinger et al., 1999; Devic et al., 1996). Similarly, we found that Xenopus Rasip1 is expressed in the developing vasculature throughout development, including early expression in emerging angioblasts (stage 25) and during vessel coalescence (Fig.5E-G). In addition, Rasip1 is expressed strongly during angiogenesis, in sprouting ISVs (Fig.5G), and remains expressed after initiation of heart beat (st.34) and blood circulation (Fig.5H).
Figure 5. Rasip1 knockdown in frog embryos results in failure of blood vessel formation.
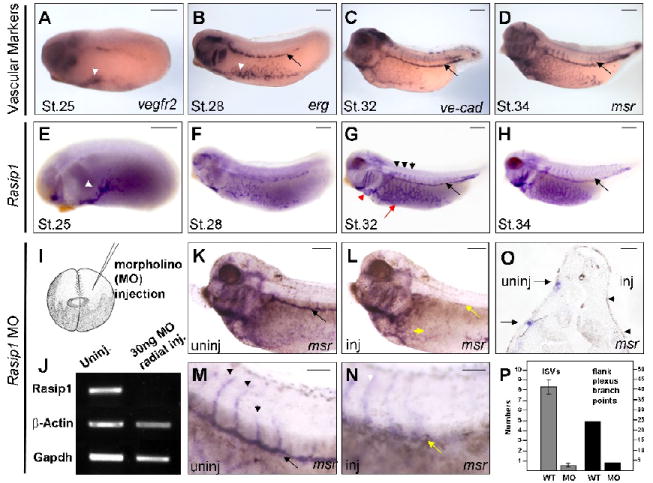
Expression of Xenopus tropicalis Rasip1 transcripts in frog embryos by in situ hybridization marks the developing embryonic blood vessels (in all panels anterior is to the left). A-D) Vascular markers reveal the embryonic vasculature at stages indicated, including angioblasts (white arrowheads in A, B) and developing blood vessels, such as the posterior cardinal vein (black arrow, B-D). A) vegfr2; B) erg, vascular ETS factor; C) ve-cad, vascular endothelial cadherin; D) msr, vascular G-protein coupled receptor. E-H) Rasip1 initially marks scattered angioblasts (E, st.25), but progressively marks aggregating flank vessels (F, st.28). G) As vessels form, Rasip1 marks all embryonic frog vessels examined, including the flank plexus (red arrow), endocardium (red arrowhead), cardinal veins (black arrow) and ISVs (black arrowheads). H) Expression of Rasip1 declines slightly in vessels as they mature (including data not shown). I) Schematic of microinjection of Rasip1 morpholino injections into blastomeres of early cleavage stage embryo. J) Semi-quantitative RT-PCR shows the knockdown of Rasip1 transcript expression in radially injected embryos (24 cycles). (K-N) msr in situ hybridization of injected embryo, outlining vasculature on either uninjected (K,M) or injected (L,N) sides. K,M) Uninjected side of embryo displays major blood vessels, including the prominent cardinal vein (black arrow) and ISVs. M) Higher magnification of embryo in panel K, showing ISVs sprouting from the cardinal vein on the uninjected side (black arrowheads). L,N) MO injected side of embryos shows a severe reduction in vascular structures, including absence of cardinal vein (yellow arrow) and reduced plexus vessels (yellow arrowhead). N) Higher magnification of embryo in panel L, showing complete absence of both ISVs (white arrowhead) and cardinal vein (yellow arrow). Note that faint outline of ISVs in panel N results from ISVs from uninjected side appearing through the dorsal fin tissue (white arrowhead). O) Transverse sections through injected embryos show that cardinal veins and ISVs are lost on the injected side (arrowheads) but not the uninjected side (arrows). P) Quantification of observations in K-O. Total number of ISVs and flank plexus branch points were counted from the injected versus uninjected sides of MO-injected embryos. Y-axis for ISVs is to left; Y-axis for flank branch points is to right. The scale bars represent 250μm (A-H), 100μm (K, L), and 50μm (M, N, O), respectively.
