

Abstract
Purpose
There are distinctive areas of colocalization of matrix metalloproteinase (MMP)-2 and -14 on trabecular meshwork (TM) cells that resemble podosomes or invadopodia. Studies were conducted to determine whether TM cells exhibit podosome- or invadopodia-like structures (PILS) and whether they produce focal extracellular matrix (ECM) turnover.
Methods
Porcine and human TM cells and perfused anterior segment organ cultures were studied. Localization of PILS components on TM cells and in sections from anterior segments was determined by immunohistochemistry and confocal microscopy. Cells were grown on type I collagen labeled with fluorescein isothiocyanate (FITC) for degradation analysis. Confocal time lapse images were taken of labeled TM cells on FITC-collagen.
Results
Immunostaining for MMP-2, MMP-14, and the typical PILS components cortactin, caldesmon, α-actinin, N-WASP, Arp-3, and cdc42 colocalized on these distinctive structures. Integrin-αV and -β1, fibronectin, and versican colocalized with PILS components. TM cells on FITC-conjugated collagen developed focal regions of degradation. Time-lapse imaging showed dramatic and controlled movement of TM cell processes during this ECM degradation and fragment internalization. MMP-2, MMP-14, and cortactin colocalized at regions that appear to be PILS on cells within the outflow pathway in sections of human anterior segments.
Conclusions
TM cells exhibit areas where PILS components colocalize with MMP-2 and -14. Similar structures are found in sections, suggesting that PILS occur in situ in the outflow pathway. The collagen degradation suggests that PILS may serve as focal sites for targeted ECM turnover, an event linked to modifications of aqueous outflow resistance and intraocular pressure homeostasis.
Glaucoma is a major blinding disease, affecting well over 67 million people worldwide.1,2 Intraocular pressure (IOP) elevation, resulting from a poorly understood obstruction to the outflow of aqueous humor through the trabecular meshwork (TM), is a major risk factor for primary open-angle glaucoma.1,3,4 The fluid resistance to aqueous humor outflow appears to reside within the cribriform or juxtacanalicular (JCT) region of the TM in both normal and glaucomatous eyes.5–7 However, definite identification of the actual site or of the specific component(s) responsible for the normal or the glaucomatous resistance remains uncertain.8–11 Evidence has been presented that both the trabecular ECM and cellular cytoskeleton contribute to the outflow resistance.8,10–20 Because of the tight coupling that exists between cells and their ECMs, separation of their individual contributions has been difficult.
Homeostatic IOP regulation by the TM has been reported.21–23 One hypothetical mechanism involves TM cells' sensing the mechanical distortion and stretching of the JCT region produced by increased IOP and responding by initiating ECM turnover.11,21 TM cells have been shown to sense and respond to mechanical stretching.21–32 In anterior segment organ culture, increasing the levels or activity of MMPs within the TM increases outflow facility while inhibiting the endogenous MMP activity decreases outflow facility.14 This fact argues strongly that ongoing ECM turnover is necessary to maintain the outflow resistance and that changing ECM turnover can modify the resistance. In addition to increasing MMP activity and thus initiating ECM degradation, biosynthetic replacement of the degraded ECM with new ECM components with a slightly modified composition seems necessary to facilitate modest homeostatic adjustment of the outflow resistance.11 This process would have to be highly controlled and accurately directed to keep from disorganizing the outflow resistance and disrupting the structure and function of the JCT region. Intuitively, it seems likely that ECM turnover would occur at or near the JCT cells' surface and in a very focal and controlled manner. To date, no specific mechanism for such focal ECM turnover control in the TM has been postulated.
Structures involved in focal ECM degradation has been identified on several types of cells.33 These structures, commonly referred to as podosomes or invadopodia, are specialized cell adhesion sites that are also involved in ECM degradation. They are central to macrophage ECM degradation, osteoclast bone resorption, metastatic cell invasion, diapedesis, cell motility, and migration.34–40 Similar structures have been observed under some conditions in fibroblasts, endothelial cells, smooth muscle cells, and epithelial cells.37,38 The size, shape, and molecular organization of these structures in various cell types are relatively diverse.36–38,41 Simple podosomes, as are commonly found on the ventral surface of macrophages, are approximately 500 nm in diameter, with a vertical F-actin core and numerous actin-binding proteins and are surrounded by a ring of talin, paxillin, and vinculin.37,38 Their formation is thought to be associated with cortactin clustering at discrete microdomains on the ventral surface of cells, perhaps recruited directly or indirectly by cdc42 and sometimes near sites where stress fibers insert into adhesion plaques or focal adhesions. N-WASP (neural-Wiscott-Aldrich syndrome protein) and Arp 2/3 (actin-related protein) are involved in branching of F-actin microfilaments and play important roles in podosome or invadopodia formation.42,43 Invadopodia have longer lives and may evolve from simple podosomes,36 which normally last only minutes to an hour. In some types of cells, 5-μm circular rosettes and larger complex aggregates or clusters of podosomes with less uniform structures are observed.36–38,41 Components common to PILS include: cortactin, α-actinin, caldesmon, gelsolin, dynamin, F-actin, ARP 2/3, N-WASP, MMP-2, MMP-14, MMP-9, TIMP-2, phosphotyrosine, Src, cdc42, cofilin, vinculin, talin, paxillin, tubulin, and in some cases, integrin-αVβ3. All these components are also found in other cellular or cell-associated structures, but their confluence is generally considered indicative of podosomes or invadopodia.34–40 However, because of the rapidly evolving structures of podosomes and invadopodia, the relative temporal distributions of these components is not well defined and is probably variable.
