

Abstract
The early chick embryo contains subpopulations of cells that express lineage-specific transcription factors. We have developed protocols to examine the role of these cells during development that involve labeling them for cell tracking purposes and ablating them within the epiblast. The procedures take advantage of the fact that subpopulations of epiblast cells differentially express cell surface antigens recognized by monoclonal antibodies. Embryos are removed from the shell and incubated on the yolk with an antibody. Cells that bind the antibody are either tagged with a fluorescent secondary antibody or lysed with complement. For long-term analyses, embryos are returned to a host shell and placed in an incubator. This method of whole embryo manipulation ex-ovo and incubation in-ovo supports normal development into the fetal period.
Indexing terms: Chick Embryo, Staining and Labeling
INTRODUCTION
The epiblast of the chick embryo contains small numbers of cells that contain mRNAs for the basic helix-loop-helix transcription factors MyoD and NeuroM that are markers for the skeletal muscle and neuronal lineages, respectively (1-6). All MyoD positive epiblast cells synthesize a cell surface antigen recognized by the G8 monoclonal antibody (MAb) (3-6). The E12 MAb binds to a cell surface antigen present on a subpopulation of NeuroM expressing cells (4). In order to examine the roles of MyoD- and NeuroM-positive epiblast cells in embryonic tissues, we have developed protocols to label and ablate these subpopulations in living embryos prior to the onset of gastrulation. Fluorescent secondary antibodies were used to visualize G8 and E12 labeled cells. Lysis of antibody labeled cells was carried out by incubating embryos in complement. A critical component of these experiments was the development of a method that would support the growth of embryos manipulated at the blastula stage into the fetal period.
An ex-ovo whole embryo culture system was developed over 50 years ago to facilitate experimentation with the early chick embryo (7). The method involves removing the embryo from the yolk and culturing it on a glass ring in albumen. Several modifications have been made to the New culture method, including replacing the medium with a mixture of thin albumen and Bactoagar (8) and placing the embryos on a nucleopore or filter paper raft (9,10). The Early Chick Method of Chapman et al. (10) permits the establishment of more cultures in less time and supports the development of younger embryos compared to the New culture system. The visibility and accessibility of the embryo in these ex-ovo culture systems facilitates tissue manipulations (11). However, the limitation of these culture systems is that development progresses normally for only 72 hours (10).
Longer term evaluation of chick embryo development can be carried out by the “shell-less method” originally developed in the laboratory of Dr. Judah Folkman (12). This method involves culturing embryos on the yolk in petri dishes. Modifications to the original procedure include suspending embryos on a tripod in plastic wrap (13), or growing them in plastic cups (14,15) or hexagonal polycarbonate weigh boats (16,17). Survival is low if cultures are established with embryos younger than 38 hours of incubation (Jean-Marie Gasc, personal communication). Addition of eggshell as a source of calcium to shell-less cultures increases the survival of 48 hour embryos beyond day 14 (18-21).
Chick embryos can also be cultured in surrogate eggshells (22-25). The original procedure was a three step process in which fertilized ova recovered from the oviduct were cultured in a glass jar for 24 hours, transferred to a windowed host shell for three days and transferred again to a second larger shell through hatching (22). Under these conditions the maximum rate of hatching was 23%. Hatching increased to 63% when the process began with the second step using blastoderm stage embryos.
Embryos can be manipulated directly in the shell. In this procedure, a window is cut in the shell and the embryo on the yolk is stabilized for manipulation by removing some of the albumen (26). As with the shell-less culture methods, survival rates increase when these in–ovo cultures are established after 36 hours of development (Dr. Matthew Korn, personal communication).
Our procedure is a hybrid of the shell-less and in-ovo culture methods. The procedure involves placing the contents of the egg into a tissue culture dish, labeling or ablating cells in the embryo while it resides on the yolk and transferring the embryo to a single host shell for growth into the fetal period. Our technique for labeling or ablating cells with complement depends on the existence of antibodies that specifically recognize surface antigens differentially expressed on subpopulations of cells. The advantages of this method are the ability to precisely target specific cell types in the blastula and the high rate of survival of manipulated embryos into the fetal period.