To determine if Rasip1 function is required for vessel development in vivo, we targeted the sequence in Xenopus tropicalis embryos with antisense Rasip1 morpholino oligos (Rasip1-MO) designed to inhibit splicing of Rasip1 transcripts (Suppl. Fig.7). RT-PCR analysis with primers spanning the exon-intron boundary confirmed that Rasip1 splicing, and hence expression of mature transcripts, was effectively abolished in embryos radially injected with 30ng of Rasip1-MO (Fig.5J). To specifically examine the effect of Rasip1 knockdown on embryonic blood vessel development, we injected the Rasip1-MO (16 ng), into one side of the embryos at the 2-cell stage and then assayed them by in situ hybridization at stage 32 with the endothelial marker msr (Fig.5K-O).
We found that blood vessel development was severely inhibited in Rasip1-MO injected embryos as detected by msr staining (compare Fig.5K to L). Most strikingly, we found that the posterior cardinal vein failed to form on the injected side in 77% of injections that were assayed at later stages of development (st.33-35) (Suppl.Fig.8). In addition, we noted a significant reduction in both the number of ECs, as marked by msr, along the flank of the embryo, and the organization of these cells into the vitelline plexus (reduction of average branch points from 23 to 4) (Fig.5L,P). The uninjected side, in contrast, remained unaffected and displayed normal vascular structures, including a normal posterior cardinal vein (Fig.5K,M). We also observed that sprouting ISVs failed to appear, which was not surprising as they originate from the posterior cardinal vein (Fig.5N,P). Sectioning of injected embryos revealed the distinct absence of the posterior cardinal vein and ISVs on the injected side (Fig.5O). Quantification of these observations showed that the cardinal vein and ISVs were lost over 90% of the time, while the plexus EC branching was reduced by over 80%. We detect these effects on the vasculature using the endothelial markers msr, flk1, erg or ve-cadherin (Suppl.Fig.8). Interestingly, assays at earlier stages of development (st.20-21), using msr, do not reveal reduction in the number of angioblasts at the location of the posterior cardinal vein, indicating that vasculogenesis, not specification, is affected in MO-injected embryos. These experiments provide evidence that Rasip1 is required in vivo for proper vessel development.
Discussion
This report provides the first evidence that Rasip1 is both expressed and required in the endothelium of the embryonic vasculature. We show that Rasip1 is specifically expressed in the endothelium of the developing blood vessels of both mouse and frog embryos. Additionally, we demonstrate that this expression initiates early, in angioblasts prior to their aggregation into vessels, and continues in established vessels, which grow and remodel via angiogenesis. We also demonstrate that Rasip1 is fundamentally required in ECs, both in vitro and in vivo. Knocking down Rasip1 in cultured ECs inhibits their migration and coalescence into vessel-like structures, while knocking down Rasip1 in amphibian embryos results in failure of vessel development. These findings establish Rasip1 as a novel and robust marker of embryonic ECs, throughout their specification and differentiation, and as a likely important regulator of vascular development.
Rasip1 is a novel endothelial marker
Few vascular genes have proven useful as specific markers of the endothelium. VEGFR2, PECAM, Tie2 and VE-Cadherin are the most frequently used markers available to date, in that they are highly enriched in ECs and are often used as specific markers. However, they are not completely endothelial specific, as they are often transiently expressed in other tissues, at some point during development. For instance, VEGFR2 is also detected in hematopoietic cells (Yamaguchi et al., 1993), PECAM is also in macrophages (Lee, 1991), Tie2 is found in mesenchymal cells of heart outflow tracts (Kisanuki et al., 2001), VE-Cadherin is also expressed in liver hematopoietic stem cells (Kim et al., 2005) and both VEGFR2 and PECAM are also found in lymphatic vessels (Enholm et al., 2001). In addition, most other commonly used endothelial markers such as Dll4, Egl7, ephrin-B2, EphB4, Jagged1, Notch1 and many more, are widely expressed in a number of other organs and tissues (Conway et al., 2001; Eichmann et al., 2005; Torres-Vazquez et al., 2003).