The ECM degradation component of the IOP homeostatic mechanism involving mechanical stretching of the JCT region is mediated at least in part by MMP-2 and -14.14,21,22 MMP-2 (also known as gelatinase A or type IV collagenase) is different from the other MMPs, in that its activation involves recruitment to the cell surface by TIMP-2 where it is activated by MMP-14 (also known as membrane type 1 MMP).44–49 While evaluating modulation of MMP-2 and -14 by agents that affect outflow facility, we noted that these two proteinases colocalized on the ventral surface of TM cells. These regions of colocalization were unusual but very distinctive, often appearing as a circle, uneven oval or a skewed irregular triangle or rhombus. They were common on TM cells and, subjectively, appeared to be increased by mechanical stretching or the addition of TNFα, TGFβ, or serum (not shown). Somewhat similar structures had been reported for podosomes and invadopodia.37,50–53 We therefore investigated whether these MMP-2 and -14 colocalization regions could be podosome- or invadopodia-like structures (PILS) and whether they exhibited dynamic properties that could be related to their putative ECM turnover function in the JCT.
Materials and Methods
Porcine eyes were obtained from Carlton Packing Company (Carlton, OR) 2 to 3 hours postmortem. Human eyes were obtained from the Oregon Lions Eye Bank (Portland, OR) within 48 hours postmortem. The eyes were obtained and managed in compliance with the guidelines of the Declaration of Helsinki regarding research involving human tissue. Alexa Fluor 488- and 595-conjugated secondary antibodies and Alexa Fluor 350-, 488-, and 594-conjugated phalloidin (blue, green, and red, respectively), a red fluorescent tracer (CellTracker Red CMTPX), and DNA (PicoGreen) assay kits were from Invitrogen-Molecular Probes (Eugene, OR). Antibody to fibronectin was from BD-Transduction Laboratories (Franklin Lakes, NJ); antibodies to MMP-2 were from Abcam (Cambridge, MA) and Chemicon (Temecula, CA); antibodies to caldesmon were from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma-Aldrich (St Louis, MO); antibodies to MMP-14 were from EMD/Calbiochem (La Jolla, CA), Triple Point Biologics (Forest Grove, OR), and Abcam; antibodies to TIMP-2, N-WASP, and the versican neoepitope (aa 436-441) that is unmasked by ADAMTS-1/4 cleavage of versican were from Abcam; antibody to versican's G3 domain was from Kamiya Biomedical (Seattle, WA); antibodies to cortactin and α-actinin were from Upstate Biotechnology (Lake Placid, NY); antibodies to cdc42, Arp-3, and integrin-β1 were from Santa Cruz Biotechnology; and antibodies to integrin-αV and -β3 were from Chemicon. Collagen type I–coated 6-well plates (Bioflex) were from Flexcell (Hillsborough, NC); high- and low-glucose Dulbecco's modified Eagle's medium (DMEM), antibiotics, and antimycotic (penicillin, streptomycin and amphotericin B) were from Invitrogen-Gibco (Grand Island, NY); fetal bovine serum (FBS) was from Hyclone (Logan, UT); Draq5 was from Axxora (San Diego, CA); normal goat serum, Fluoromount G mounting medium, and Labtek II chamber slides were from Fisher Scientific (Pittsburgh, PA); and porcine skin gelatin and normal sera for various other species were from Sigma-Aldrich.
TM Cell Culture, Treatments, and Extractions
Porcine and human TM cells were cultured as previously detailed54–59 in medium-glucose (a 1:1 mix of high- and low-glucose) DMEM supplemented with 10% FBS and 1% antibiotic and antimycotic mix. At passages 2 to 4, cells were plated at approximately 50% confluence (150,000 cells per well) onto collagen type I–coated membranes in six-well plates (Bioflex; Flexcell). To coat chamber slides with collagen type I, we mixed porcine skin gelatin at 1% in phosphate-buffered saline (PBS), autoclaved the mixture, and applied it to the slides. The slides were then incubated overnight under a UV light in a laminar flow hood to enhance attachment. Cells were maintained serum free for 48 hours before and during the various treatments. Mechanical stretching of membrane inserts or membranes was conducted as detailed previously, using small glass beads of defined size and pressure on the lid of the six-well plates.21,22
Perfused Anterior Segment Organ Culture and Sectioning
Perfused anterior segment organ culture was conducted as previously detailed,14,21,60–63 with human or porcine anterior segments. Perfused porcine organ culture was initiated within 5 hours postmortem with a constant pressure of 8.89 mm Hg. Human anterior segments were recovered in stationary organ culture for 3 to 7 days64 before perfusion was begun at a similar constant pressure head. Flow rates were measured gravimetrically. For elevated IOP treatments, the pressure head was doubled. After treatment, anterior segments were removed from the flow chambers and immediately immersion-fixed in 10% neutral-buffered formalin for at least 24 hours. Tissues were decalcified overnight in decalcifying solution (Fisher Scientific) to allow easier sectioning through the sclera.65 Each eye was cut into approximately 8 to 10 wedges, each of which was embedded into the same paraffin block. Five-micrometer radial serial sections were then cut at the pathology and histology core facility of the OHSU Cancer Center (Oregon Health & Science University, Portland, OR). Hematoxylin and eosin (H&E)–stained slides of each eye were examined to ensure the presence of cells and tissue organization (data not shown). At least three eyes per treatment were analyzed.
Immunofluorescence and Confocal Microscopy
For tissue samples, sections were deparaffinized and rehydrated before immunostaining. Slides were blocked with 2% normal goat serum in PBS before addition of primary antibodies diluted in PBS, which were incubated for 3 hours at room temperature. After three 5-minute washes with PBS, secondary antibody diluted in PBS was added to each well. After incubation at room temperature for 1 hour, the slides were washed in PBS (three times for 5 minutes), and coverslips were mounted (Fluoromount G; Fisher Scientific). Each slide was examined by immunofluorescence and confocal microscopy. Negative control slides containing both secondary but no primary antibodies were also included.