MATERIALS AND METHODS
Labeling and Ablating Cells Ex-Ovo
The contents of the egg were gently emptied into a Petri dish. Embryos residing on the yolk were staged according to the method of Hamburger and Hamilton (27). Stages 2 and 4 embryos were incubated in 100 µl of the G8 or E12 MAb diluted 1:20 in Dulbecco’s phosphate buffered saline (PBS) (Invitrogen) for 45 minutes at room temperature and rinsed three times with PBS. The G8 and E12 IgM MAbs bind to uncharacterized antigens present on the surface of cells expressing MyoD or NeuroM mRNA, respectively (3-6). Control embryos were incubated in PBS lacking primary antibody. For cell tracking experiments, rinsing was followed by incubation at room temperature for 45 minutes in goat anti-mouse IgM conjugated with rhodamine (Chemicon) diluted 1:600 in PBS. Embryos were rinsed three times in PBS. Diffusion of primary and secondary antibodies was limited by overlying the solutions with a piece of parafilm as described in the Protocols section.
Cell ablation within the stage 2 epiblast was carried out by incubating cells with the G8 or E12 MAb as described above, rinsing in PBS and applying 100 µl of baby rabbit complement (Cedar Lane, Inc.) diluted 1:40 in ice cold, sterile Hanks' buffered saline containing 0.1% bovine serum albumen. Embryos were incubated in complement for 30 minutes at room temperature. Control embryos were incubated in Hanks’ buffer solution instead of primary antibody and then treated with complement. Dead cells were visualized by incubating embryos in 0.2% trypan blue (Sigma Aldrich) in PBS for 15 minutes. Embryos that were not incubated in trypan blue were transferred to host shells as described below.
Culturing Cells In Ovo
Host shells were prepared under sterile conditions by cutting a window in the egg and removing its contents. Embryos that were labeled with antibodies or treated with complement on the yolk were poured into the host shell. The thick albumen was poured into the shell first, followed by the embryo on the yolk. Thin albumen was used to fill the shell until the embryo was approximately 0.5-1 cm below the opening. The window was covered with a sterile piece of plastic wrap. Treated and control embryos were incubated at 37oC for a maximum of 17 days.
Analyzing Embryos
Embryos were fixed overnight in 4% formaldehyde. Whole stage 2 and 14 embryos and transverse 10 µm paraffin sections of 5 and 6 day embryos were counterstained with Hoechst nuclear dye (Sigma Aldrich) and mounted in Elvanol (Dupont). Fluorescent cells were visualized with a Nikon Eclipse 800 epifluorescence microscope (Optical Apparatus) equipped with a 530-560 nm excitation and 573-648 nm barrier filters for rhodamine and 330-380 nm excitation and 435-485 nm barrier filters for Hoechst dye. Some sections were stained with hematoxylin and eosin Y (Richard Allen Scientific). Photographs were taken under 4x NA 0.2 and 60x oil NA 1.4 objectives. Images were produced with the Evolution QE Optronics video camera (Media Cybernetics) and the Image Pro Plus image analysis software program (Phase 3 Imaging Systems). Figures were annotated and adjusted for brightness and contrast using Adobe Photoshop 6.0.
RESULTS AND DISCUSSION
Our ex-ovo/in-ovo procedure is a precise and efficient method for labeling and ablating cells in blastula stage chick embryos that supports development into the fetal period. We have used this procedure to fluorescently label subpopulations of epiblast cells that express myogenic and neurogenic transcription factors with the G8 and E12 MAbs, respectively (4-6). In the stage 2 embryo, the G8 and E12 MAbs bind to separate populations of cells in the posterior epiblast (Fig. 1A and B) (4). A second and larger subpopulation of E12-positive cells is present in the anterior epiblast (4,5).
Fig. 1. Labeling Embryos Ex-Ovo with Fluorescent Antibodies.

Stage 2 embryos were labeled with the G8 and E12 MAbs and fluorescent secondary antibodies (red). Nuclei were stained with Hoechst dye (blue). The inset in panels A and B illustrates the location of cells labeled with the G8 (A) and E12 MAbs (B) in the posterior epiblast of unsectioned stage 2 embryos. Labeled embryos developed normally after returning them to the shell and incubating them for three (C), five (D), or 13 days (E). The region of the somite outlined in the hematoxylin and eosin stained section of the 5 day embryo (F) is shown at higher magnification in the fluorescence photomicrograph (G). Epiblast cells stained with the G8 MAb were incorporated into the dorsomedial region of the dermomyotome and myotome (G). The locations of epiblast cells labeled with the E12 MAb at stage 4 are outlined in the drawing of the unsectioned stage 14 embryo (H). Cells stained with E12 were visible in the brain (I) and neural tube (J). Bar = 9 µm in A, B and C and 54 µM in F-I.