This report adds Rasip1 to the short list of useful endothelial-enriched sequences, which can be used to study the developing cardiovascular system. Like VEGFR2, Rasip1 transcript levels are highly enriched in embryonic ECs. This is in contrast to PECAM and Tie2 whose transcript levels are lower, and thus difficult to visualize by in situ hybridization. While we have assayed commercially available antibodies to RASIP1 protein, both in vitro and in vivo, none have proven useful for either immunofluoresence or immunohistochemistry. Until effective antibody reagents become available, assays for Rasip1 transcripts will be useful for studies ranging from examination of early angioblast specification to vasculogenesis and angiogenic vessel formation. Given its expression conservation across species, in both frog and mouse, it will very likely be more widely applicable to vascular studies in other species as well.
Rasip1 is essential for endothelial cell function
Knockdown of Rasip1 levels in cultured ECs reveals a critical role for Rasip1 in basic cellular functions, such as cell motility and angiogenesis. When Rasip1 function is reduced, both these basic endothelial behaviors are severely abrogated. siRasip1-treated cells, but not siHPRT-treated cells, display a reduction in their propensity to aggregate and form cords or vessels, in a matrigel angiogenesis assay. In addition, whereas untreated or control siRNA-treated ECs will normally migrate actively across tissue culture-treated plastic in vitro, such as in a ‘wound healing’ assay, cells lacking Rasip1 function fail to migrate. We propose that this reflects a direct effect on EC motility, since we detect no decrease in the rate of endothelial proliferation in siRasip1-treated cells when we use Ki67 to assay dividing cells.
Given that these basic cellular behaviors are likely to comprise the foundations of vessel formation, we predicted that endothelial cells in emerging blood vessels would also require Rasip1 function. Indeed, reduction of Rasip1 in vivo leads to a dramatic failure of embryonic vessel formation. Using a MO-based approach, we knocked down endogenous expression of Rasip1 in Xenopus tropicalis embryos, which led to a clear failure of the posterior cardinal vein and associated ISVs to form. These results demonstrate that Rasip1 is required for the proper formation of vascular structures that develop via both vasculogenesis (posterior cardinal vein) and angiogenesis (ISVs). Interestingly, vessels of the flanking vitelline plexus, within the lateral plate mesoderm, while disorganized when Rasip1 function is reduced, were not completely abrogated. The basis for the difference in the response of these different vascular beds to the absence of Rasip1 is unclear. However, it has been proposed that inherently different populations of hematopoietic, and perhaps endothelial, cells arise within these different embryonic regions (Kau and Turpen, 1983; Maeno et al., 1985). It is possible that they may represent ‘primitive’ (ventral/flank) versus ‘definitive’ (dorsal lateral plate/somite) ECs, and that these two populations have a differential requirement for Rasip1 function.
Rasip1 is not required for angioblast specification
Significantly, despite disruption of vessels when Rasip1 function is knocked down in frog embryos, angioblasts still emerge within the mesoderm. This finding indicates that Rasip1 plays a role sometime after initial angioblast specification. This observation is supported by the timing of Rasip1 expression initiation during vessel development in frogs, as angioblast specification is conveniently separated in time from the process of vessel formation via vasculogenesis. In frogs, angioblasts are specified within the mesoderm of late neurula stage embryos (st.18-22), many hours prior to vessel formation that occurs at the late tailbud stage (st.30-32). We find that Rasip1 expression in frogs initiates later (st.22) than VEGFR2 (st.18) (Cleaver et al., 1997), thus displaying a marked delay and appearing distinctly later than the earliest known angioblast markers. In addition, when Rasip1 function is knocked down using MOs, we observe that early angioblasts emerge relatively normally as assayed by msr expression at st.20-21. We therefore suggest that Rasip1 is likely to function in angioblasts and ECs following their initial specification, possibly during their migration, cord formation or tubulogenesis.
