

Abstract
A range of extrinsic signals, including afferent activity, affect neuronal growth and plasticity. Neuronal activity regulates intracellular Ca2+, and activity-dependent calcium signaling has been shown to regulate dendritic growth and branching (Konur and Ghosh, 2005). NMDA receptor (NMDAR) stimulation of Ca2+/calmodulin-dependent protein kinase signaling cascades has, moreover, been demonstrated to regulate neurite/axonal outgrowth (Wayman et al., 2004). We used a sodium channel activator, brevetoxin (PbTx-2), to explore the relationship between intracellular [Na+] and NMDAR-dependent development. PbTx-2 alone, at a concentration of 30 nm, did not affect Ca2+ dynamics in 2 d in vitro cerebrocortical neurons; however, this treatment robustly potentiated NMDA-induced Ca2+ influx. The 30 nm PbTx-2 treatment produced a maximum [Na+]i of 16.9 ± 1.5 mm, representing an increment of 8.8 ± 1.8 mm over basal. The corresponding membrane potential change produced by 30 nm PbTx-2 was modest and, therefore, insufficient to relieve the voltage-dependent Mg2+ block of NMDARs. To unambiguously demonstrate the enhancement of NMDA receptor function by PbTx-2, we recorded single-channel currents from cell-attached patches. PbTx-2 treatment was found to increase both the mean open time and open probability of NMDA receptors. These effects of PbTx-2 on NMDA receptor function were dependent on extracellular Na+ and activation of Src kinase. The functional consequences of PbTx-2-induced enhancement of NMDAR function were evaluated in immature cerebrocortical neurons. PbTx-2 concentrations between 3 and 300 nm enhanced neurite outgrowth. Voltage-gated sodium channel activators may accordingly represent a novel pharmacologic strategy to regulate neuronal plasticity through an NMDA receptor and Src family kinase-dependent mechanism.
Introduction
Neuronal activity plays a key role in the regulation of dendritic development (McAllister, 2000; Cline, 2001; Chen and Ghosh, 2005). Neuronal activity regulates intracellular Ca2+, and activity-dependent calcium signaling has been shown to regulate dendritic growth and branching (Konur and Ghosh, 2005). Ca2+/calmodulin-dependent protein kinases (CaMKs) and mitogen-activated protein kinase (MAPKs) appear to be key mediators of calcium-dependent neurite outgrowth (Redmond et al., 2002). CaMK kinase (CaMKK) has been demonstrated to be an upstream regulator of both CaMK- and MAPK-signaling pathways. NMDA receptor (NMDAR)-dependent CaMKK/calmodulin kinase I-signaling cascades have, moreover, been shown to regulate neurite/axonal outgrowth (Wayman et al., 2004), activity-dependent synaptogenesis (Saneyoshi et al., 2008), and Ca2+-dependent extracellular regulated kinase activation and dendritic outgrowth (Schmitt et al., 2004; Wayman et al., 2006). NMDARs, therefore, play a critical role in activity-dependent development and plasticity, dendritic arborization, spine morphogenesis, and synapse formation by stimulating these calcium-dependent signaling pathways (Rajan and Cline, 1998; Sin et al., 2002; West et al., 2002; Wong and Ghosh, 2002; Miller and Kaplan, 2003; Tolias et al., 2005; Ultanir et al., 2007).
Recent studies have indicated that changes in intracellular sodium concentration ([Na+]i) produced in the soma and dendrites as a result of neuronal activity may act as a signaling molecule and play a role in activity-dependent synaptic plasticity. It has been shown that synaptic stimulation causes [Na+]i increments of 10 mm in dendrites and of up to 35–40 mm in dendritic spines (Rose et al., 1999; Rose and Konnerth, 2001). In hippocampal neurons, intracellular [Na+] increments have been demonstrated to increase NMDAR-mediated whole-cell currents and NMDAR single-channel activity by increasing both channel open probability and mean open time. This [Na+]i-mediated upregulation of NMDAR function has been shown to require Src kinase activation (Yu and Salter, 1998). Src family kinases act as a crucial point of convergence for signaling pathways that enhance NMDAR activity, and, by upregulating the function of NMDARs, Src gates the production of NMDAR-dependent synaptic potentiation and plasticity (Salter and Kalia, 2004).
We reasoned that [Na+]i may act as a positive regulator of developmental plasticity in immature cerebrocortical neurons. In the present study, we used brevetoxin (PbTx-2), a voltage-gated sodium channel (VGSC) activator, to manipulate [Na+]i in immature murine cerebrocortical neurons. Brevetoxins interact with neurotoxin site 5 on the α-subunit of VGSCs (Catterall and Gainer, 1985; Poli et al., 1986) and augment sodium influx through VGSCs by shifting the activation potential to more negative values, increasing the mean open time of the channel and inhibiting channel inactivation (Jeglitsch et al., 1998). Using immature cerebrocortical neurons, we now demonstrate that a VGSC activator elevates [Na+]i levels, enhances NMDA-induced Ca2+ influx, increases both NMDAR channel open probability and mean open time, and augments neurite outgrowth. These results provide direct evidence to support the hypothesis that NMDA receptor function is upregulated by elevated [Na+]i, which, in turn, promotes neurite outgrowth.
Materials and Methods
Materials
Trypsin, penicillin, streptomycin, heat-inactivated fetal bovine serum, horse serum, and soybean trypsin inhibitor were obtained from Atlanta Biologicals. Minimum essential medium, deoxyribonuclease, poly-l-lysine, poly-d-lysine hydrobromide, cytosine arabinoside, NMDA, protease inhibitor mixture, MK-801, d(-)-2-amino-5-phosphono-pentanoic acid (APV), and nifedipine were purchased from Sigma. Pluronic acid, fluo-3 acetoxymethyl ester (AM), and sodium-binding benzofuran isophthalate (SBFI)-AM were purchased from Invitrogen. 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3, 4-d] pyrimidine (PP2), 4-amino-7-phenylpyrazol [3, 4-d] pyrimidine (PP3), and STO-609 were purchased from Calbiochem. ECL Plus kits were purchased from GE Healthcare. Neurobasal and B-27 supplement were purchased from Invitrogen. FLIPR (fluorometric imaging plate reader) membrane potential assay kit was purchased from Molecular Devices. Anti-phospho Src (416) and anti-Src antibodies were purchased from Cell Signaling Technology, rabbit anti-human protein gene product 9.5 (PGP 9.5) from AbD SeroTec, and fluorescein (FITC) affiniPure goat anti-rabbit IgG from Jackson ImmunoResearch Laboratories. Brevetoxin-2 (PbTx-2) was isolated and purified from Karinia breve cultures at the Center for Marine Sciences at the University of North Carolina (Wilmington, NC).
Methods
Cerebrocortical neuron culture.
Primary cultures of cerebrocortical neurons were harvested from embryos of Swiss–Webster mice on embryonic day 16 and cultured as described previously (Cao et al., 2008). Cells were plated onto poly-l-lysine-coated 96-well (9 mm), clear-bottomed, black-well culture plates (Costar) at a density of 1.5 × 105 cells per well, 24-well (15.6 mm) culture plates at a density of 0.05 × 106 cells per well, 12-well (22 mm) culture plate (TPP-Midscience) at a density of 1.8 × 106 cells per well or 6-well (35 mm) culture dishes at a density of 4.5 × 106 cell per well, respectively, and incubated at 37°C in a 5% CO2 and 95% humidity atmosphere. Cytosine arabinoside (10 μm) was added to the culture medium on day 2 after plating to prevent proliferation of non-neuronal cells. The culture media was changed on days 4 and 7 using a serum-free growth medium containing neurobasal medium supplemented with B-27, 100 I.U./ml penicillin, 0.10 mg/ml streptomycin, and 0.2 mm l-glutamine. All animal use protocols were approved by the Institutional Animal Care and Use Committee.
Excitotoxicity assays.
The growth medium of neurons grown on 12 well plates was collected and saved, and the neurons washed thrice in 1 ml of sterile filtered Locke's incubation buffer (154 mm NaCl, 5.6 mm KCl, 1.0 mm MgCl2, 2.3 mm CaCl2, 8.6 mm HEPES, 5.6 mm glucose, 0.1 mm glycine, pH 7.4). The neurons were then exposed to varying concentrations of NMDA in 1 ml of Locke's buffer for 2 h at 37°C in a 5% CO2 and 95% humidity atmosphere. At the termination of NMDA exposure, the incubation medium was collected for later analysis of lactate dehydrogenase (LDH) activity, and the neurons were washed thrice in 1 ml of fresh Locke's buffer followed by replacement with previously collected growth medium that had been filtered and supplemented with 1.25 mg/ml d-glucose. The cell cultures were then incubated at 37°C in a 5% CO2 and 95% humidity atmosphere. At 24 h after treatment exposure, the growth medium was collected and saved for analysis of LDH activity. The LDH activity was assayed according to a previously described method (Koh and Choi, 1987).
Neuronal injury was also assessed morphologically by exposing cerebrocortical neurons for 5 min to the vital dye fluorescein diacetate (5 μg/ml). After 5 min incubation in dye, the neurons were washed three times in fresh Locke's buffer. Three random 20× field images were taken per well. Uniform sector areas (n = 6) were counted for viable neurons on each 20× field image and were normalized to the number of viable neurons per area of the well.
Immunocytochemistry.