Embryos were incubated in host shells in order to analyze the sites of incorporation of G8 and E12 labeled epiblast cells as development progressed. Approximately 75% of embryos were visible through the window in the shell for the first three days after transfer (Fig. 1C). All embryos became visible through the window by the fourth day in-ovo, thereby permitting daily observations of morphogenesis. The survival rate of embryos labeled with antibodies during stages 1-4 and returned to host shells and J). E12-positive cells were observable in whole embryos for an additional 48 hours of development by epifluorescence microscopy, although confocal microscopy or sectioning the embryos is required for quantitative analyses. These experiments demonstrate that epiblast cells can be labeled with non-function perturbing antibodies ex-ovo and cultured in host shells without adversely affecting development. Detection of antigens sensitive to endogenous protease digestion may require incubations at 4oC instead of room temperature.
The technique of complement-induced cell lysis was utilized over 50 years ago to examine antigenicity in the chick embryo (28). We have taken advantage of complement’s ability to lyse cells bound with antibodies to eliminate specific subpopulations within the epiblast that express different cell surface antigens (5). Incubation in baby rabbit complement alone at a dilution of 1:40 did not increase cell death in the epiblast (Fig. 2A). Labeling cells with the G8 and E12 MAbs followed by incubation in complement resulted in lysis of separate populations of epiblast cells, as determined by the uptake of trypan blue (Fig. 2B and C) (5). Testing the specificity of lysis of antibody labeled cells with different concentrations of complement purified from various sources is recommended for each primary antibody and stage of development.
Fig. 2. Ablating Cells in the Epiblast with Antibodies and Complement.
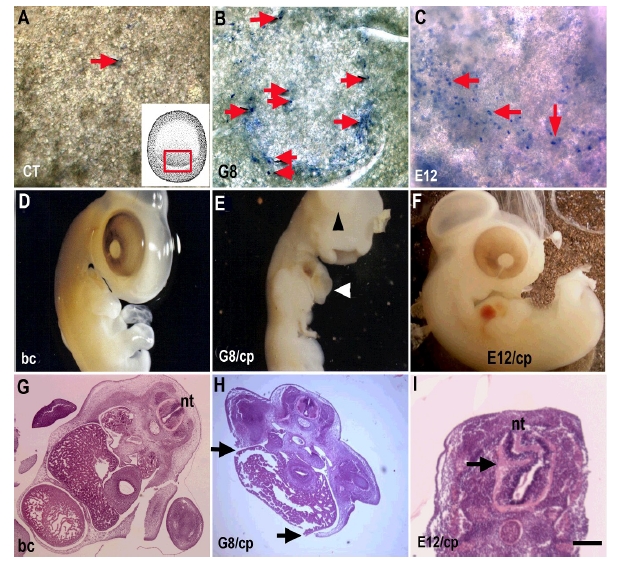
Stage 2 embryos were incubated with PBS (CT), the G8 or E12 MAb and treated with complement. Lysed cells were visible in the epiblast after staining with trypan blue (arrows). Only a few dead cells were visible in the epiblast after incubation with PBS and complement (arrows in A). The inset in panel A illustrates the location of lysed cells in panels B and C. Subpopulations of dead cells were visible in the posterior epiblast after lysing cells bound with the G8 (B) and E12 (C) MAbs. Six days after treatment, embryos treated with complement alone appeared normal upon gross inspection (D) and in sections stained with hematoxylin and eosin (G). Ablation of G8 labeled cells resulted in herniation of organs through the ventral body wall (E and H). Incubation with E12 and complement resulted in neural tube (nt) defects visible in tissue sections (I). Bar = 135 µM in A-C and G-I.