In contrast, we observe that Rasip1 expression in mouse appears to be initiated remarkably early, around the same time that VEGFR2 and other vascular markers begin transcription within the earliest endothelial precursors of the yolk sac. This is not surprising since most murine endothelial markers initiate almost simultaneously during a relatively short timeframe, making it difficult to establish the order of vascular gene onset (Drake and Fleming, 2000). It will therefore be necessary to carry out more detailed expression analyses of vascular markers, or functional epistases experiments, during this initial phase of vasculogenesis to determine when Rasip1 may exert its function and to place it within potential regulatory genetic cascades.
Summary
Data presented here supports a requirement for Rasip1 function within developing ECs during blood vessel formation. Knockdown of Rasip1 in frog embryos results in failure of the posterior cardinal vein and ISVs to develop and in disorganization of the vitelline plexus. However, at this point, the cellular and molecular mechanisms that prevent the proper formation of these vessels remain unclear. At the cellular level, it is likely that the observed vascular defects are a result of either a reduction of angioblast survival, or a failure of cord formation via vasculogenesis or vascular tube formation, with subsequent cell death or dedifferentiation. At the molecular level, it is not yet clear how, when and whether Rasip1 modulates Ras downstream of VEGF signaling, or whether it impacts alternative pathways. Studies are currently underway to clarify these molecular relationships during vascular development.
Together, our studies demonstrate the critical role of the Ras effector Rasip1 for proper vessel formation and normal endothelial cell behavior. In addition, we identify Rasip1 as a novel tool for studies of embryonic vessel development. In vitro experiments demonstrate that Rasip1 is required for EC migration and coalescence of angioblasts into vessels, while in vivo experiments show that it is required for formation of blood vessels in frog embryos. Due to its conservation, we predict that Rasip1 will also be required for proper formation of the vasculature in other species, including mammals. Further studies of Rasip1 will open novel and exciting questions regarding the role of intracellular signaling molecules in vascular development, including members of the Ras family, both areas which to date have been relatively unexplored. Given the central importance of blood vessels during many diseases, such as cancer and tumor angiogenesis, we propose that further understanding of the mechanism and impact of Ras and Rasip1 signaling in ECs will prove to be of great clinical relevance.
Supplementary Material
Table 1. Rasip1 is required for proper blood vessel formation.
Listed in table are the numbers of embryos with absent/severely reduced angioblasts (st.20-27) or posterior cardinal vein (st.33-37) observed on injected side of embryo (x), out of the total number assayed (y): x/y. Controls include uninjected embryos (top row) and uninjected side of injected embryos (data not tabulated).
| msr(st.20-21) | flk1(st.25-27) | erg(st.33-35) | ve-cadherin (st.33-35) | msr (st.35-37) | |
|---|---|---|---|---|---|
| uninjected | 1/21 | 1/17 | 0/13 | 0/16 | 0/25 |
| 16ng Rasip1-MO | 1/19 | 10/14 | 12/15 | 11/15 | 4/5 |
Acknowledgments
We thank Paul Krieg, Eric Olson and Alethia Villasenor for critical reading of the manuscript and discussions. We thank M.K. Chiang and D. Melton for the PECAM clone. We thank E. Keshet (and J. Rossant) for the Flk1(VEGFR2)-lacZ mice. AMZ is supported by NIH R01 grant DK070858. O.C. is supported by AHA Grant-in-Aid 0755054Y, NIH R01 grant DK079862-01 and the Basil O’Connor March of Dimes Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aitsebaomo J, Wennerberg K, Der CJ, Zhang C, Kedar V, Moser M, Kingsley-Kallesen ML, Zeng GQ, Patterson C. p68RacGAP is a novel GTPase-activating protein that interacts with vascular endothelial zinc finger-1 and modulates endothelial cell capillary formation. J Biol Chem. 2004;279:17963–72. doi: 10.1074/jbc.M311721200. [DOI] [PubMed] [Google Scholar]