To assess the influence of PbTx-2 on neuronal morphogenesis, cells grown on poly-lysine coated cover glass placed inside 24-well culture plates were used. PbTx-2 at concentrations ranging from 0.1 to 1000 nm were added to the culture medium at 3 h after plating. In some experiments, these concentrations of PbTx-2 were coincubated with tetrodotoxin (TTX) (1 μm), MK-801 (1 μm), nifedipine (1 μm), or STO-609 (2.6 μm). Cultures were fixed for 20 min at room temperature at 15, 24, 40, or 108 h after plating using 4% paraformaldehyde in PBS. After fixation, neurons were blocked and permeabilized by incubation for 30 min with PBS containing 2% fetal bovine serum and 0.15% Triton X-100. The coverslips were then inverted onto 100 μl droplets of blocking buffer containing the protein gene product 9.5 (anti-PGP 9.5) primary antibody for 60 min at room temperature or overnight at 4°C. After three successive washes in blocking buffer, coverslips were incubated with a secondary antibody [FITC (anti-rabbit IgG)] for 60 min at room temperature. Coverslips were washed and mounted on microscope slides and analyzed by fluorescence microscopy on an Olympus IX 71 inverted microscope with a Nikon camera. Digital images of individual neurons were captured and total neurite length quantified using IP Lab 3.6.5 software (Scanalytics). At least 30 randomly chosen neurons from different cultures were evaluated for each treatment group.
Intracellular Ca2+ monitoring.
Cerebrocortical neurons grown in 96-well plates were used for intracellular Ca2+ concentration ([Ca2+]i) measurements as described previously (Dravid et al., 2005). Briefly, the growth medium was removed and replaced with dye-loading medium (100 μl per well) containing 4 μm fluo-3 AM and 0.04% pluronic acid in Locke's buffer. After 1 h of incubation in dye-loading medium, the neurons were washed four times in fresh Locke's buffer (200 μl per well, 22°C) using an automated microplate washer (Bio-Tek Instruments) and transferred to a FLEX Station II (Molecular Devices) benchtop scanning fluorometer chamber. The final volume of Locke's buffer in each well was 150 μl. Fluorescence measurements were performed at 37°C. The neurons were excited at 488 nm and Ca2+-bound fluo-3 emission was recorded at 538 nm at 1.2 s intervals. After recording baseline fluorescence for 60 s, 50 μl of a 4× concentration of NMDA, PbTx-2, or both were added to the cells at a rate of 26 μl/s, yielding a final volume of 200 μl/well; the fluorescence was monitored for an additional 140–240 s. The fluo-3 fluorescence was expressed as (Fmax − Fmin)/Fmin where Fmax was the maximum, Fmin the minimum fluorescence measured in each well.
Intracellular sodium concentration ([Na+]i) measurement.
[Na+]i measurement and full in situ calibration of SBFI fluorescence ratio was performed as described previously (Cao et al., 2008). Cells grown in 96-well plates were washed four times with Locke's buffer (in mm: 8.6 HEPES, 5.6 KCl, 154 NaCl, 5.6 glucose, 1.0 MgCl2, 2.3 CaCl2, 0.1 glycine, pH 7.4) using an automated microplate washer (Bio-Tek Instruments). The background fluorescence of each well was measured and averaged before dye loading. Cells were then incubated for 1 h at 37°C with dye-loading buffer (100 μl/well) containing 10 μm SBFI-AM and 0.02% pluronic F-127. After 1 h incubation in dye-loading medium, cells were washed five times with Locke's buffer, leaving a final volume of 150 μl in each well. The plate was then transferred to the chamber of a FLEXstation II (Molecular Devices). Cells were excited at 340 and 380 nm, and Na+-bound SBFI emission was detected at 505 nm. Fluorescence readings were taken once every 4 s for 60 s to establish the baseline, and then 50 μl PbTx-2 at final concentrations ranging from 5 to 1000 nm were added to each well from the compound plate at a rate of 26 μl/s, yielding a final volume of 200 μl/well. The raw emission data at each excitation wavelength were exported to an Excel work sheet and corrected for background fluorescence. The SBFI fluorescence ratios (340 of 380) versus time were then analyzed, and time–response and concentration–response graphs were generated using GraphPad Prism (GraphPad Software).
Full in situ calibration of the SBFI fluorescence ratio was done using calibration media containing the following (in mm): 0.6 MgCl2, 0.5 CaCl2, 10 HEPES, Na+ and K+ such that [Na+] plus [K+] = 130, 100 gluconate, and 30 Cl− (titrated with 10 mol/l KOH to pH 7.4). Gramicidin D (5 μm) (Na+ ionophore), monensin (10 μm) (Na+/H+ carrier), and ouabain (100 μm) (Na+/K+-ATPase inhibitor) were added to equilibrate the intracellular and extracellular sodium concentration. After five washes, the Locke's buffer was replaced by 150 μl sodium-containing calibration solution (0–130 mm). The plate was then loaded onto the FLEXstation chamber for recording of emitted fluorescence during excitation at 340 and 380 nm. Fluorescence data were converted to a ratio (340 of 380) after background correction. To convert the fluorescence ratio of emitted SBFI signals into a [Na+]i value, the following equation was used: [Na+] = βKd [(R − Rmin)/(Rmax − R)] (1), where β is the ratio of the fluorescence of the free (unbound) dye to bound dye at the second excitation wavelength (380 nm), Kd is the apparent dissociation constant of SBFI for Na+, R is the background-subtracted SBFI fluorescence ratio, and Rmin and Rmax are, respectively, the minimum and maximum fluorescence values. The data relating [Na+]i to R were fitted by a three-parameter hyperbolic equation having the following form: R = Rmin + [a ([Na+])/(b + [Na+])] (2), where a and b are constants and equal to Rmax − Rmin and βKd, respectively (Diarra et al., 2001; Cao et al., 2008). These data relating [Na+]i to R (see Fig. 5B) were well described (r2 = 0.98) by Equation 2. The derived parameters were Rmin = 1.45 ± 0.03, a = 2.073 ± 0.06, and b = 30.75 ± 1.9. The value for Rmin obtained by this method was identical to the value of Rmin derived experimentally at [Na+] = 0 mm. The corresponding values for Rmax and βKd were, therefore, Rmax = 3.52 ± 0.09 and βKd = 30.75 ± 1.9 mm. We compared the values of Rmax and βKd obtained from a Hanes plot (Cao et al., 2008) to those derived from the three-parameter hyperbolic fit. The equation was rearranged to generate a Hanes plot such that [Na+]/(R − Rmin) = {[Na+]/(Rmax − Rmin)} + [βKd/(Rmax − Rmin)] (3).
Figure 5.

PbTx-2 increases intracellular sodium levels in DIV-2 cerebrocortical neurons. A, In situ calibration of SBFI fluorescence ratio (340 of 380). Time–response data show stepwise changes in SBFI fluorescence ratio values evoked by successive increments in extracellular sodium ion concentration. B, Three parameter hyperbolic curve fit adequately describes calibration data. SE bars are contained within each data point. C, Time–response profile for PbTx-2-induced [Na+]i elevation as represented by the SBFI fluorescence ratio. Data represent the mean ± SEM of three separate experiments, each with six to eight replicates. The scale on the right ordinate depicts the calibrated [Na+]i corresponding to SBFI fluorescence ratio values. The basal [Na+]i was found to be 8.1 ± 0.32 mm. A maximum elevation of [Na+]i to 16.9 ± 1.5 mm was determined after addition of PbTx-2 (30 nm) corresponding to a [Na+]i increment of 8.8 ± 1.8 mm. (*p < 0.001, unpaired t test). D, Nonlinear regression analysis of the PbTx-2 concentration–response data (EC50 = 43.3 nm; 23.5–79.9 nm, 95% CI).
The plotting of [Na+]/(R − Rmin) versus [Na+]i as a Hanes function yielded a straight line (r2 = 0.997) (data not shown). The slope {1/(Rmax − Rmin)} provides a means to estimate of Rmax, whereas the intercept on the abscissa is equal to −βKd. The value for Rmin was obtained from the experimental data. The values of Rmax and βKd calculated from Hanes plot were 3.55 ± 0.09 and 32.96 ± 1.9 mm, respectively, and were not significantly different from the values derived from the three-parameter hyperbolic fit which were 3.52 ± 0.09 (Rmax) and 30.75 ± 1.9 mm (βKd).
Western blotting.
Western blot analysis was performed in cells grown in 12-well plates as described previously (Cao et al., 2007). Cells were washed three times with Locke's buffer and then allowed to equilibrate in Locke's buffer for 30 min. Cultures were then treated with the indicated drugs at 37°C for specified times. Cultures were then transferred to ice slurry to terminate drug exposure. After washing with ice-cold PBS, cells were harvested in ice-cold lysis buffer containing 50 mm Tris, 50 mm NaCl, 2 mm EDTA, 2 mm EGTA, 1% NP-40, 0.1% SDS, 2.5 mm sodium pyrophosphate, and 1 mm sodium orthovanadate. Phenylmethylsulfonyl fluoride (1 mm) and 1× protease inhibitor mixture were then added and the lysate incubated for 20 min at 4°C. Cell lysates then underwent sonication and were centrifuged at 13,000 × g for 15 min at 4°C. The supernatant was assayed by Bradford method to determine protein content. Equal amounts of protein were mixed with the Laemmli sample buffer and boiled for 5 min. The samples were loaded onto a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane by electroblotting. The membranes were blocked in TBST (20 mm Tris, 150 mm NaCl, 0.1% Tween 20) with 5% skim milk for 1 h at room temperature. After blocking, membranes were incubated overnight at 4°C in primary antibody diluted in TBST containing 5% skim milk. The blots were washed and incubated with the secondary antibody conjugated with horseradish peroxidase for 1 h, washed four times in TBST, and exposed with ECL Plus for 4 min. Blots were exposed to Kodak hyperfilm and developed. Blots were subsequently stripped (63 mm Tris base, 70 mm SDS, 0.0007% 2-mercaptoethanol, pH 6.8) and reprobed for further use. Western blot densitometry data were obtained using MCID Basic 7.0 software (Imaging Research). ANOVA and graphing were completed using GraphPad Prism (GraphPad Software).
Membrane potential assay fluorescence monitoring.