Membranes were cut from the plates or cell inserts and mounted on microscope slides before immunostaining. For immunohistochemistry of most cultured cells, mild extraction and permeabilization buffer (ice cold PBS with 0.1% Tween-20) was used before fixation in 2% paraformaldehyde in PBS. In a few cases, as indicated, harsh extraction and permeabilization buffer (mild buffer with 0.5% Triton X-100, 4% polyethyleneglycol, and 100 mM PIPES [pH 6.9], 1 mM magnesium chloride and 1 mM EGTA) was used before fixation in 2% paraformaldehyde in PBS. Both types of extraction and permeabilization were for 1 to 2 minutes. The slides were blocked with filtered 2% nonfat milk or 2% goat serum in PBS and incubated with the indicated primary antibodies. They were again rinsed in PBS and incubated with the appropriate Alexa Fluor 488- and Alexa Fluor 595-conjugated secondary antibodies. In some cases, Draq5, a DNA binding dye, was added to the secondary antibody mixture at a dilution of 1:500. Phalloidin conjugated with one of the Alexa Fluor dyes was used to label F-actin microfilaments.
Immunostained tissue sections and chamber slides were visualized with an differential interference contrast (DIC) and fluorescence microscope (model BX51; Olympus, Lake Success, NY) equipped with a camera (model DC500; Leica, Bannockburn, IL). Confocal microscopy was also performed (FluoView 1000; Olympus). Randomly selected fields from each slide were imaged, and color channels were scanned sequentially to maximize separation. Confocal images were processed using one of three image-analysis programs (FluoView FV1000 software; Olympus; ImageJ software; available by ftp at zippy.nimh.nih.gov/ or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; or Photoshop, Adobe Systems, San Jose, CA).
ECM Degradation and Live Cell Motility
Collagen-coated chamber slides or membranes (Bioflex; Flexcell) were labeled with fluorescein isothiocyanate (FITC) for degradation studies. FITC (30 μg/mL) in 100 mM sodium tetraborate and 80 mM NaCl [pH 93] was incubated with collagen-coated slides or membranes for 2 hours at 37°C in the dark. The slides were then washed extensively in sterile PBS. For direct degradation studies, the cells were plated at ∼50% confluence and allowed to attach for 2 to 24 hours. Where indicated, the cells were made serum free for 48 hours and then subjected to mechanical stretching for 24 hours. Membranes or slides were treated with mild extraction and permeabilization buffer, fixed with 2% paraformaldehyde in PBS, rinsed extensively with PBS, blocked with 2% skim milk, and probed with primary and secondary antibodies as indicated. Slides were then analyzed by standard DIC and fluorescence or confocal microscopy.
For time-lapse live-cell motility to visualize ECM degradation, the cells were plated on FITC-labeled collagen-coated slides and allowed to attach overnight. They were then loaded with red fluorescent tracer (Celltracker Red; Invitrogen-Molecular Probes) for 1 hour as directed by the manufacturer, the medium was exchanged, and confocal microscopy was conducted in a 37°C chamber with 100% humidity and 5% CO2. This procedure was conducted with an image restoration system (CORE Deltavision Image Restoration; Applied Precision Inc, Issaquah, WA). A set of Z-stack images was collected at 6 to 10 different focal levels (steps of 0.2, 1, or 2 μm) every 10 minutes for 4 hours. Images were analyzed and converted to movies (QuickTime, using softWoRx Explorer 1.2; Applied Precision Inc., and QuickTime Pro; Apple Computer, Cupertino, CA).
Results
Colocalization of MMP-2 and -14
When TM cells were immunostained for MMP-2 and -14, they exhibited numerous regions of discrete colocalization, as well as areas of separate distributions. TM cells that were grown on collagen type I–coated membranes and then subjected to mechanical stretching for 24 hours (Fig. 1) normally showed 3 to 10 regions per cell where MMP-2 (green) and MMP-14 (red) immunostaining were colocalized (yellow or orange area). Phalloidin (blue) staining of F-actin is shown to help define the shape and position of individual cells. A portion of the immunostaining of each of the MMPs was separate. For instance, note the numerous discrete regions of either green or red in the various panels (Fig. 1). Small dots of MMP-2 staining were dispersed over much of the cell surface, with minimal staining in the region of the nucleus. MMP-2 regions also appeared to surround the yellow colocalization regions, and the separate MMP-14 (red) staining was often near or interspersed through the yellow colocalization regions. The yellow colocalization regions were of several shapes, including uniform circular rosettes approximately 5 μm in diameter (Fig. 1D). Most regions of colocalization appeared as irregularly shaped blotches, with some forming small irregular circular or oval structures. Some of the colocalization areas were near the center of the cell adjacent to the nucleus, but many of the larger patches of colocalization were more peripheral. Based on observations of the individual confocal Z-stack images of the optical sections (not shown), the regions of colocalization were on the ventral surface of the TM cells. Although the images shown are of TM cells that had been mechanically stretched, control specimens that were not stretched showed similar areas of colocalization. Subjectively, these areas were more abundant after stretching (not shown). When human or porcine TM cells were cultured on standard polycarbonate insert membranes, pronectin-coated membranes, or directly on chamber slides, similar patterns of immunostaining and colocalization were observed (not shown).
Figure 1.