Lysis of subpopulations of epiblast cells labeled with the G8 or E12 MAbs produced different malformations as development progressed. Elimination of G8-positive cells in the stage 2 epiblast led to a herniation of organs through the ventral body wall (Fig. 2E and H). Loss of integrity of the body wall resulted from a dramatic reduction in skeletal muscle in the myotome (5). The mechanism whereby G8/MyoD-positive epiblast cells regulate myogenesis in the somites involves the production of the bone morphogenetic inhibitor Noggin (5). These embryos died between the sixth and seventh day of development.
Lysis of the subpopulation of NeuroM expressing cells recognized by the E12 MAb led to neural tube defects (Fig. 2I). The role of E12-positive epiblast cells during neural tube development has not yet been characterized. Embryos treated with E12 and complement remained viable for at least nine days. Exposure to baby rabbit complement alone did not produce malformations in the embryo (Fig. 2D and G).
We have used this ex-ovo/in-ovo procedure to follow the fate of cells labeled in the epiblast and to determine the effects of ablating them on differentiation and morphogenesis (5,6). Our procedure requires no special equipment other than a dissecting microscope and a fiber optic light source for illumination from above. A significant advantage of this ex-ovo/in-ovo procedure over previously published culture methods, including the technique of manipulating embryos through a window in the shell, is that experiments can be initiated prior to the onset of gastrulation and subsequently grown to fetal stages with a high rate of survival. The application of antibodies for labeling or ablation is achieved without perturbing the embryo.
The procedures we have developed for cell tracking and ablation depend on the existence of antibodies that recognize cell surface antigens expressed on specific subpopulations in the early embryo. Other methods of marking cells that could be used with our ex-ovo/in-ovo system include transplantation of quail cells into chick hosts (29), microinjection of vital dyes (30-33) and microinjection or electroporation of genes coding for fluorescent proteins (34,35). Microinjection of fluorescent dyes followed by laser ablation was used to eliminate cells in stages 12-18 embryos grown in shell-less cultures (17). This approach could be attempted with blastula stage embryos using the ex-ovo/in-ovo protocol.
An important advantage of our ex-ovo/in-ovo method is that it supports the development of embryos manipulated at the blastula stage into the fetal period. The success of this method may be attributed, in part, to returning the embryo to the shell that maintains the appropriate tension on the yolk and provides calcium to the embryo. Although we have not yet extended our experiments beyond day 17, the host shell is expected to support development until hatching.
Acknowledgments
We thank Dr. Karen Knudsen and Justin Elder for critically reading the manuscript. This work was supported by NIH HD043157 and AR52326 (MGW).
Abbreviations
- Mab
Monoclonal antibody; PBS
- EGFP
Phosphate buffered saline; BSA
- Bovine serum albumen
Protocols
Supplies and Reagents
100 mm tissue culture dishes
Egg cartons
70% ethanol
Plastic wrap, cut in 7.5 x 7.5 cm squares, wiped with 70% ethanol and dried before use. Only store brands of plastic wrap and not name brands such as Saran wrap should be used in these experiments.
Dissecting microscope with external light source for illumination from above
Kimwipes
Sterile Dulbecco’s phosphate buffered saline (PBS) (Invitrogen)
Parafilm cut in 1.5 x 1.5 cm squares
Sterile forceps
Sterile tubes with caps
Sterile glass pipettes and pipet tips
Sterile primary and fluorescent secondary antibodies (Chemicon)
Sterile baby rabbit complement (Cedar Lane, Inc.)
Hoechst 33258 dye (Sigma Aldrich)
Sterile 0.1% bovine serum albumen (BSA) in Hanks’ buffered saline (Invitrogen)
Trypan blue (Sigma Aldrich)
Methods
-
Removing embryos from the shell
Eggs were removed from the incubator and cooled to room temperature. Unincubated eggs may be used cold or at room temperature.
Eggs were turned on their side, rinsed with 70% ethanol and air dried.
Some eggs were set aside for the preparation of host shells as described in step 3.
Eggs were cracked and the contents poured into 100 mm tissue culture dishes without breaking the yolk. The side of the scissors was used to gently stroke the yolk to move the embryo to its upper surface.
Albumen was gently teased away from each side of the embryo with a Kimwipe (Fig. 3A) creating a small depression over the embryo. In stages 10-15 embryos, the albumen was not as thick and the depression was less obvious than over the younger embryos. The yolks of older embryos are more fragile than younger embryos and greater care must be taken to remove the albumen.