Membrane potential in the cerebrocortical neuron cultures was determined using the FLIPR membrane potential (FMP) assay (Molecular Devices). In this method, we used the FMP blue dye to assess the membrane potential of neurons in culture. Quantification of the changes in membrane potential was derived using KCl as a reference. The FMP blue dye is a lipophilic, anionic, bis-oxonol-based dye that distributes across the cell membrane as a function of membrane potential and displays enhanced fluorescence emission after binding to intracellular proteins (Whiteaker et al., 2001; Baxter et al., 2002). This kit also contains a proprietary extracellular fluorescence quencher that eliminates the need to wash neurons after the dye-loading incubation. In preliminary experiments, we determined that, with cerebrocortical neurons in culture, an optimum dye concentration was one-eighth of the suggested manufacturer concentration. The dye stock solution (1×) was prepared by dissolving 10 ml Locke's buffer to the content of each vial. For loading the cells, eightfold diluted stock solutions were used. After removing the culture medium, 180 μl of assay buffer was added to the neurons, and the plate was incubated at 37°C in a 5% CO2 and 95% humidity atmosphere for 30 min. For KCl calibration measurements, varying concentrations of 10× KCl standard solutions in assay buffer were prepared. After 30 min equilibration incubation, the plate was transferred to a Flex Station II chamber, and the fluorescence measurements were performed at 37°C. Neurons were excited at 530 nm, and emission was recorded at 565 nm at 2 s intervals. After recording the baseline for 60 s, either 20 μl KCl or PbTx-2 was added to a final volume of 200 μl at a rate of 26 μl/s, and the fluorescence was monitored for an additional 440 s. This protocol was executed by alternate column-by-column addition of KCl and PbTx-2 in the same 96-well plate using the Flex Station II pipettor.
A linear regression analysis of the log concentration of K+ versus FMP blue fluorescence area under the curve (AUC) was generated. Because the membrane potential of isolated neurons is mainly the result of the K+ equilibrium potential (Hille, 1992), we used the Goldman–Hodgkin–Katz equation to generate a standard curve for the estimation of membrane potential (EM) at various concentrations of extracellular K+:
 |
where EM is membrane potential, R is universal gas constant, T is temperature using the Kelvin scale, and PK, PNa, and PCl are permeabilities for K+, Na+, and Cl−, respectively. [K+]out, [Na+]out, and [Cl−]out, and [K+]in, [Na+]in, and [Cl−]in are the respective extracellular and intracellular concentrations of K+, Na+, and Cl−. A 2 d in vitro (DIV-2) neuronal [Cl−]in value of 140 mm was used for these calculations (Kuner and Augustine, 2000). The regression for the [K+]out versus Δ fluorescence and EM was used for estimating PbTx-2-induced change in membrane potential.
Electrophysiology.
Single-channel currents were recorded at 23°C in the cell-attached configuration (Hamill et al., 1981). Patch pipettes were pulled from borosilicate glass capillaries (Warner Instruments), coated with Sylgard 184 (Dow Corning) and fire-polished to a resistance of 10–15 MΩ when filled with the pipette solution. The external recording solution consisted of Mg2+-free Locke's buffer with 20 μm EDTA to chelate trace amounts of divalent cations. PbTx-2 was always applied in the bath. The patch pipette solution consisted of extracellular Locke's buffer without MgCl2 and with either 3 or 10 μm NMDA and 100 μm glycine. In some experiments, 10 μm strychnine, 10 μm bicuculline methiodide and 10 μm DNQX were included in the external solution to block nonspecific components. All recordings were done from DIV-2 cerebrocortical neurons. Cell-attached patch recordings were done using an Axopatch 200B amplifier (Molecular Devices), filtered at 8 kHz (−3 dB, 8-pole Bessel), and digitized at 40 kHz digitized with Axon pClamp 10.2 software. The pipette potential was +60 mV. Records were idealized with a segmental k-means algorithm (Qin, 2004) using QUB software (www.qub.buffalo.edu). All conductance levels were assumed to be equal for the analysis. Dwell-time histograms were generated and fitted using Channelab (Synaptosoft) with an imposed dead time of 100 μs. The open probability (Po), mean open time, and amplitude were compared by paired t test. For representation in figures, the Po, mean open time, and amplitude were normalized to the average of respective control values. The corresponding PbTx-2-treated values were normalized to their paired control values.
Results
Immature cerebrocortical neurons show decreased vulnerability to NMDA-induced excitotoxicity
Because NMDA receptors have been shown to be a crucial mediator of excitotoxicity (Berman and Murray, 2000; Hardingham and Bading, 2003) and immature neurons exhibit decreased vulnerability to excitotoxicity both in vivo (Liu et al., 1996) and in vitro (Choi et al., 1987; Mizuta et al., 1998; Cheng et al., 1999; Marks et al., 2005; King et al., 2006), we examined the influence of cerebrocortical culture age on vulnerability to NMDA-induced excitotoxicity. We assessed NMDA-induced LDH efflux, a biochemical index for neuronal injury, as a function of development in culture. Cerebrocortical neurons were exposed to a range of NMDA concentrations for 2 h and assayed for LDH efflux at 2 and 24 h after the initiation of exposure. At 2, 4, and 6 DIV, cerebrocortical cultures showed little sensitivity to NMDA-induced LDH efflux assessed immediately after either the 2 h exposure or at 24 h after exposure; however, DIV-9 cerebrocortical neurons showed sensitivity to NMDA. In DIV-9 cultures, the assessment of NMDA toxicity at 24 h after initiation of exposure resulted in substantial levels of LDH efflux as reflected in a maximal LDH efflux value of 247 ± 10.5% of control (Fig. 1). In DIV-9 neurons, the NMDA EC50 value with 95% confidence intervals (CI) for LDH efflux was 53.1 μm (27.9–101 μm). The insensitivity to NMDA-induced excitotoxicity in DIV-2, -4, and -6 cultures agrees with previous reports. Studies with primary cerebrocortical cultures have reported a decreased vulnerability to excitotoxicity in 2–7 DIV cultures compared with 11–14 DIV cultures after challenge with glutamate or NMDA (Choi et al., 1987; Mizuta et al., 1998; Cheng et al., 1999; King et al., 2006). Similarly, sensitivity to excitatory amino acid-induced toxicity in neocortical neurons has been shown to emerge at DIV-7–9 but not in DIV-2 (Frandsen and Schousboe, 1990; Griffiths et al., 1997). To confirm our LDH efflux data, fluorescein diacetate staining was used to allow visualization and quantification of viable neurons. As depicted in Figure 1, in DIV-2 and DIV-9 cerebrocortical neurons, the viable cell count data derived from fluorescein diacetate staining was well correlated with the LDH efflux data and provided confirmation of NMDA-induced excitotoxicity in DIV-9 but not DIV-2 cerebrocortical neurons. The NMDA EC50 value for decreasing the live neuron count was 82.2 μm (95% CI 38.4–176 μm). Based on this insensitivity to NMDA-induced excitotoxicity, DIV-2 cerebrocortical cultures were selected as a model system to explore the influence of sodium on NMDA receptor signaling.
Figure 1.

NMDA-induced excitotoxicity in immature cerebrocortical neurons. A, B, Comparison of LDH efflux and estimation of viable neurons by using fluorescein diacetate staining in DIV-2 (A) and DIV-9 (B) cerebrocortical neurons at 24 h after initiation of a 2 h NMDA exposure. Mean ± SEM of LDH efflux values and viable cell count are represented as percentage of control. Experiment was repeated six times each with triplicate values. C, Representative images of fluorescein diacetate staining of cerebrocortical neurons at 2 and 24 h time points.
PbTx-2 augments NMDA-induced Ca2+ influx in immature cerebrocortical neurons
Previous studies have indicated that activity-dependent neuronal development is primarily triggered through Ca2+-dependent signaling pathways (Chen and Ghosh, 2005; Konur and Ghosh, 2005). Although NMDA receptors represent a key source of Ca2+ entry, the underlying mechanisms responsible for NMDAR regulation of activity-dependent development remain to be fully delineated. Because synaptic activity has been shown to elevate [Na+]i (Rose and Konnerth, 2001) and sodium acts as a positive regulator of NMDAR function (Yu and Salter, 1998), we used the sodium channel activator, PbTx-2, as a probe to further explore the influence of sodium on NMDAR signaling. A quantitative assessment of the increments in neuronal sodium concentration produced by an array of VGSC activators has demonstrated that brevetoxins such as PbTx-2 elevate [Na+]i to concentrations as high as 50 mm (Cao et al., 2008). Before the investigation of the influence of PbTx-2 on NMDA-induced Ca2+ influx, we first assessed the effect of PbTx-2 alone on Ca2+ dynamics in DIV-2 cerebrocortical neurons. The concentration dependence of the increase in [Ca2+]i stimulated by PbTx-2 was examined in fluo-3-loaded DIV-2 cerebrocortical neurons. PbTx-2 produced a rapid and concentration-dependent increase in [Ca2+]i (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Nonlinear regression analysis of the concentration dependence of the PbTx-2-induced integrated fluo-3 response indicated that the EC50 for PbTx-2-stimulated increase in [Ca2+]i was 244 nm (119–498 nm, 95% CI). A concentration of 30 nm PbTx-2 was found to be subthreshold in DIV-2 neurons in that this concentration did not increase fluo-3 fluorescence (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). This PbTx-2 concentration response profile generalized to DIV-4, -6, and -9 neurons in that a 30 nm concentration of PbTx-2 did not alter [Ca2+]i (data not shown).