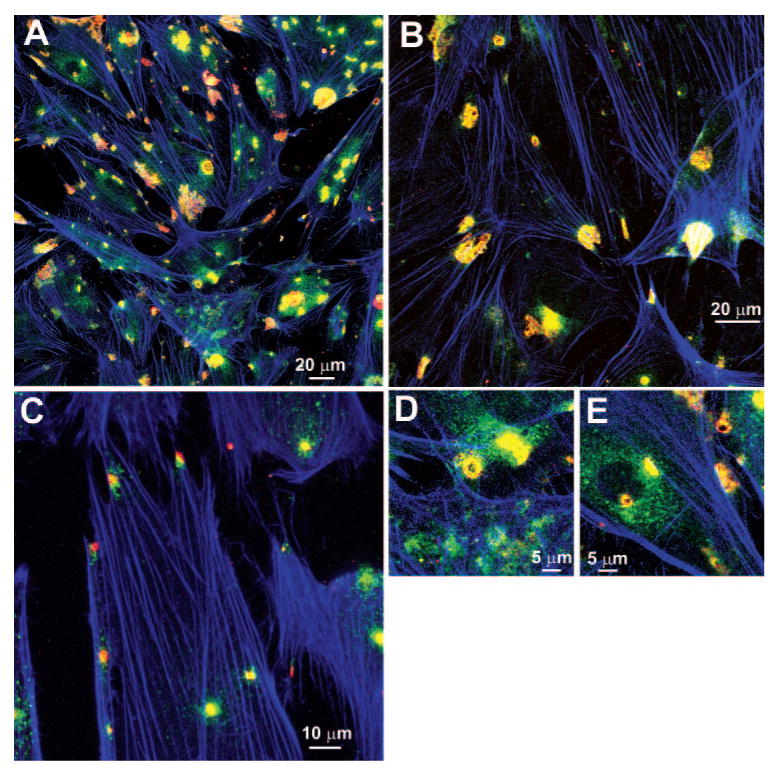
Immunohistochemical localization of MMP-14 and -2. Porcine TM cells, cultured on collagen type I–coated membranes, were made serum free 48 hours before mechanical stretching for 24 hours. Images show immunostaining for MMP-14 (red), MMP-2 (green), and F-actin (phalloidin, blue) for several experiments at different magnification levels (A–E).
Colocalization of MMPs and Cortactin
Because these structures resembled images of podosomes or invadopodia in the literature,37,50–53 we looked at the relationship between MMP-2 (red) and cortactin (green), which is a typical component of podosomes and invadopodia (Fig. 2). Strong yellow colocalization was apparent at numerous regions on TM cells. In addition, each of these proteins showed significant areas where they did not colocalize. Discrete MMP-2 staining was seen as mentioned earlier, and intense MMP-2 immunostaining was often seen surrounding areas of colocalization. Cortactin immunostaining was also seen separate from MMP-2 at different areas, typically showing several fairly distinctive patterns. Small rings of intense green cortactin immunostaining, approximately 5 μm in diameter and resembling structures often called rosettes of podosomes,38 were seen surrounding unstained or lightly stained central cores (note particularly Figs. 2A, 2B, 2F, 2H). Areas of intense yellow colocalization often surrounded and in some cases appeared to overrun these central rings. Numerous other more amorphous blobs or roughly triangular- or rhomboid-shaped areas of colocalization were also seen. These regions were often larger, 10 to 15 μm across, and resembled structures called podosome clusters.38 Larger, irregular rings (Figs. 2A, 2I) and various amorphous lattice-like structures (Figs. 2C, 2D, 2G) were also apparent. Similar regions of intense cortactin and MMP-14 colocalization, with significant areas of noncoincidence, were also observed (data not shown).
Figure 2.

Immunohistochemical localization of cortactin and MMP-2. Porcine TM cells, cultured on collagen type I–coated membranes were made serum free 48 hours before mechanical stretching for 24 hours. (A–I) Immunostaining for cortactin (green) and MMP-2 (red). Scale bar, 10 μm.
Localization of Other Typical PILS Components
Localization patterns of several other typical PILS components were then determined on stretched porcine TM cells. Immunostaining patterns for α-actinin (green) and cortactin (red) showed strong colocalization at regions that resembled those for the MMPs and cortactin (Fig. 3A). Discrete regions of separate immunostaining were also seen for each component. At the sites of some apparent PILS, regions of red, green, and yellow were often interspersed in close proximity (note 2× magnification inset). Blue phalloidin staining of F-actin stress fibers was also shown to help define the shape and positions of the cells. Caldesmon (red) and cortactin (green) immunostaining also showed similar colocalization patterns with regions of separate staining and interspersed spots of red, green, and yellow (Fig. 3B). Note also the latticelike structures of cortactin with mixed colocalization in the two insets (Fig. 3B). N-WASP (red) and cortactin (green) immunostaining showed similar colocalization patterns (Fig. 3C), although the regions of separate red and green appeared to be more abundant. Numerous discrete red dots of N-WASP immunostaining were particularly notable on both the central and peripheral regions of these cells. Immunostaining for the small Rho family GTPase and cdc42 (green) and for cortactin (red) showed similar patterns of discrete and coincident spots (Fig. 3D). In addition to the apparent rosettes, the patterns of these immunostaining regions were intricate and complex as were apparent in these insets.
Figure 3.

Immunohistochemical localization of α-actinin, caldesmon, N-WASP, and cdc42 with cortactin and F-actin in cultured TM cells. Porcine TM cells, cultured on collagen type I–coated membranes, were made serum free 48 hours before mechanical stretching for 24 hours. Images show immunostaining for (A) α-actinin (green), cortactin (red), and F-actin (phalloidin, blue); (B) caldesmon (red), cortactin (green), and F-actin (phalloidin, blue); (C) N-WASP (red), cortactin (green), and F-actin (phalloidin, blue); and (D) cdc42 (green), cortactin (red), and F-actin (phalloidin, blue). Insets: various higher resolution images of structures.