-
Labeling cells with antibodies and lysing with complement
The G8 and E12 MAbs were diluted 1:20 in PBS.
The protective paper was carefully removed from a square of parafilm without touching its sterile surface. The parafilm was held with a forceps with the sterile side facing down. With the other hand, 100 µl of the primary antibody solution was added to the embryo (Fig. 3B). The solution was placed just lateral to one edge of the embryo and not directly onto the middle of the embryo. The embryo was immediately covered with the parafilm to spread the solution over the embryo (Fig. 3C). The tissue culture dish was covered with a lid to avoid drying.
Embryos were incubated in primary antibody solution for 45 minutes at room temperature.
The parafilm was gently lifted off of the embryo with a forceps.
The embryo was rinsed three times with 100 µl PBS. The PBS was added to the side of the embryo. Care was taken to remove as much PBS as possible between rinses.
In cell tracking experiments, the secondary antibody diluted 1:600 in PBS was added as described in step 2b. Embryos were incubated for 45 minutes at room temperature.
In cell tracking experiments, the secondary antibody diluted 1:600 in PBS was added as described in step 2b. Embryos were incubated for 45 minutes at room temperature. for 30 minutes at room temperature.
Embryos incubated with a secondary antibody or treated with complement were rinsed as described in 2e.
Lysed cells were visualized by incubating embryos in 0.2% trypan blue in PBS for 15 minutes. Embryos were rinsed in PBS. Embryos incubated in trypan blue were discarded after photography
Other embryos labeled with antibodies or lysed with complement were incubated in host shells as described below.
-
Preparing host shells
The egg was held horizontally. A scissors was used to poke a very small hole in the upper surface of the shell close to the blunt end of the egg.
The tip of the scissors was inserted into the hole and an opening was cut in the shell just large enough for the yolk to fit through (Fig. 3D and E).
The entire contents of the egg were poured out of the shell.
Ten ml of the thin albumen was saved from a few eggs in a sterile tube.
-
Placing the embryo in the host shell
Three to five ml of thin albumen was removed from the culture dish containing the embryo on the yolk to insure that the contents did not exceed the capacity of the host shell.
The host shell was held in one hand. With the other hand, the thick albumen was poured into the shell followed by the embryo on the yolk (Fig. 3F). Embryos with broken yolks were discarded.
The thin albumen saved in step 3d was used to fill the shell until the embryo was approximately 0.5-1 cm below the opening in the shell (Fig. 3G).
A small amount of thin albumen was drizzled onto the shell around the perimeter of the opening to seal the plastic wrap to the shell.
The opening in the shell was covered with plastic wrap pulled as taut as possible (Fig. 3H). Borwompinyo et al. (36) determined that windows covered with Handi Wrap supported a higher rate of hatching than Saran Wrap. We found that store brands of plastic wrap (ACME) were more porous than brand name wraps and allowed for better air exchange. Only the upper surface of the shell should be covered with plastic wrap.
The egg was laid horizontally in an egg carton for support with the covered window facing up (Fig. 3H). Embryos were grown at 37.5°C in a humidified incubator. The humidity within the incubator was monitored regularly as drying adversely affects survival.
Fig. 3. Culturing Embryos in Host Shells after Ex-Ovo Manipulation.

Embryos on the yolk were placed in a Petri dish. Prior to manipulation, excess albumen was teased away from each side of the embryo with a Kimwipe (A). A solution was delivered to the lateral edge of the embryo with a pipet (B). A piece of parafilm was used to spread the solution over the embryo (B and C). Host shells were prepared by cutting an opening in the top of the shell large enough for the yolk to fit through (D and E). The thick albumen and embryo on the yolk were poured into the empty host shell (F). Thin albumen was added to approximately 0.5 cm below the opening in the shell (G). The opening was covered with plastic wrap (H).