Given that 30 nm PbTx-2 was without effect on Ca2+ dynamics in cerebrocortical neurons, we investigated NMDA-induced Ca2+ influx in the presence and absence of this concentration of PbTx-2 in DIV-2, -4, -6, and -9 cerebrocortical cultures. NMDA produced a concentration-dependent elevation of [Ca2+]i in all four ages of cerebrocortical neuron cultures. Analysis of the NMDA-concentration response data, in the absence of 30 nm PbTx-2, for DIV-2, -4, -6, and -9 revealed a leftward shift in these concentration–response curves as a function of development (Fig. 2). The EC50 value for NMDA-induced Ca2+ influx ranged from 19.2 μm in DIV-4 neurons to 11.1 μm in DIV-9 neurons. The presence of 30 nm PbTx-2 produced a robust potentiation of NMDA-induced Ca2+ influx in cultures of all ages (Figs. 2, 3; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The NMDA concentration–response curves for Ca2+ influx were significantly leftward shifted by 30 nm PbTx-2. Nonlinear regression analysis of these data indicated that NMDA EC50 values were significantly lower in the presence of PbTx-2 (Fig. 2E). These data suggest that PbTx-2 sensitizes cerebrocortical neurons to NMDA-induced Ca2+ influx. The PbTx-2 potentiation of NMDA-induced Ca2+ influx was most prominent at an NMDA concentration of 3 μm in DIV-2 neurons as depicted in Figure 3. NMDA at a concentration of 3 μm alone did not affect [Ca2+]i but in the presence of 30 nm PbTx-2 elicited a robust increment in [Ca2+]i. Although not as strong as the effect in DIV-2 cultures, 30 nm PbTx-2 augments NMDA-induced Ca2+ influx in all ages of cerebrocortical cultures (the response of DIV-9 neurons is shown in supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Based on these results, a concentration of 30 nm PbTx-2 was selected to further probe the potential influence of sodium on NMDAR signaling.
Figure 2.

PbTx-2 augments NMDA-induced Ca2+ influx in immature cerebrocortical neurons as a function of culture age. A–D, Nonlinear regression analysis of the fractional increase [(Fmax − Fmin)/Fmin] in Ca2+ influx as a function of NMDA concentration in the culture ages of 2, 4, 6, and 9 DIV. Each point represents mean ± SEM of sextuplicate values. Experiment was repeated six times with sextuplicate determinations in six independent cultures. E, EC50 and 95% CI derived from the NMDA concentration–response (depicted in A–D) in the presence and absence of 30 nm PbTx-2.
Figure 3.

Effect of 30 nm PbTx-2 on NMDA-induced Ca2+ influx in immature cerebrocortical neurons. Representative time–response data using DIV-2 cerebrocortical neurons. Cerebrocortical neurons were treated with a range of NMDA concentrations in the presence and absence of 30 nm PbTx-2. The presence of 30 nm PbTx-2 produced a robust augmentation of NMDA-induced Ca2+ influx in DIV-2 neurons.
PbTx-2 potentiation of NMDA-induced Ca2+ influx requires VGSCs and activation of a Src family kinase
To confirm the involvement of VGSCs and NMDARs in the PbTx-2 enhancement of NMDA-induced [Ca2+]i, we conducted a pharmacological evaluation of the response to a fixed concentration of 3 μm NMDA in DIV-2 cerebrocortical neurons. The role of NMDARs and VGSCs in the response to NMDA in the presence and absence of PbTx-2 was assessed by pretreating cerebrocortical neurons with 100 μm d-APV, a competitive NMDA receptor antagonist or 1 μm TTX, a VGSC pore blocker. These compounds both abrogated (p < 0.01) NMDA-induced Ca2+ influx in the presence of PbTx-2 (Fig. 4A,B). These results, therefore, confirm the requirement for activation of NMDARs and VGSCs in this response. Because the upregulation of NMDAR function by [Na+]i has been shown to involve Src kinase activation, we next examined the role of Src family kinases in the PbTx-2 potentiation of NMDA-induced Ca2+ influx using the specific Src family kinase inhibitor, PP2 (10 μm) (Fig. 4C,D). PP2 dramatically reduced (p < 0.01) the PbTx-2-induced potentiation of NMDA-induced Ca2+ influx in cerebrocortical neurons. In contrast, PP3 (10 μm), the inactive congener of PP2, did not affect the PbTx-2 potentiation of NMDA-induced Ca2+ influx, supporting the involvement of a Src family kinase in this response. The activation of Src family kinases are tightly and dynamically controlled by autophosphorylation and dephosphorylation of specific tyrosine residues. The phosphorylation of a tyrosine residue (Y416) within the activation loop of Src kinase has been identified as important for its activation (Salter and Kalia, 2004). We therefore assessed tyrosine phosphorylation of Src using an anti-phospho-Y416 Src antibody. Western blot analysis revealed that the combination of 30 nm PbTx-2 and 3 μm NMDA produced an activation of Src kinase as reflected by a significant increase in the phosphorylation of tyrosine 416 (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 4.

Pharmacological evaluation of PbTx-2-induced potentiation of NMDA-induced Ca2+ influx in DIV-2 cerebrocortical neurons. A, Time–response data are from representative experiment performed in sextuplicate and repeated four times. Cerebrocortical neurons were treated with either 1 μm TTX or 100 μm APV before the addition of 3 μm NMDA and 30 nm PbTx-2. B, Each bar represents mean ± SEM (n = 4). Both 1 μm TTX and 100 μm APV completely blocked PbTx-2 potentiation of NMDA-induced Ca2+ influx. *p < 0.01 (ANOVA followed by Dunnett's multiple comparison test). C, Effect of PP2, a specific Src-family kinase inhibitor, on PbTx-2-induced potentiation of NMDA-induced Ca2+ influx in DIV-2 cerebrocortical neurons. Time–response data are from representative experiment performed in sextuplicate and repeated six times. Cerebrocortical neurons were exposed to 10 μm PP2 or PP3 15 min before the addition of 3 μm NMDA and 30 nm PbTx-2. D, Each bar represents mean ± SEM (n = 6). *p < 0.01, paired t test.
PbTx-2 increases intracellular sodium levels in immature cerebrocortical neurons
Given the role of [Na+]i as a putative regulator of NMDAR-mediated signaling, it was important to quantify the magnitude of PbTx-2-induced elevation of [Na+]i in immature cerebrocortical neurons. We therefore assessed PbTx-2-induced elevation of [Na+]i in DIV-2 cerebrocortical neurons loaded with SBFI. As described previously for the assay of [Na+]i in mature cerebrocortical neurons, we performed a full in situ calibration of the relationship between the ratiometric SBFI signal and [Na+]i in DIV-2 neurons (Cao et al., 2008). The emitted fluorescence intensities were recorded during excitation at 340 and 380 nm and were converted to a ratio (340 of 380) after background correction (Fig. 5A). These calibration data relating SBFI fluorescence ratio to [Na+]i were adequately described by a three-parameter hyperbolic curve fit as described in Materials and Methods (Fig. 5B). PbTx-2 treatment of DIV-2 cerebrocortical neurons produced a concentration-dependent decrease in SBFI fluorescence emitted by excitation at 380 nm, whereas emitted fluorescence during excitation at 340 nm was unaffected, indicating no significant cell swelling after PbTx-2 exposure in DIV-2 neurons (data not shown). As shown in Figure 5C, PbTx-2 produced concentration-dependent increments in [Na+]i. The in situ SBFI calibration showed that the basal [Na+]i in DIV-2 cerebrocortical neurons was 8.1 ± 0.32 mm. This value is in reasonable agreement with those determined previously in more mature neuronal cultures: a basal level [Na+]i of 8.9 mm in cultured hippocampal neurons (Rose and Ransom, 1997), 9 ± 2 mm in either cultured spinal dorsal horn or hippocampal neurons (Yu and Salter, 1998), and 10.3 ± 0.22 mm in DIV-9 cerebrocortical neurons (Cao et al., 2008). Further analysis of the concentration–response profile for PbTx-2-induced elevation of [Na+]i indicated that the EC50 value for PbTx-2 was 43.3 nm (23.5–79.9 nm, 95% CI), and the maximum elevation of [Na+]i was 36.6 ± 0.9 mm (Fig. 5D). Because a 30 nm concentration of PbTx-2 was sufficient to augment NMDA-induced Ca2+ influx, it was important to quantify the [Na+]i increment associated with this treatment. The 30 nm PbTx-2 treatment produced a maximum [Na+]i of 16.9 ± 1.5 mm, representing an increment of 8.8 ± 1.8 mm over basal. Previous reports in hippocampal neurons suggested that an increment of [Na+]i of 10 mm was sufficient to produce significant increases in NMDAR channel activity (Yu and Salter, 1998; Yu, 2006). It has, moreover, been reported that increments of [Na+]i of >5 mm may represent a critical threshold required to regulate NMDAR-mediated Ca2+ influx in primary cultures of hippocampal neurons (Xin et al., 2005). Consistent with these findings, the increment of [Na+]i detected in immature cerebrocortical neurons appears sufficient to upregulate NMDAR signaling.