- Baltzinger M, Mager-Heckel AM, Remy P. Xl erg: expression pattern and overexpression during development plead for a role in endothelial cell differentiation. Dev Dyn. 1999;216:420–33. doi: 10.1002/(SICI)1097-0177(199912)216:4/5<420::AID-DVDY10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Cleaver O, Krieg PA. Molecular Mechanisms of Vascular Development. In: Harvey RP, R N, editors. Heart Development. Academic Press; San Diego: 1999. pp. 221–244. [Google Scholar]
- Cleaver O, Tonissen KF, Saha MS, Krieg PA. Neovascularization of the Xenopus embryo. Dev Dyn. 1997;210:66–77. doi: 10.1002/(SICI)1097-0177(199709)210:1<66::AID-AJA7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Connolly JO, Simpson N, Hewlett L, Hall A. Rac regulates endothelial morphogenesis and capillary assembly. Mol Biol Cell. 2002;13:2474–85. doi: 10.1091/mbc.E02-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–21. doi: 10.1016/s0008-6363(00)00281-9. [DOI] [PubMed] [Google Scholar]
- Costa RM, Mason J, Lee M, Amaya E, Zorn AM. Novel gene expression domains reveal early patterning of the Xenopus endoderm. Gene Expr Patterns. 2003;3:509–19. doi: 10.1016/s1567-133x(03)00086-3. [DOI] [PubMed] [Google Scholar]
- Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–45. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28:488–94. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- Devic E, Paquereau L, Vernier P, Knibiehler B, Audigier Y. Expression of a new G protein-coupled receptor X-msr is associated with an endothelial lineage in Xenopus laevis. Mech Dev. 1996;59:129–40. doi: 10.1016/0925-4773(96)00585-0. [DOI] [PubMed] [Google Scholar]
- Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–9. [PubMed] [Google Scholar]
- Drake CJ, LaRue A, Ferrara N, Little CD. VEGF regulates cell behavior during vasculogenesis. Dev Biol. 2000;224:178–88. doi: 10.1006/dbio.2000.9744. [DOI] [PubMed] [Google Scholar]
- Eichmann A, Yuan L, Moyon D, Lenoble F, Pardanaud L, Breant C. Vascular development: from precursor cells to branched arterial and venous networks. Int J Dev Biol. 2005;49:259–67. doi: 10.1387/ijdb.041941ae. [DOI] [PubMed] [Google Scholar]
- Enholm B, Karpanen T, Jeltsch M, Kubo H, Stenback F, Prevo R, Jackson DG, Yla-Herttuala S, Alitalo K. Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ Res. 2001;88:623–9. doi: 10.1161/01.res.88.6.623. [DOI] [PubMed] [Google Scholar]
- Ferkowicz MJ, Yoder MC. Blood island formation: longstanding observations and modern interpretations. Exp Hematol. 2005;33:1041–7. doi: 10.1016/j.exphem.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Fryer BH, Field J. Rho, Rac, Pak and angiogenesis: old roles and newly identified responsibilities in endothelial cells. Cancer Lett. 2005;229:13–23. doi: 10.1016/j.canlet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Garnaas MK, Moodie KL, Liu ML, Samant GV, Li K, Marx R, Baraban JM, Horowitz A, Ramchandran R. Syx, a RhoA guanine exchange factor, is essential for angiogenesis in Vivo. Circ Res. 2008;103:710–6. doi: 10.1161/CIRCRESAHA.108.181388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA. Nf1 has an essential role in endothelial cells. Nat Genet. 2003;33:75–9. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Rossi DJ, Holmyard DP, Puri MC, Mbamalu G, Harpal K, Shih TS, Jacks T, Pawson T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- Kau CL, Turpen JB. Dual contribution of embryonic ventral blood island and dorsal lateral plate mesoderm during ontogeny of hemopoietic cells in Xenopus laevis. J Immunol. 1983;131:2262–6. [PubMed] [Google Scholar]