REFERENCES
- George-Weinstein M, Gerhart J, Gerhart R, Flynn J, Callihan B, Mattiacci M, Miehle C, Foti G, Lash JW, Weintraub W. Skeletal myogenesis: The preferred pathway of chick embryo epiblast cells in vitro. Dev Biol. 1996;173:279–291. doi: 10.1006/dbio.1996.0023. [DOI] [PubMed] [Google Scholar]
- Gerhart J, Baytion M, DeLuca S, Getts R, Lopez C, Niewenhuis R, Nilsen T, Olex S, Weintraub W, George-Weinstein George-Weinstein. DNA dendrimers localize MyoD mRNA in presomitic tissues of the chick embryo. J Cell Biol. 2000;149:825–833. doi: 10.1083/jcb.149.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J, Neely C, Stewart B, Perlman J, Beckmann D, Wallon M, Knudsen K, George-Weinstein M. Epiblast cells that express MyoD recruit pluripotent cells to the skeletal muscle lineage. J Cell Biol. 2004;164:739–746. doi: 10.1083/jcb.200309152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strony R, Gerhart J, Tornambe D, Perlman J, Neely C, Dare J, Stewart B, George- Weinstein M. NeuroM and MyoD are expressed in separate subpopulations of cells in the pregastrulating epiblast. Gene Exp Patterns. 2005;5:387–395. doi: 10.1016/j.modgep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- SchefflerGerhart J, Elder J, Neely C, Schure J, Kvist T, Knudsen K, George-Weinstein M. MyoD positive epiblast cells regulate skeletal muscle differentiation in the embryo. J Cell Biol. 2006;175:283–292. doi: 10.1083/jcb.200605037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J, Neely C, Elder J, Pfautz J, Perlman J, Narciso L, Narciso KK, Knudsen K, George-Weinstein M. Cells that express MyoD mRNA in the epiblast are stably committed to the skeletal muscle lineage. J Cell Biol. 2007;278:649–660. doi: 10.1083/jcb.200703060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New DAT. A new technique for the cultivation of the chick embryo in vitro. J Embryol Exp Morphol. 1955;3:326–331. [Google Scholar]
- Bortier H, Vakaet LCA. Fate mapping the neural plate and the intraembryonic mesoblast in the upper layer of the chicken blastoderm with xenografting and time-lapse videography. 1992:suppl:93–suppl:97. [PubMed] [Google Scholar]
- Lash J, Holtzer S, Holtzer H. An experimental analysis of the development of the spinal column. Exp Cell Res. 1957;13:292–303. doi: 10.1016/0014-4827(57)90008-3. [DOI] [PubMed] [Google Scholar]
- Chapman SC, Collignon J, Schoenwolf GC, Lumsden A. Improved method for chick whole-embryo culture using a filter paper carrier. Dev Dyn. 2001;220:284–289. doi: 10.1002/1097-0177(20010301)220:3<284::AID-DVDY1102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Callebaut M, Nueten E, Bortier H, Harrison F, Nassauw L. Map of the anlage fields in the avian unincubated blastoderm. Eur J Morphol. 1996;34:347–361. doi: 10.1076/ejom.34.5.347.13056. [DOI] [PubMed] [Google Scholar]
- Auerbach R, Kubai L, Knighton D, Folkman J. A simple procedure for the long-term cultivation of chicken embryos. Dev Biol. 1974;41:391–394. doi: 10.1016/0012-1606(74)90316-9. [DOI] [PubMed] [Google Scholar]
- Dunn BE, Fitzharris TP, Barnett BD. Effects of varying chamber construction and embryo pre-incubation age on survival and growth of chick embryos in shell-less culture. Anat Rec. 1981;199:33–43. doi: 10.1002/ar.1091990105. [DOI] [PubMed] [Google Scholar]
- Jakobson AM, Hahnenberger R, Magnusson A. A simple method for shell-less cultivation of chick embryos. . Pharmacol Toxicol. 1989;64:193–195. doi: 10.1111/j.1600-0773.1989.tb00629.x. [DOI] [PubMed] [Google Scholar]
- Dugan JD, Lawton MT, Glaser B, Brem H. A new technique for explantation and in vitro cultivation of chicken embryos. Anat Rec. 1991;229:125–128. doi: 10.1002/ar.1092290114. [DOI] [PubMed] [Google Scholar]
- Yelbuz TM, Waldo KL, Kumiski Kumiski, Stadt HA, Wolfe RR, Leatherbury L, Kirby ML. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–510. doi: 10.1161/01.cir.0000023044.44974.8a. [DOI] [PubMed] [Google Scholar]
- Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetraology of Fallot and pulmonary atresia. Dev Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Tuan RS. Calcium transport and related functions in the chorioallantoic membrane of the cultured shell-less chick embryos. Dev Biol. 1980;74:196–204. doi: 10.1016/0012-1606(80)90061-5. [DOI] [PubMed] [Google Scholar]
- Tuan RS. Supplemented eggshell restores calcium transport in chorioallantoic membrane of cultured shell-less chick embryos. J Embryol Exp Morphol. 1983;74:119–131. [PubMed] [Google Scholar]
- Dunn BE, Dunn NB, Scharf KE. Effect of calcium supplementation on growth of shell-less cultured chick embryos. J Exp Zool Suppl. 1987;1:33–37. [PubMed] [Google Scholar]
- Ono T, Waksugi N. Mineral content of quail embryos cultured in mineral-rich and mineral-free conditions. Poultry Sci. 1983;63:159–166. doi: 10.3382/ps.0630159. [DOI] [PubMed] [Google Scholar]
- Perry MM. A complete culture system for the chick embryo. . Nature. 1988;331:70–72. doi: 10.1038/331070a0. [DOI] [PubMed] [Google Scholar]
- Naito M, Perry MM. Development in culture of the chick embryo from cleavage to hatch. Br Poult Sci. 1989;30:251–256. doi: 10.1080/00071668908417145. [DOI] [PubMed] [Google Scholar]
- Naito M, Nirasawa K, Oishi T. Development in culture of the chick embryo from fertilized ovum to hatching. J Exp Zool. 1990;254:322–326. doi: 10.1002/jez.1402540311. [DOI] [PubMed] [Google Scholar]
- Naito M, Nirasawa K, Oishi T. An in vitro method for chick embryos obtained from the anterior portion of the magnum of oviduct. Br Poult Sci. 1995;36:161–164. doi: 10.1080/00071669508417762. [DOI] [PubMed] [Google Scholar]
- Korn MJ, Cramer KS. Windowing chicken eggs for developmental studies. . doi: 10.3791/306. JoVE 2007;8. Available at http://dx.doi.org/10.3791/306. [DOI] [PMC free article] [PubMed]
- Hamburger V, Hamburger HL. A series of normal stages in the development of the chick embryo. Dev Dyn. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Mun AM. Toxic effects of normal sera and homologous antisera on the chick embryo. Biol Bulletin. 1957;115:239–256. [Google Scholar]
- LeDouarin N. A biological cell labeling technique and its uses in experimental embryology. Dev Biol. 1973;30:217–222. doi: 10.1016/0012-1606(73)90061-4. [DOI] [PubMed] [Google Scholar]
- Stern CD, Fraser SE, Keynes RJ, Primmett DR. A cell lineage analysis of segmentation in the chick embryo. Development. 1988;104Suppl:231–244. doi: 10.1242/dev.104.Supplement.231. [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M, Bronner-Fraser SE. Cell lineage analysis reveals multipotency of some avian neural crest cells. . Nature. 1988 Sep;335:161–164. doi: 10.1038/335161a0. [DOI] [PubMed] [Google Scholar]
- Schoenwolf GC, Sheard P. Shaping and bending of the avian neural plates as analysed with a fluorescent-histochemical marker. . Development. 1989;105:17–25. doi: 10.1242/dev.105.1.17. [DOI] [PubMed] [Google Scholar]
- Br Poult Sci GN, Bronner-Fraser M, Fraser SE. A vital dye analysis of the timing and pathways of avian trunk neural crest cell migration. . Development. 1989;106:809–816. doi: 10.1242/dev.106.4.809. [DOI] [PubMed] [Google Scholar]
- Okada A, Lansford R, Weimann JM, Fraser SE, Mcconnell SK. Imaging cells in the developing nervous system with retrovirus expressing modified green fluorescent protein. . Exp Neurol. 1999:394–406. doi: 10.1006/exnr.1999.7033. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. . Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Borwompinyo S, Brake J, Mozdziak PE, Petitte JN. Culture of chicken embryos in surrogate eggshells. . Culture of chicken embryos in surrogate eggshells. . 2005;84:1477–1482. doi: 10.1093/ps/84.9.1477. [DOI] [PubMed] [Google Scholar]