PbTx-2 augmentation of NMDA receptor signaling does not involve depolarization-induced relief of Mg2+ blockade
The ability of PbTx-2 to potentiate NMDA-induced Ca2+ influx could be a consequence of either the elevation of [Na+]i or neuronal depolarization with attendant relief of the Mg2+ block of NMDAR. To ascertain the magnitude of PbTx-2-induced membrane depolarization, we assessed PbTx-2-induced membrane potential changes in DIV-2 cerebrocortical neurons using a membrane-potential sensitive fluorescence dye, FMP blue. The changes in membrane potential measured with FMP blue are well correlated with those determined by patch-clamp analysis (Baxter et al., 2002). To document that FMP blue behaved as a Nernstian fluorescent indicator of membrane potential in cerebrocortical neurons, we determined the relationship between extracellular K+ concentration and fluorescence intensity. To equate changes in membrane potential to alteration in FMP blue fluorescence, a KCl concentration–response calibration curve was generated to establish that fluorescent intensity was directly proportional to the extracellular K+ concentration (Fig. 6A). Extracellular K+ produced a concentration-dependent increase in maximum FMP blue fluorescence consistent with a depolarization-induced redistribution of the lipophilic anion dye and attendant increase in fluorescence quantum efficiency. As depicted in Figure 6B, the regression analysis of the K+ concentration-dependent changes FMP blue fluorescence showed marked linear correlation (r2 = 0.99). For a Nernstian fluorescent indicator of membrane potential, the ratio of fluorescence inside to the outside of the cell should be related to the membrane potential as described by the Nernst equation (Ehrenberg et al., 1988). This prediction is based on the principal that the membrane potential of isolated neurons is mainly the result of the K+ diffusion potential (Hille, 1992). We, therefore, used the Goldman–Hodgkin–Katz equation to generate a standard curve for the estimation of membrane potential (EM) at various concentrations of extracellular K+. The membrane potential of cerebrocortical neurons was dependent on the external concentration of K+ (Hille, 1992). The concordance of the [K+]out versus membrane fluorescence and [K+]out versus EM regressions indicates that changes in cerebrocortical neuron FMP blue fluorescence can be used to estimate membrane potential. Therefore, the relationship between fluorescence change and EM depicted in Figure 6B was generated to determine PbTx-2-induced changes in membrane potential of cerebrocortical neurons. The resting membrane potential of DIV-2 cerebrocortical neurons was found to be −29.5 ± 0.01 mV. This is consistent with previous demonstrations of relatively depolarized resting membrane potentials of immature neurons that later become more hyperpolarized as neurons mature (Ramoa and McCormick, 1994; Kim et al., 1995). As shown in Figure 6C, PbTx-2 produced a rapid and concentration-dependent increment in FMP blue fluorescence in DIV-2 cerebrocortical neurons. Nonlinear regression analysis of the PbTx-2 concentration–response relationship yielded an EC50 value of 96.7 nm (71.2–131.5 nm, 95% CI) (Fig. 6D). Because the 30 nm concentration of PbTx-2 was sufficient to augment NMDA-induced Ca2+ influx and to elevate [Na+]i, it was important to assess the membrane potential changes associated with this treatment. The 30 nm PbTx-2 treatment produced a transient increase in FMP blue fluorescence that was roughly equivalent to the fluorescence change produced by an extracellular K+ concentration of 7.6 mm. The corresponding membrane potential change was accordingly found to be negligible, representing only a 0.2 ± 0.02 mV depolarization (from 29.5 ± 0.01 to 29.3 ± 0.02 mV). This change in membrane potential would, therefore, not be sufficient to influence voltage-dependent Mg2+ block of NMDARs (Mayer et al., 1984). We confirmed the membrane potential and lack of influence of PbTx-2 (30 nm) in DIV-2 neurons using noninvasive single-channel recordings (Tyzio et al., 2003). Measurement of single-channel NMDA receptor currents in cell-attached mode indicated that the membrane potential was −28 mV (19–38, 95% CI), and 30 nm PbTx-2 did not affect this measure in DIV-2 neurons (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 6.

PbTx-2-evoked change in membrane potential in DIV-2 cerebrocortical neurons. A, Concentration–response profile for KCl-evoked FMP blue fluorescence change as a function of time. Each point represents mean ± SEM of 12 values. Experiment was repeated three times on independent cultures. B, The integrated time–response data for the increment in FMP blue fluorescence (AUC) was plotted as a function of K+ concentration. The displayed regression and correlation coefficient (r2 = 0.998) were derived from linear regression analysis. The right ordinate scale shows membrane potential for each [K+] which was calculated using Goldman–Hodgkin–Katz equation as described in Materials and Methods. The resting membrane potential was −29.5 ± 0.01 mV. C, Concentration–response of PbTx-2-induced changes in membrane potential as determined by changes in FMP blue fluorescence. Each point represents the mean ± SEM of 12 values. This experiment was repeated three times on independent cultures. D, Nonlinear regression analysis of the integrated time–response data for the increment in FMP blue fluorescence (AUC) as a function of PbTx-2 concentration. The membrane potential values were determined by performing K+ calibration regressions in the same culture plate. The membrane potential change evoked by 30 nm PbTx-2 was 0.2 ± 0.02 mV.
PbTx-2 increases NMDA single-channel open probability and mean open time
To gain insight into the effect of PbTx-2 on single-channel properties of NMDA receptors, unitary currents were recorded from DIV-2 cerebrocortical neurons. Cell-attached patch recording was performed with 3 μm NMDA and 100 μm glycine in the patch pipette at a patch potential of +60 mV. Experiments were performed in the nominal absence of extracellular Mg2+ in the recording buffer supplemented with 20 μm EDTA to chelate trace amounts of divalent ions. In the majority of patches, we observed only single openings with no apparent simultaneous double openings. The absence of double openings was presumably attributable to the submaximal concentration of NMDA used in the patch pipette and the low expression of NMDA receptors in immature cerebrocortical neurons. Patches in which we observed simultaneous double openings were not further analyzed. Single-channel recordings were idealized using the QUB and analyzed using ChanneLab with an imposed resolution of 100 μs. Bath application of 30 nm PbTx-2 significantly increased the open probability (Po) of NMDA receptors from 0.0054 ± 0.002 under control conditions to 0.031 ± 0.008 (443 ± 102% of control) after 30 nm PbTx-2 (n = 5, p < 0.05, paired t test) (Fig. 7A,B). The mean open time was also increased by PbTx-2 application from 1.99 ± 0.26 ms without PbTx-2 to 2.95 ± 0.34 ms (153 ± 20% of control) after PbTx-2 (n = 5, p < 0.05, paired t test) (Fig. 7B). PbTx-2 did not modulate the amplitude of single-channel currents (control, 6.4 ± 0.3 pA; PbTx-2, 6.8 ± 0.4 pA; n = 5) (Fig. 7B). The composite open and shut dwell-time histograms were generated and fitted using Channelab. The open time histogram could be fitted by the sum of two exponential components with time constants of 1.2 (81%) and 3.1 (19%). The time constants after PbTx-2 application were 1.6 (65%) and 4.1 (35%). Thus, PbTx-2 increased the area of the longer time constant. The composite shut time histograms could be fitted by sum of four exponential components with time constants of 0.63 (47%), 7.4 (12%), 211 (22%), and 1410 (19%). The time constants were essentially same after PbTx-2 application 0.63 (45%), 10.0 (16%), 180 (28%), and 1361 (11%), except that the area of the slowest time constant was lowered by PbTx-2, which may underlie the increase in Po by PbTx-2 (Fig. 7C).
Figure 7.

Increase in NMDA receptor channel open probability by PbTx-2. A, Cell-attached patch recording from DIV-2 cerebrocortical neurons. NMDA receptor unitary currents were evoked by 3 μm NMDA and 100 μm glycine in the patch pipette (pipette potential = +60 mV, filtered at 5 kHz for representation, digitized at 40 kHz). Enhancement of NMDA receptor activity by bath application of 30 nm PbTx-2. B, Bath application of 30 nm PbTx-2 increased the NMDA receptor channel open probability (Po) and mean open time (MOT) (n = 5, *p < 0.05, paired t test) but not the mean amplitude (Amp) (n = 5). C, Pooled dwell-time histograms were fitted using Channelab. The open time histogram was fitted by the sum of two Gaussian components, and the shut time histogram was fitted by the sum of four Gaussian components. The time constants and area are described in Results.
To address the requirement for Na+ influx in the PbTx-2-mediated enhancement of NMDA receptor function, PbTx-2 was applied when external Na+ was substituted by equimolar concentrations of the impermeant cation N-methyl d-glucamine (NMDG). PbTx-2 failed to enhance NMDA receptor function under these conditions (Fig. 8A). The Po before and after PbTx-2 application were 0.004 ± 0.002 and 0.005 ± 0.002, respectively (n = 4). The mean open time was also unaltered (before: 1.21 ± 0.14 ms; after: PbTx-2, 1.28 ± 0.22 ms; n = 4). Resubstitution with a Na+-containing extracellular solution with PbTx-2 lead to a significant increase in both Po and mean open time compared with control condition [Po, 0.011 ± 0.003 (275 ± 36% of control); mean open time, 1.97 ± 0.22 ms (166 ± 20% of control); p < 0.05, paired t test)]. These results suggest that PbTx-2 (30 nm)-induced influx of Na+ is crucial for augmentation of NMDA receptor function.
Figure 8.