- Kim I, Yilmaz OH, Morrison SJ. CD144 (VE-cadherin) is transiently expressed by fetal liver hematopoietic stem cells. Blood. 2005;106:903–5. doi: 10.1182/blood-2004-12-4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–42. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Ruoslahti E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat Med. 2005;11:1346–50. doi: 10.1038/nm1324. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, Gebbink MF, Voest EE. Stimulation of angiogenesis by Ras proteins. Biochim Biophys Acta. 2004;1654:23–37. doi: 10.1016/j.bbcan.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Kuijper S, Turner CJ, Adams RH. Regulation of angiogenesis by Eph-ephrin interactions. Trends Cardiovasc Med. 2007;17:145–51. doi: 10.1016/j.tcm.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Lee SH. Phenotypic analysis of human bone marrow macrophages. Blood Cells. 1991;17:45–54. discussion 54-8. [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeno M, Tochinai S, Katagiri C. Differential participation of ventral and dorsolateral mesoderms in the hemopoiesis of Xenopus, as revealed in diploid-triploid or interspecific chimeras. Dev Biol. 1985;110:503–8. doi: 10.1016/0012-1606(85)90108-3. [DOI] [PubMed] [Google Scholar]
- Meadows KN, Bryant P, Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem. 2001;276:49289–98. doi: 10.1074/jbc.M108069200. [DOI] [PubMed] [Google Scholar]
- Merajver SD, Usmani SZ. Multifaceted role of Rho proteins in angiogenesis. J Mammary Gland Biol Neoplasia. 2005;10:291–8. doi: 10.1007/s10911-006-9002-8. [DOI] [PubMed] [Google Scholar]
- Mitin N, Konieczny SF, Taparowsky EJ. RAS and the RAIN/RasIP1 effector. Methods Enzymol. 2006;407:322–35. doi: 10.1016/S0076-6879(05)07027-8. [DOI] [PubMed] [Google Scholar]
- Mitin NY, Ramocki MB, Zullo AJ, Der CJ, Konieczny SF, Taparowsky EJ. Identification and characterization of rain, a novel Ras-interacting protein with a unique subcellular localization. J Biol Chem. 2004;279:22353–61. doi: 10.1074/jbc.M312867200. [DOI] [PubMed] [Google Scholar]
- Moser M, Patterson C. Bone morphogenetic proteins and vascular differentiation: BMPing up vasculogenesis. Thromb Haemost. 2005;94:713–8. doi: 10.1160/TH05-05-0312. [DOI] [PubMed] [Google Scholar]
- Park C, Lavine K, Mishina Y, Deng CX, Ornitz DM, Choi K. Bone morphogenetic protein receptor 1A signaling is dispensable for hematopoietic development but essential for vessel and atrioventricular endocardial cushion formation. Development. 2006;133:3473–84. doi: 10.1242/dev.02499. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Anderson AL, Hidaka M, Swetenburg RL, Patterson C, Stanford WL, Bautch VL. A vascular gene trap screen defines RasGRP3 as an angiogenesis-regulated gene required for the endothelial response to phorbol esters. Mol Cell Biol. 2004;24:10515–28. doi: 10.1128/MCB.24.24.10515-10528.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007;21:2511–24. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- Serban D, Leng J, Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res. 2008;102:1350–8. doi: 10.1161/CIRCRESAHA.107.169664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Sosnowski RG, Feldman S, Feramisco JR. Interference with endogenous ras function inhibits cellular responses to wounding. J Cell Biol. 1993;121:113–9. doi: 10.1083/jcb.121.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–38. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- Torres-Vazquez J, Kamei M, Weinstein BM. Molecular distinction between arteries and veins. Cell Tissue Res. 2003;314:43–59. doi: 10.1007/s00441-003-0771-8. [DOI] [PubMed] [Google Scholar]
- Walls JR, Coultas L, Rossant J, Henkelman RM. Three-dimensional analysis of vascular development in the mouse embryo. PLoS ONE. 2008;3:e2853. doi: 10.1371/journal.pone.0002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG. In situ hybidization: A practical approach. Oxford University Press; Oxford, New York, USA: 1999. [Google Scholar]
- Yamaguchi TP, Dumont DJ, Conlon RA, Breitman ML, Rossant J. flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development. 1993;118:489–98. doi: 10.1242/dev.118.2.489. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.