When Arp-3 (green) and cortactin (red) were localized in TM cells (Figs. 4A, 4B), numerous regions of punctate immunostaining, both separate and coincident, were also observed. Arp-3 was often distributed in clumps near the nucleus, but also showed spots distributed just back from the peripheral cortactin-rich regions (note Fig. 4B, inset). When Arp-3 (green) and N-WASP (red) were localized together (Figs. 4C, 4D), a similar pattern was observed. Much yellow colocalization was apparent, as were a considerable number of discrete green Arp-3 dots and numerous red N-WASP dots. Colocalization of cdc42 (green) with N-WASP (red) provided additional green central structures surrounded by red dots with considerable yellow colocalization between them (Fig. 4E).
Figure 4.

Immunohistochemical localization of Arp, cortactin, N-WASP, and cdc42 in cultured TM cells. Porcine TM cells, cultured on collagen type I–coated membranes, were made serum free 48 hours before mechanical stretching for 24 hours. Images show immunostaining for (A) and (B) Arp-3 (green), cortactin (red), and F-actin (phalloidin, blue); (C) and (D) Arp-3 (green), N-WASP (red), and F-actin (phalloidin, blue); and (E) cdc42 (green), N-WASP (red), and F-actin (phalloidin, blue).
Colocalization of PILS with Integrins and ECM Components
Since PILS are typically cell–ECM adhesion structures, we evaluated select integrin and ECM molecules with PILS components. When integrin-β1 (red) and cortactin (green) were immunolocalized (Figs. 5A, 5B), strong areas of coincidence at PILS were apparent. Integrin-β1 was also distributed as small punctate dots at the periphery of these cells. Figure 5A included F-actin staining and Figure 5B did not, to facilitate identification of this pattern. In addition to the strong areas of intense colocalization, numerous areas of discrete green cortactin staining were apparent. Most of these show interspersed small red and yellow dots. Lighter immunostaining for integrin-αV (red) and cortactin (green; Figs. 5C, 5D) showed similar patterns of colocalization and individual staining. Notably, the discrete αV immunostaining was found more centrally in the cells, in contrast to the more peripheral β1 staining pattern. Integrin-β3 showed minimal colocalization with cortactin (not shown).
Figure 5.

Immunochemical localization of integrin-β1 and -αV with cortactin and F-actin in cultured TM cells. Porcine TM cells, cultured on collagen type I–coated membranes, were made serum free 48 hours before mechanical stretching for 24 hours. Images show immunostaining for (A) integrin-β1 (red), cortactin (green), and F-actin (phalloidin, blue); (B) integrin-β1 (red) and cortactin (green); (C) integrin-αV (red), cortactin (green), and F-actin (phalloidin, blue); and (D) integrin-αV (red) and cortactin (green).
Immunostaining for fibronectin (green) and MMP-2 (red; Figs. 6A, 6B) also showed regions of separate and some areas of coincident staining. Fibronectin staining showed numerous thick and some fine extracellular fibrils, which became highly coincident with MMP-2 at small regions within the PILS. In some areas, fine dots of coincident immunostaining were apparent in the MMP-2 staining zones where fibronectin was in fine fibrils or not, but was clearly not in thick fibrils. Cortactin showed some colocalization with syndecan-2 but not with syndecan-4 at PILS (not shown).
Figure 6.

Immunohistochemical localization of fibronectin, MMP-2, versican and cortactin on cultured TM cells. Porcine TM cells, cultured on collagen type I–coated membranes, were made serum free 48 hours before mechanical stretching for 24 hours. Images show immunostaining for (A) MMP-2 (red) and fibronectin (green); (B) MMP-2 (red), fibronectin (green), and F-actin (phalloidin, blue); (C) versican (red), cortactin (green) and F-actin (phalloidin, blue); (D) versican (red) and F-actin (phalloidin, blue); (E) versican (red), cortactin (green), and F-actin (phalloidin, blue); (F) versican neoepitope (red), cortactin (green), and F-actin (phalloidin, blue); and (G) versican neoepitope (red) and cortactin (green).
Immunostaining for the large chondroitin sulfate proteoglycan, versican, and for the neoepitope that is created when ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin domains) cleaves versican were also determined (Figs. 6C–G). Versican (red) and cortactin (green) showed strong areas of yellow colocalization at PILS with some areas of separate immunostaining for each (Fig. 6C). Several PILS showed a versican ring approximately 5 μm in diameter of very strong colocalization surrounding a central core of cortactin (red versican in Fig. 6D where green cortactin is not shown or yellow in Fig. 6E where both red and green are shown). The versican neoepitope (Figs. 6F, 6G) showed a similar pattern of colocalization between cleaved versican and cortactin. In some cases, similar rings of colocalizing versican surrounding central cores with only cortactin immunostaining were also observed. The versican was also seen as a red halo surrounding the cortactin centers of the PILS.
Colocalization of PILS Components in Human TM In Situ
To determine whether PILS might exist in situ, we compared immunolocalization of MMP-14 and MMP-2 with cortactin in 5-μm paraffin-embedded sections of human anterior segments, which had been perfused at 8.89 mm Hg for 48 hours (Fig. 7). Both MMP-14 (green; Figs. 7A, 7D) and MMP-2 (green; Fig. 7C) showed small intense yellow areas of colocalization with cortactin (red) throughout the sections. These regions of colocalization were observed in cells lining the trabecular beams throughout the TM, in cells within the JCT region, occasionally in cells of the inner or outer walls of Schlemm's canal, and even in some cells beneath the outer wall. These colocalization regions were usually approximately 5 to 10 μm, which is roughly similar to the sizes we observed for PILS in cell culture. Separate cell-associated cortactin and MMP-14 or -2 immunostaining was also apparent throughout these sections. Sections incubated with both of the secondary but no primary antibodies showed only limited nonspecific immunostaining or autofluorescence (Fig. 7B).