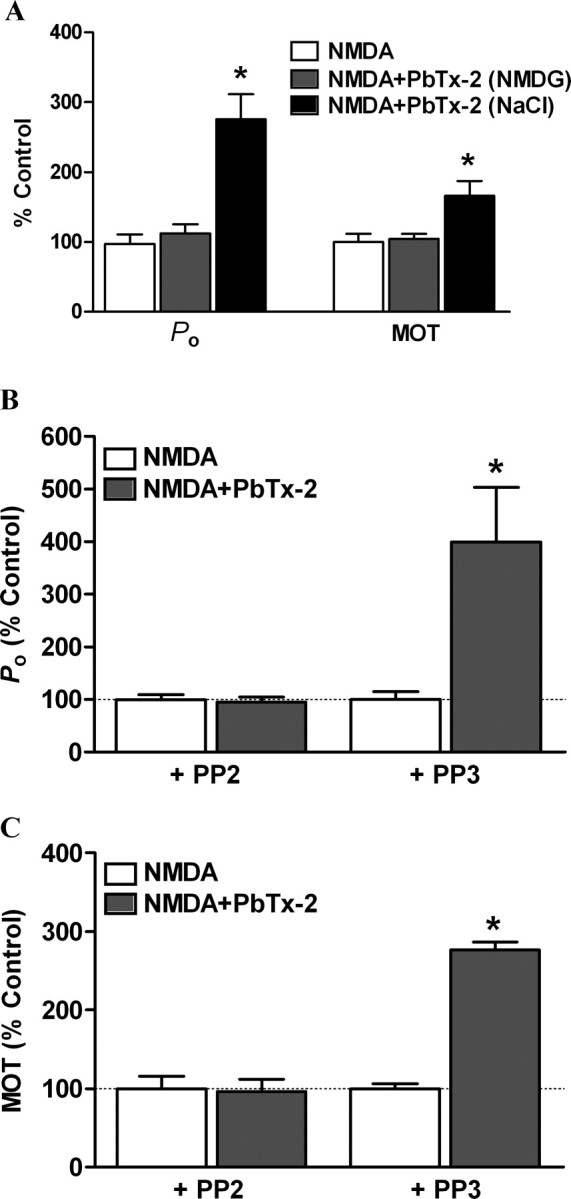
Augmentation of NMDA receptor unitary currents by PbTx-2 is dependent on sodium influx and Src kinase. Single-channel NMDA receptor currents were recorded as described in Figure 7. A, Replacement of extracellular Na+ with impermeant cation NMDG prevented PbTx-2-induced increase in the Po and mean open time of NMDA receptors (n = 4). Resubstitution with Na+-containing Locke's buffer restored PbTx-2-mediated increase in the Po and mean open time of NMDA receptors (n = 4, *p < 0.05, paired t test). B, A 15 min preincubation with the Src kinase inhibitor PP2 (10 μm), but not the inactive analog PP3 (10 μm), blocked PbTx-2-induced enhancement of NMDA receptor Po (n = 4). C, PP2, but not PP3, blocked PbTx-2-induced enhancement of NMDA receptor mean open time (n = 4, *p < 0.05, paired t test).
We further addressed the role of a Src family kinase in PbTx-2-induced augmentation of NMDA receptor function. Recordings were performed after 15 min preincubation with PP2 (Fig. 8B,C). PbTx-2 failed to increase Po [control (PP2), 0.0047 ± 0.0005; PbTx-2 (PP2), 0.0045 ± 0.0005; n = 4)] (Fig. 8B) or mean open time [control (PP2), 1.95 ± 0.32 ms; PbTx-2 (PP2), 1.89 ± 0.29; n = 4)] (Fig. 8C) when Src kinase was inhibited by PP2. To address any nonspecific effects of PP2, single-channel recordings were performed using PP3. PbTx-2-mediated enhancement of NMDA receptor function persisted in the presence of PP3 with both the Po [control (PP3), 0.0032 ± 0.0005; PbTx-2 (PP3), 0.013 ± 0.003; n = 4; p < 0.05, paired t test)] (Fig. 8B) and mean open time [control (PP3), 1.39 ± 0.09 ms; PbTx-2 (PP3), 3.87 ± 0.15; n = 4; p < 0.05, paired t test)] (Fig. 8C) showing significant increases compared with PP3 controls. These results suggest a critical role for a Src family kinase in the Na+-mediated regulation of NMDA receptor activity.
PbTx-2 enhances neurite outgrowth in immature cerebrocortical neurons
We next sought to determine the functional consequences of PbTx-2 augmentation of NMDAR function in immature cerebrocortical neurons. We, therefore, examined the influence of PbTx-2 on neurite outgrowth. A range of PbTx-2 concentrations were added to cerebrocortical cultures 3 h after plating, and the total neurite length was assessed at 15, 24, 40, or 108 h later. The PbTx-2 concentration–response for total neurite outgrowth at the 15, 24, 40, and 108 h time points exhibited a bidirectional, or hormetic, profile. The 24-h exposure data depicted in Figure 9A reveal that PbTx-2 concentrations between 1 and 30 nm produced a graded increase in neurite outgrowth, whereas concentrations >30 nm had progressively smaller responses. In all experiments, the 30 nm PbTx-2 concentration produced the largest effect on neurite outgrowth (Fig. 9A–C). This dramatic effect of 30 nm PbTx-2 on neurite length could be seen at all time points (∼1.5-fold at 15 h, p < 0.05; 2.0-fold at 24 h, p < 0.01; 1.8-fold at 40 h, p < 0.01; 1.4-fold at 108 h, p < 0.05).
Figure 9.

Effect of PbTx-2 on neurite outgrowth of cerebrocortical neurons. A, Concentration–response profile of PbTx-2 on neurite outgrowth at 24 h after plating. Various concentrations of PbTx-2 were added to the culture medium at 3 h after plating. The maximum enhancement of neurite extension was seen with 30 nm PbTx-2. Each value represents mean ± SEM of 120 cells. *p < 0.05–0.01 (ANOVA followed by Dunnett's multiple comparison test). PbTx-2-enhanced neurite extension displayed a hormetic concentration–response relationship with the peak response at 30 nm. B, C, Representative images (B) and quantification (C) of neurite length of cerebrocortical neurons at 15, 24, 40, and 108 h after plating.
Using selective pharmacological inhibitors, we next sought to explore the signaling mechanisms underlying the enhanced neurite outgrowth produced by PbTx-2. We used the selective VGSC antagonist TTX to document the role of sodium channels in this response. Coincubation of TTX (1 μm) with 30 nm PbTx-2 completely blocked (control, 187.5 ± 15.5 μm; PbTx-2, 265.8 ± 20.86 μm; TTX, 185 ± 13.2 μm; TTX plus PbTx-2, 190.2 ± 10.7 μm) the influence of PbTx-2 on total neurite length (Fig. 10A,B), indicating that the PbTx-2 response is mediated by activation of VGSCs. The role of NMDARs in the response to PbTx-2 was assessed using the uncompetitive NMDA receptor antagonist, MK-801. MK-801 pretreatment similarly abrogated PbTx-2-enhanced neurite development (control, 101.5 ± 7.51 μm; PbTx-2, 166.5 ± 17.9 μm; PbTx-2 plus MK-801, 83.1 ± 5.26 μm). Inhibition of Src kinase with PP2 also eliminated the effect of PbTx-2 on neurite outgrowth (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). In contrast, nifedipine, an L-type calcium channel blocker, did not affect (156.6 ± 12.5 μm) PbTx-2-enhanced neurite length (Fig. 10C,D).
Figure 10.

Pharmacological evaluation of signaling pathways involved in PbTx-2-induced neurite outgrowth. A, Representative images (scale bar, 10 μm) and respective quantification (B) of neurite extension at 24 h. The cerebrocortical neurons were treated with 30 nm PbTx-2 in the presence and absence of 1 μm TTX at 3 h after plating. Bars represent mean ± SEM of 30 cells. *p < 0.05, unpaired t test. C, D, Representative images (scale bar, 10 μm) (C) and quantification (D) of neurite extension at 24 h after plating. The 30 nm PbTx-2 exposure was examined in the presence or absence of MK-801 (1 μm), STO-609 (2.6 μm), or nifedipine (1 μm) beginning at 3 h after plating. Each bar represents the mean ± SEM of 30 cells. PbTx-2-enhanced neurite extension was significantly blocked by MK-801 and STO-609 (*p < 0.05, ANOVA followed by Dunnett's multiple comparison test).
Because CaMKK represents a common upstream activator of both calmodulin kinase I (CaMKI), CaMKIV, and MAPKs, the major signaling mediators for Ca2+-dependent stimulation of neurite development, we determined the influence of the selective, cell-permeable CaMKK inhibitor, STO-609 (1,8-naphthoylene benzimidazole-3-carboxylic acid) (Tokumitsu et al., 2002, 2003; Wayman et al., 2004) on the response to PbTx-2. STO-609 (2.6 μm) treatment completely blocked PbTx-2-enhanced neurite outgrowth (Fig. 10C,D). These results suggest that PbTx-2-induced neurite outgrowth is dependent on NMDAR-mediated Ca2+ entry with subsequent activation of a CaMKK pathway. Together, these data indicate that PbTx-2 enhancement of the neurite outgrowth sequentially involves an increase in Na+ influx, upregulation of NMDAR function, and engagement of a Ca2+-dependent CaMKK pathway (supplemental Fig. S7, available at www.jneurosci.org as supplemental material).
Discussion
Activation of NMDA receptors plays an essential role in brain development including the control of dendritic growth (Cline, 2001; Ewald et al., 2008). NMDA receptor-mediated responses promote dendritic arbor growth, whereas pharmacological blockade of NMDA receptors reduce dendritic growth rate (Rajan and Cline, 1998; Lee et al., 2005). Additionally, the effects of neuronal activity on dendritic development are mediated by calcium-dependent signaling events (Konur and Ghosh, 2005). In the present study, we used a sodium channel activator to mimic the influence of neuronal activity in an effort to explore the relationship between intracellular [Na+]- and NMDAR-dependent development. The findings of this study in immature murine cerebrocortical cultures provide compelling evidence in support of a role for [Na+]i in activity-dependent processes of neuronal development.
VGSCs are vital for normal CNS functioning, and recent studies have additionally shown that intracellular sodium may act as a signaling molecule. Based on the original work of Hodgkin and Huxley (1952) with squid axons, a single action potential was calculated to minimally change the Na+ electrochemical gradient (Hille, 1992). The situation in mammalian neurons with fine axons, dendrites, and spines is, however, much different, attributable to greater surface-to-volume ratios. Thus, a single action potential may elevate [Na+]i substantially (Hille, 1992). Using two-photon imaging to measure Na+ transients in spines and dendrites of CA1 pyramidal neurons in hippocampal slices, Rose et al. (1999) demonstrated action potential-induced [Na+]i increments reached values of 3–4 mm after a train of just 20 action potentials.