Figure 7.

Immunohistochemical localization of MMP-2, MMP-14, and cortactin in the TM of human anterior segment organ cultures. Human anterior segments were perfused at 8.9 mm Hg for 48 hours before processing for immunohistochemistry. Confocal images show immunostaining for (A) MMP-14 (red) and cortactin (green); (B) control with no primary but both secondary antibodies: (C) MMP-2 (red) and cortactin (green); and (D) MMP-14 (red) and cortactin (green). Schlemm's canal (SC) is as labeled with the TM to the right in each case.
Degradation of Collagen by TM Cells and Real-Time Imaging of Degradation
To evaluate the possibility that PILS are involved in ECM turnover in the TM, porcine TM cells were plated on chamber slides for imaging. These slides had been coated with collagen type I, which was then labeled with FITC. After the cells had been allowed to attach overnight and had begun degrading the labeled collagen, the TM cells were loaded with red fluorescent dye (CellTracker Red; Invitrogen-Molecular Probes) and placed in an imaging chamber at 37°C in 5% CO2 and 100% humidity. A Z-stack of confocal images was taken at one x–y position and at different focal levels, beginning low and moving upward in 1-μm steps at 10-minute intervals for approximately 4 hours. Figure 8 shows a select time sequence of images taken at one x, y, and z position that was extracted from Movies S1–S4, online at http://www.iovs.org/cgi/content/full/49/12/5353/DC1. FITC-collagen was degraded extensively, forming large dark craters with jagged edges by the time imaging was initiated, approximately 24 hours after plating. During the 4-hour imaging period, these degradation craters were enlarged beneath areas where TM cell processes were moving. Active TM cell processes moved relatively rapidly and bright FITC-fragments appeared to be treadmilling up from below and across the dorsal surface of these processes toward the central cell body. Many strong yellow colocalization vacuoles were apparent within the cells where the green FITC-collagen fragments were coincident with the red dye. These cytoplasmic vacuoles were seen primarily surrounding the nuclei and in the central portion of the cell body, with fewer present in the active cellular projections. The cell bodies were extremely pliable, shifting shape and orientation dramatically over the 4-hour filming time. However, the cell bodies did not migrate from their original positions, and the degradation craters became enlarged but did not develop as migration tracks. In Figure 8, the white arrowhead marks one area that had degraded across this period. At 10 minutes, this region had been invaded by a projection but no degradation was apparent. By 110 minutes, approximately five very fine cellular tips had moved beyond the arrowhead and at the arrowhead the cell had thickened into what could be a PILS. By 210 or 260 minutes, this thickened cellular process had retracted, and two new ones had formed on either side of the original region. The FITC-collagen had been degraded and removed from the original region, leaving a darkened area shaped much like the PILS-shaped projection at 110 minutes. The arrow on the other side of this cell shows a different region that began with intact FITC-collagen, progressed with a thickened cellular region and ended with the cellular process partially withdrawing and leaving a region with reduced FITC-collagen. Movies S1–S4 show images taken at 10-minute intervals divided into 4-hour sequences taken at one focal level, then repeated several times moving up to 1-μm higher focal levels, with similar sequences taken at different positions on different slides.
Figure 8.
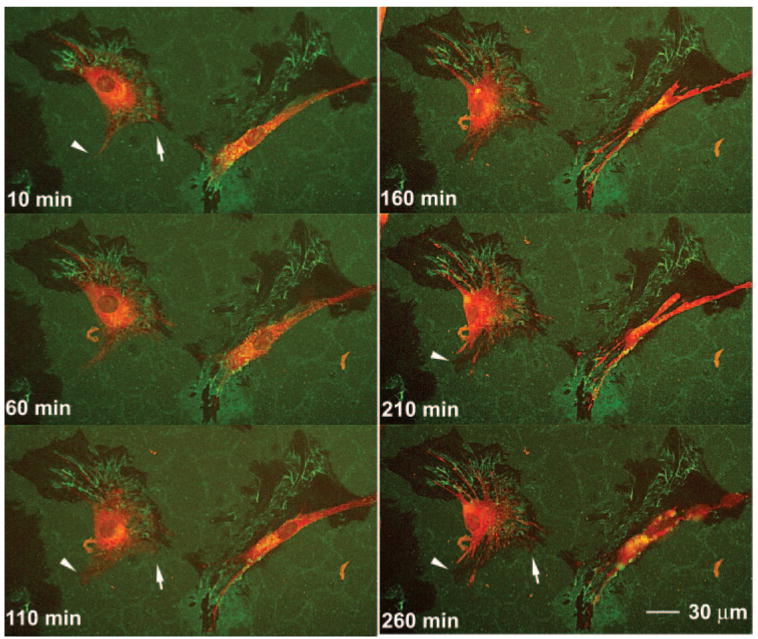
Time-lapse confocal images of TM cells degrading FITC-labeled collagen type I. Porcine TM cells were plated on FITC-labeled collagen type I–coated chamber slides and allowed to adhere overnight. The dye (red) was then incubated with cells for 1 hour, and time lapse confocal images were collected at six focal levels every 10 minutes for 4 hours. The cells were maintained at 37°C in 5% CO2 and 100% humidity throughout. Images were selected showing two cells at 50-minute intervals from one focal plane (Z-stack). Arrowheads and arrows: regions where cellular projections extended, widened, and then withdrew, leaving regions of diminished FITC-labeling in the last frames. Full QuickTime movies of all time points and focal planes for this and several other experiments are included online as Movies S1–S4, http://www.iovs.org/cgi/content/full/49/12/5353/DC1.
Discussion
The distinctive structures to which MMP-2 and -14 colocalized have the appearance of podosomes or invadopodia as observed on several other types of cells.37,50–53 Individual simple podosomes are small, ∼500 nm in diameter, and we have not attempted specialized high-resolution imaging66 of their complicated and somewhat controversial substructures.35,37–39,41,67 On their ventral surface, some specialized cells like macrophages often exhibit hundreds of separate simple podosomes, which have a vertical actin core associated with numerous specific actin-binding proteins and surrounded by a vinculin, talin, and paxillin ring.37,38 On TM cells, we can see some colocalization dots that are in this very approximate size range and appear to have a central actin core (not shown). In TM cells, these are not as extensive as reported for macrophages or similar cells. Individual simple podosomes are reported to have relatively short half-lives, a few minutes to an hour, and usually evolve quickly into more complex aggregate structures or clusters.36–38 Although there is considerable variability in the shape and apparent structure of podosome or invadopodia clusters reported in the literature,36–38 TM cells appear to exhibit PILS clusters resembling many of them. In Figure 1D and several panels of Figure 2, particularly 2F and 2H, TM cells exhibited typical 5-μm circular podosome rosettes, as observed in v-Src-transformed fibroblasts or TGFβ-treated aortic endothelial cells.38,68 These rosettes can enlarge and become somewhat uneven68 beginning to resemble the osteoclasts podosome sealing rings, that are involved in bone resorption.37,69 The shape and size of these rings resembles the uneven cortactin ring we observed in Figures 2A and 2I. The most common PILS clusters in the TM were the amorphous blobs or the irregular triangular or rhomboid shapes typical of PILS clusters observed on v-Src-transformed fibroblasts, human umbilical vein endothelial cells, invasive melanoma cells, and vascular smooth muscle cells.35,37–39,67 The detailed functions of each type of podosome or invadopodia are also fairly different, with osteoclasts forming sealing rings for bone resorption, macrophages forming general ECM degradation zones with many individual podosomes covering most of the ventral surface of the cell, and metastatic cells forming specialized invadopodia to facilitate penetration into new tissue regions.36 Common to them all are ECM adhesive and degradative functions.
The clear colocalization of cortactin, α-actinin, caldesmon, MMP-2, MMP-14, N-WASP, cdc42, Arp-3, and integrin-β1 and -αV at these distinctive structures provides strong support for their identification as TM cell PILS. It should be noted that we have colocalized most of these components with each other in experiments not shown here due to space limitations. Colocalization of these various components with each other are in agreement with the colocalization data shown herein. It is well established that all of these macromolecules have other functions in most cells and all also show distributions to additional areas of TM cells that are clearly not currently PILS. However, their confluence at these typical structures is considered indicative of podosomes or invadopodia formation.36–38,41 The exact organization of the various molecular components within PILS—that is, which ones bind directly to each other—remains somewhat uncertain, suggesting that there may be organizational differences between the various cell types. Nearly 50 separate macromolecules have been associated with PILS. In addition, the highly dynamic nature of PILS suggests that some of these components are regulatory and transiently involved in formation and recruitment, whereas others are involved in actual PILS functions or in PILS dissolution or movement within the cell, which was also apparent from our observations. For instance, in Figure 2, there are several regions of apparent PILS where it appears that cortactin (green) is involved in formation of rosettes or other types of PILS clusters, but where MMP-2 has yet to be recruited or is only partially recruited to the PILS. Of course, without a detailed time course, either observing individual PILS over time or somehow synchronizing the PILS formation process, this staging of PILS structural development and evolution remains conjectural on our part.
One established PILS function is cell-ECM adhesion, often the specialized adhesion that facilitates cell movement.36 The localization of integrin-β1 and -αV to TM cell PILS suggests a role for these molecules in specialized TM cell adhesion to ECM via PILS. Integrin-β3 did not show particular colocalization at PILS (data not shown). As with the other PILS components, integrin-β1 and -αV were also localized to other regions of the cell. The portion of integrin-β1 that did not colocalize with cortactin was generally distributed to the cell periphery, whereas the noncolocalizing integrin-αV distributed more centrally. In separate studies (data not shown) the PILS attachment sites are clearly separate from TM cell focal adhesions or focal contacts.
Fibronectin also showed intense regions of colocalization with some PILS, although most of the fibronectin immunostaining was totally separate from PILS. Fibronectin appears to be predominantly present as thick ECM fibrils,70,71 which exhibit only select intersections with PILS. Both versican and the ADAMTS-4 cleaved form of versican showed clear associations with PILS, particularly the notable rings of versican and cortactin colocalization. Since the epitope for the full-length versican antibody that we used is to the C-terminal G3 domain of versican (data not shown) and the neoepitope antibody is to the C-terminal portion of the ADAMTS-4 cleavage site, we cannot actually differentiate between full-length and cleaved forms with the G3 antibody. It is likely that both are included in the G3 domain staining in Figures 6C–E. Although we can only speculate, this distinctive relationship between cortactin and both forms of versican has intriguing implications.