We have previously found that the sodium channel activator PbTx-2 augments NMDA receptor-mediated Ca2+ influx in both spontaneously oscillating mature and nonoscillatory immature cerebrocortical neurons (Dravid at al., 2005). PbTx-2 also enhanced the effect of bath applied NMDA on extracellular signal-regulated kinase 2 activation in cerebrocortical neurons. The influence of [Na+]i dynamics on NMDA receptor function has been demonstrated in hippocampal neurons where elevation of [Na+]i increased the open probability of NMDA receptors (Yu and Salter, 1998; Yu, 2006). An increment of [Na+]i of 10 mm was sufficient to produce significant increases in NMDA receptor single-channel activity. This Na+-dependent regulation of NMDA receptor function was, moreover, shown to be controlled by Src-induced phosphorylation of the receptor (Yu and Salter, 1998; Yu, 2006). These results were confirmed and extended in the present study using the sodium channel activator PbTx-2 as a probe to elevate intracellular Na+. To unambiguously demonstrate the enhancement of NMDA receptor function by PbTx-2, we recorded single-channel currents from cell-attached patches. The shut time histogram with slow time constants resemble NR2B-containing receptors (Erreger et al., 2005), consistent with the expression of NR1/NR2B-containing receptors in immature neurons (Williams et al., 1993). PbTx-2 treatment increased both the mean open time and open probability of NMDA receptors. These effects of PbTx-2 on NMDA receptor function were dependent on extracellular Na+ and activation of Src kinase. An increase in intracellular Na+ and Src activation have previously been shown to increase the mean open time and open probability of NMDA receptors in hippocampal neurons (Yu et al., 1997; Yu and Salter, 1998). An increase in apparent mean open time may represent increased stability of the open conformation rather than a change in agonist affinity, because the apparent mean open time appears to be independent of agonist concentration at NR1/NR2A receptors (Schorge et al., 2005). PbTx-2 exposure changed the area of certain shut time constants with no apparent shift in the time constants themselves, suggesting that the sodium channel activator may act by reducing the probability that the channel enters a long-lived shut state. Because the single-channel recordings were done in the absence of extracellular Mg2+, these results additionally argue against relief of Mg2+-dependent block in the actions of PbTx-2. These data, therefore, confirm the regulatory influence of Na+ on NMDAR channel activity in hippocampal neurons described previously (Yu and Salter, 1998) and extend this relationship between [Na+]i and NMDA receptor function to cerebrocortical neurons.
A key finding of this study is that a functional consequence of the regulatory influence of [Na+]i on NMDAR channel activity was established. Using immature cerebrocortical neurons, we found that PbTx-2 concentrations between 3 and 300 nm enhance neurite outgrowth. Thus, the ability of PbTx-2 to augment NMDAR channel activity translated into an enhancement of the trophic influence of NMDAR on developing cerebrocortical neurons. Accepting the premise that the effects of neuronal activity on the development and structural plasticity of dendritic arbors are primarily mediated by engagement of NMDA receptors (Tolias et al., 2005), our results suggest that PbTx-2 activation of sodium channels with attendant enhancement of NMDA receptor signaling mimics the response to neuronal activity.
An inverted-U model describes the relationship between NMDA receptor activity and neuronal survival and outgrowth (Lipton and Nakanishi, 1999; Hardingham and Bading, 2003). This inverted-U concentration–response relationship has primarily, but not exclusively, been regressed to intracellular Ca2+ regulation. An optimal window for [Ca2+]i is required for activity-dependent neurite extension and branching, with lower levels stabilizing growth cones and higher levels stalling them, in both cases preventing extension (Gomez and Spitzer, 2000; Hui et al., 2007). It is noteworthy that the concentration dependence for KCl-induced neurite outgrowth in PC12 cells also displays an inverted-U profile (Solem et al., 1995).
The PbTx-2 concentration–response profile for neurite outgrowth reproducibly produced a comparable bidirectional or hormetic relationship (Fig. 9). Although we have reported previously that the PbTx-2 concentration–response relationship for neuronal Ca2+ influx does not produce an inverted-U response (Berman and Murray, 2000), the pathways for Ca2+ influx did differ for low (100 nm) versus high (1000 nm) concentrations of PbTx-2 in DIV-2 cerebrocortical neurons (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The Ca2+ influx pathway stimulated by 100 nm PbTx-2 was primarily through NMDA receptors, whereas 1000 nm PbTx-2 stimulates influx through both L-type calcium channels and NMDA receptors (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The more sustained depolarization associated with exposure to 1000 nm PbTx-2 may produce greater activation of L-type calcium channels in these immature neurons (Fig. 6C). NMDA receptor-dependent and L-type Ca2+ channel-dependent Ca2+ influx pathways may differentially contribute to the observed stimulation of neurite outgrowth. Ca2+-signaling pathways resulting from L-type Ca2+ channels and NMDA receptors have been shown to differ (Bading et al., 1993). Wayman et al. (2006) have shown that activity-dependent dendritic arborization in DIV-9 hippocampal neurons involves a Ca2+-signaling pathway downstream of the NMDA receptor through activation of a CaMKK and CaMKI. These authors used 16 mm KCl to induce neuronal activity and demonstrated that the observed increase in total dendritic length and branching was eliminated by treatment with an NMDA receptor antagonist. In contrast, evidence for the involvement of L-type Ca2+ channels in dendritic growth and arborization of DIV-4 cortical neurons exposed to 50 mm KCl has been reported previously (Redmond et al., 2002). The explanation for the discordant results of these previously published studies most likely resides in the differing strengths of depolarizing stimuli produced by 16 versus 50 mm KCl: only the latter would be sufficient to activate L-type Ca2+ channels in mature cultures. Similarly, only 1000 nm PbTx-2 in the present study was capable of stimulating Ca2+ influx through L-type Ca2+ channels. Given that the resting membrane potential of DIV-2 cerebrocortical neurons was found to be in the −28 to −30 mV range and that L-type Ca2+ channels have an activation threshold at membrane voltages positive to −30 mV (Trombley and Westbrook, 1991), the small depolarization produced by 1000 nm PbTx-2 may have been sufficient to activate L-type channels in these immature cultures. Although these data collectively do not provide an explanation for the inverted-U concentration–response curve for neurite outgrowth, they do illustrate the importance of the strength of a depolarizing stimulus as a determinant of the Ca2+-signaling pathways activated.
Although the exact mechanism underlying the inverted-U response remains to be determined, high concentrations of PbTx-2 might promote slow inactivation of VGSCs with attendant reduction in sodium influx (Mitrovic et al., 2000; Ong et al., 2000). Alternatively, high concentrations of PbTx-2 could increase VGSC internalization, which has been shown to be a consequence of Na+ influx in immature neuronal tissue (Dargent et al., 1994). Consistent with these provisional explanations for the inverted-U PbTx-2 concentration–response relationship, quantification of cerebrocortical [Na+]i after 24-h exposure to PbTx-2 also exhibited a hormetic profile (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). These data suggest that cerebrocortical neuron [Na+]i may shape the PbTx-2 concentration–response curve by upregulating NMDAR signaling.
We have recently shown that an array of sodium channel-gating modifiers produce [Na+]i increments that are of sufficient magnitude to increase NMDA receptor channel activity (Cao et al., 2008). Sodium channel activators, therefore, appear capable of mimicking activity-dependent control of neuronal development by upregulating NMDA receptor signaling pathways that influence neuronal growth and plasticity. Voltage-gated sodium channel activators may accordingly represent a novel pharmacologic strategy to regulate neuronal plasticity through an NMDA receptor and Src family kinase-dependent mechanism.
Footnotes
This work was supported in part by National Institutes of Health Grants ES10594 (to D.G.B.) and NS053398 (to T.F.M.).
References
- Bading et al., 1993.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Baxter et al., 2002.Baxter DF, Kirk M, Garcia AF, Raimondi A, Holmqvist MH, Flint KK, Bojanic D, Distefano PS, Curtis R, Xie Y. A novel membrane potential-sensitive fluorescent dye improves cell-based assays for ion channels. J Biomol Screen. 2002;7:79–85. doi: 10.1177/108705710200700110. [DOI] [PubMed] [Google Scholar]
- Berman and Murray, 2000.Berman FW, Murray TF. Brevetoxin-induced autocrine excitotoxicity is associated with manifold routes of Ca2+ influx. J Neurochem. 2000;74:1443–1451. doi: 10.1046/j.1471-4159.2000.0741443.x. [DOI] [PubMed] [Google Scholar]
- Cao et al., 2007.Cao Z, George J, Baden DG, Murray TF. Brevetoxin-induced phosphorylation of Pyk2 and Src in murine neocortical neurons involves distinct signaling pathways. Brain Res. 2007;1184:17–27. doi: 10.1016/j.brainres.2007.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao et al., 2008.Cao Z, George J, Gerwick WH, Baden DG, Rainier JD, Murray TF. Influence of lipid soluble gating modifier toxins on sodium influx in neocortical neurons. J Pharmacol Exp Ther. 2008;326:604–613. doi: 10.1124/jpet.108.138230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall and Gainer, 1985.Catterall WA, Gainer M. Interaction of brevetoxin A with a new receptor site on the sodium channel. Toxicon. 1985;23:497–504. doi: 10.1016/0041-0101(85)90034-0. [DOI] [PubMed] [Google Scholar]
- Chen and Ghosh, 2005.Chen Y, Ghosh A. Regulation of dendritic development by neuronal activity. J Neurobiol. 2005;64:4–10. doi: 10.1002/neu.20150. [DOI] [PubMed] [Google Scholar]
- Cheng et al., 1999.Cheng C, Fass DM, Reynolds IJ. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca(2+)](i) increases and NMDA receptor mRNA levels. Brain Res. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Choi et al., 1987.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, 2001.Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11:118–126. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Dargent et al., 1994.Dargent B, Paillart C, Carlier E, Alcaraz G, Martin-Eauclaire MF, Couraud F. Sodium channel internalization in developing neurons. Neuron. 1994;13:683–690. doi: 10.1016/0896-6273(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Diarra et al., 2001.Diarra A, Sheldon C, Church J. In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. Am J Physiol Cell Physiol. 2001;280:C1623–C1633. doi: 10.1152/ajpcell.2001.280.6.C1623. [DOI] [PubMed] [Google Scholar]
- Dravid et al., 2005.Dravid SM, Baden DG, Murray TF. Brevetoxin augments NMDA receptor signaling in murine neocortical neurons. Brain Res. 2005;1031:30–38. doi: 10.1016/j.brainres.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Ehrenberg et al., 1988.Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger et al., 2005.Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald et al., 2008.Ewald RC, Van Keuren-Jensen KR, Aizenman CD, Cline HT. Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J Neurosci. 2008;28:850–861. doi: 10.1523/JNEUROSCI.5078-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen and Schousboe, 1990.Frandsen A, Schousboe A. Development of excitatory amino acid induced cytotoxicity in cultured neurons. Int J Dev Neurosci. 1990;8:209–216. doi: 10.1016/0736-5748(90)90013-r. [DOI] [PubMed] [Google Scholar]
- Gomez and Spitzer, 2000.Gomez TM, Spitzer NC. Regulation of growth cone behavior by calcium: new dynamics to earlier perspectives. J Neurobiol. 2000;44:174–183. [PubMed] [Google Scholar]
- Griffiths et al., 1997.Griffiths R, Malcolm C, Ritchie L, Frandsen A, Schousboe A, Scott M, Rumsby P, Meredith C. Association of c-fos mRNA expression and excitotoxicity in primary cultures of mouse neocortical and cerebellar neurons. J Neurosci Res. 1997;48:533–542. doi: 10.1002/(sici)1097-4547(19970615)48:6<533::aid-jnr6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Hamill et al., 1981.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hardingham and Bading, 2003.Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hille, 1992.Hille B. Ionic channels of excitable membranes. Sunderland, MA: Sinauer; 1992. pp. 403–411. [Google Scholar]
- Hodgkin and Huxley, 1952.Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952;116:449–472. doi: 10.1113/jphysiol.1952.sp004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui et al., 2007.Hui K, Fei GH, Saab BJ, Su J, Roder JC, Feng ZP. Neuronal calcium sensor-1 modulation of optimal calcium level for neurita outgrowth. Development. 2007;134:4479–4489. doi: 10.1242/dev.008979. [DOI] [PubMed] [Google Scholar]
- Jeglitsch et al., 1998.Jeglitsch G, Rein K, Baden DG, Adams DJ. Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J Pharmacol Exp Ther. 1998;284:516–525. [PubMed] [Google Scholar]
- Kim et al., 1995.Kim HG, Fox K, Connors BW. Properties of excitatory synaptic events in neurons of primary somatosensory cortex of neonatal rats. Cereb Cortex. 1995;5:148–157. doi: 10.1093/cercor/5.2.148. [DOI] [PubMed] [Google Scholar]
- King et al., 2006.King AE, Chung RS, Vickers JC, Dickson TC. Localization of glutamate receptors in developing cortical neurons in culture and relationship to susceptibility to excitotoxicity. J Comp Neurol. 2006;498:277–294. doi: 10.1002/cne.21053. [DOI] [PubMed] [Google Scholar]
- Koh and Choi, 1987.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Konur and Ghosh, 2005.Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005;46:401–405. doi: 10.1016/j.neuron.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Kuner and Augustine, 2000.Kuner T, Augustine GJ. A genetically encoded ratiometric neurotechnique indicator for chloride: Capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Lee et al., 2005.Lee LJ, Lo FS, Erzurumlu RS. NMDA receptor-dependent regulation of axonal and dendritic branching. J Neurosci. 2005;25:2304–2311. doi: 10.1523/JNEUROSCI.4902-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton and Nakanishi, 1999.Lipton SA, Nakanishi N. Shakespeare in love–with NMDA receptors? Nat Med. 1999;5:270–271. doi: 10.1038/6481. [DOI] [PubMed] [Google Scholar]
- Liu et al., 1996.Liu Z, Stafstrom CE, Sarkisian M, Tandon P, Yang Y, Hori A, Holmes GL. Age-dependent effects of glutamate toxicity in the hippocampus. Brain Res Dev Brain Res. 1996;97:178–184. doi: 10.1016/s0165-3806(96)00141-1. [DOI] [PubMed] [Google Scholar]
- Marks et al., 2005.Marks JD, Boriboun C, Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer et al., 1984.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- McAllister, 2000.McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- Miller and Kaplan, 2003.Miller FD, Kaplan DR. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol. 2003;13:391–398. doi: 10.1016/s0959-4388(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Mitrovic et al., 2000.Mitrovic N, George AL, Jr, Horn R. Role of domain 4 in sodium channel slow inactivation. J Gen Physiol. 2000;115:707–718. doi: 10.1085/jgp.115.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta et al., 1998.Mizuta I, Katayama M, Watanabe M, Mishina M, Ishii K. Developmental expression of NMDA receptor subunits and the emergence of glutamate neurotoxicity in primary cultures of murine cerebral cortical neurons. Cell Mol Life Sci. 1998;54:721–725. doi: 10.1007/s000180050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong et al., 2000.Ong BH, Tomaselli GF, Balser JR. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J Gen Physiol. 2000;116:653–662. doi: 10.1085/jgp.116.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli et al., 1986.Poli MA, Mende TJ, Baden DG. Brevetoxins, unique activators of voltage-sensitive sodium channels, bind to specific sites in rat brain synaptosomes. Mol Pharmacol. 1986;30:129–135. [PubMed] [Google Scholar]
- Qin, 2004.Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J. 2004;86:1488–1501. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan and Cline, 1998.Rajan I, Cline HT. Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J Neurosci. 1998;18:7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoa and McCormick, 1994.Ramoa AS, McCormick DA. Developmental changes in electrophysiological properties of LGNd neurons during reorganization of retinogeniculate connections. J Neurosci. 1994;14:2089–2097. doi: 10.1523/JNEUROSCI.14-04-02089.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond et al., 2002.Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Rose and Konnerth, 2001.Rose CR, Konnerth A. NMDA receptor-mediated Na+ signals in spines and dendrites. J Neurosci. 2001;21:4207–4214. doi: 10.1523/JNEUROSCI.21-12-04207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose and Ransom, 1997.Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol. 1997;499:573–587. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose et al., 1999.Rose CR, Kovalchuk Y, Eilers J, Konnerth A. Two-photon Na+ imaging in spines and fine dendrites of central neurons. Pflugers Arch. 1999;439:201–207. doi: 10.1007/s004249900123. [DOI] [PubMed] [Google Scholar]
- Salter and Kalia, 2004.Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Saneyoshi et al., 2008.Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt et al., 2004.Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. J Biol Chem. 2004;279:24064–24072. doi: 10.1074/jbc.M401501200. [DOI] [PubMed] [Google Scholar]
- Schorge et al., 2005.Schorge S, Elenes S, Colquhoun D. Maximum likelihood fitting of single channel NMDA activity with a mechanism composed of independent dimers of subunits. J Physiol. 2005;569:395–418. doi: 10.1113/jphysiol.2005.095349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin et al., 2002.Sin WC, Haas K, Ruthazer ES, Cline HT. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419:475–480. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- Solem et al., 1995.Solem M, McMahon T, Messing RO. Depolarization-induced neurite outgrowth in PC12 cells requires permissive, low level NGF receptor stimulation and activation of calcium/calmodulin-dependent protein kinase. J Neurosci. 1995;15:5966–5975. doi: 10.1523/JNEUROSCI.15-09-05966.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumitsu et al., 2002.Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu et al., 2003.Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R. A single amino acid difference between alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. J Biol Chem. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- Tolias et al., 2005.Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Trombley and Westbrook, 1991.Trombley PQ, Westbrook GL. Voltage-gated currents in identified rat olfactory receptor neurons. J Neurosci. 1991;11:434–444. doi: 10.1523/JNEUROSCI.11-02-00435.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio et al., 2003.Tyzio R, Ivanov A, Bernard C, Holmes GL, Ben-Ari Y, Khazipov R. Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J Neurophysiol. 2003;90:2964–2972. doi: 10.1152/jn.00172.2003. [DOI] [PubMed] [Google Scholar]
- Ultanir et al., 2007.Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci U S A. 2007;104:19553–19558. doi: 10.1073/pnas.0704031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman et al., 2004.Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman et al., 2006.Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- West et al., 2002.West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- Whiteaker et al., 2001.Whiteaker KL, Gopalakrishnan SM, Groebe D, Shieh CC, Warrior U, Burns DJ, Coghlan MJ, Scott VE, Gopalakrishnan M. Validation of FLIPR membrane potential dye for high throughput screening of potassium channel modulators. J Biomol Screen. 2001;6:305–312. doi: 10.1177/108705710100600504. [DOI] [PubMed] [Google Scholar]
- Williams et al., 1993.Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- Wong and Ghosh, 2002.Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3:803–812. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]
- Xin et al., 2005.Xin WK, Kwan CL, Zhao XH, Xu J, Ellen RP, McCulloch CA, Yu XM. A functional interaction of sodium and calcium in the regulation of NMDA receptor activity by remote NMDA receptors. J Neurosci. 2005;25:139–148. doi: 10.1523/JNEUROSCI.3791-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, 2006.Yu XM. The role of intracellular sodium in the regulation of NMDA-receptor-mediated channel activity and toxicity. Mol Neurobiol. 2006;33:63–80. doi: 10.1385/MN:33:1:063. [DOI] [PubMed] [Google Scholar]
- Yu and Salter, 1998.Yu XM, Salter MW. Gain control of NMDA-receptor currents by intracellular sodium. Nature. 1998;396:469–474. doi: 10.1038/24877. [DOI] [PubMed] [Google Scholar]
- Yu et al., 1997.Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]