Based on the time lapse live cell imaging, Figure 8 and Movies S1–S4, TM cell projections or processes are highly dynamic, exhibiting considerable local cell movement without any indications of actual cell migration across the approximately 28 hours of our studies. The cells appeared to stay in the same position and enlarge the degradation craters via movement of these cellular projections without migrating appreciably from the original spot. Although this has not been studied in detail, presumably outflow pathway cells do not exhibit significant cell migration within the outflow pathway under normal conditions in situ. Specialized exceptions may be in response to extreme phagocytic challenge or putative stem cell repopulation after focal laser damage.72,73
Another fairly unique cytoskeletal structure, cross-linked actin networks (CLANs), with possible cell adhesion functions has been identified in TM cells and more recently in the TM in situ.74–76 Based on the components identified in CLANs and in PILS, the structures do not appear to be related, although this has not been established with certainty. CLANs are formed with F-actin microfilaments as the central component and are present as intersecting structures somewhat reminiscent of geodesic domes. F-actin filaments are also central to podosomes and invadopodia, but do not appear to be organized in mature TM PILS to resemble CLANS. However, as a caveat to this apparent differentiation, some of the latticelike structures in the higher magnification panels (Figs. 2C, 2D, 2G; 3B, insets) have a vague resemblance to CLANs. In the TM, CLANs that are formed by selective integrin-β1 and -β3 ligation have been reported to contain filamin, α-actinin, syndecan-4, and phosphoinositol-2-phosphate, but not cortactin, syndecan-2, or Arp-3.76 Podosomes or invadopodia in the literature and TM PILS do have α-actinin, but they also have cortactin and Arp-3. In studies not shown herein, TM cell PILS colocalized somewhat with syndecan-2, but clearly not with syndecan-4. CLANs are also found on the apical or dorsal surface of cells, whereas PILS in the TM and elsewhere are typically found exclusively on the ventral surface of cells. CLANs have been reported to contain neither integrin-β1 nor -β3, whereas TM PILS contain integrin-αV and -β1 with minimal integrin-β3. CLAN induction by dexamethasone in TM cells is coincident with diminished actin stress fibers. Stretching TM cells increases PILS and increases F-actin stress fibers (subjective observations, not shown). Therefore, TM CLANs and TM PILS appear to be different and probably unrelated structures, although additional careful analysis is necessary to establish this.
Caldesmon overexpression has been shown to increase aqueous outflow and to produce unique structural changes in TM cell F-actin cytoskeleton, which do not appear to be related to PILS.77,78 In addition to the colocalization of caldesmon with cortactin at PILS, some caldesmon immunostaining was also separate from PILS. Caldesmon clearly has cellular functions related to PILS as well as those that are unlikely to be directly related.79–82 Recently, wiskostatin, an inhibitor of N-WASP, has also been shown to rapidly increase outflow facility in perfused porcine eyes (Inoue T, et al. IOVS 2008;49:ARVO E-Abstract 1638). In addition to its role in PILS formation, N-WASP serves other roles in F-actin polymerization and regulation.83–89 This observation may therefore not relate directly to TM cell PILS. In addition, recent studies suggest additional modes of action for wiskostatin.90,91 Much of the N-WASP immunostaining that we observed was separate from PILS, although it showed distinctive distribution patterns in TM cells.
The second common function attributed to PILS is focal ECM degradation,36 normally, although not exclusively, achieved by members of the MMP family.37,38 Several other proteinases have been associated with PILS.37 MMP-2 and -14 have been commonly identified at PILS and they are certainly a central component of TM cell PILS. Previously, we have shown a unique relationship between these two MMPs and IOP homeostasis in human anterior segment organ cultures.11,14,21,22 Elevation of perfusion pressure, sensed as mechanical stretching or distortion by the JCT cells, triggers increased MMP-2 and -14 activity. Presumably, this increases ECM turnover and adjusts the outflow resistance to restore IOP to normal levels.11,14,21,22
The colocalization of MMP-2 and -14 was not totally unexpected, since the physiologic activation mechanism for secreted latent MMP-2 is thought to be by MMP-14.44–48 Our observation of the relatively sustained association of MMP-2 with the TM cell surface, particularly at PILS, suggests more than a transient binding associated with activation by MMP-14. Specifics of such an interaction have not been fully established. The association of MMP-2 and -14 with cortactin in TM tissue in situ at what appear to be PILS is also intriguing. The size and approximate shape of these regions are definitely compatible with the presence of PILS in situ in the outflow pathway. Additional studies are needed to demonstrate that they are active and functional PILS. However, this suggests a physiologic mechanism for TM cells to initiate controlled and focal ECM turnover associated with outflow facility regulation. Since we have shown previously that ongoing endogenous MMP activity is necessary to maintain the outflow resistance,14 this aspect appears to be critical in outflow pathway physiology.
The ability of TM cells, apparently using the MMPs at PILS, to degrade FITC-labeled collagen suggests that ongoing ECM turnover may be facilitated by these TM cell PILS. The active movement of the TM cell projections in Movies S1–S4 suggests that the TM cell body remains relatively fixed in position, while the projections sweep over the surrounding area degrading the ECM. Since TM JCT cells are subconfluent and exhibit similar projections,92,93 this type of active ECM degradation by cell projections may be occurring in vivo, to produce normal ongoing ECM turnover and modulate the outflow resistance. The processing of cleaved collagen fragments by TM cells in the video is rapid and dynamic. The degraded collagen fragments moving on the dorsal TM cell surface, presumably facilitated by integrins, is intriguing. The FITC-collagen fragments taken up by TM cells are apparently present in vacuoles, where they appear yellow due to coincidence with the red dye. It is also notable that FITC fluorescence intensity is very pH-dependent, declining around 10-fold between pH 7.2 and 5.8 and perhaps much more at lower pH, such as is found in endosomes.94 Thus, the amount of FITC-collagen fragments that are within degradation vesicles will probably be underestimated compared with those outside the cell at neutral pH values.
Thus, these dynamic TM cell PILS provide an appealing potential mechanism for tightly controlled focal ECM degradation in the JCT region of the outflow pathway. It is important to establish more rigorously the role of these unusual structures in the normal functional paradigm of the TM. Since our overall working hypothesis is that TM cells within the JCT are responsible for maintaining the outflow resistance and thus IOP homeostasis by modulating ECM turnover, this is exactly what we would predict, albeit retrospectively, as a mechanism for achieving and targeting this process.
Acknowledgments
The authors thank Ruth Phinney (Oregon Lions Eye Bank) for facilitating procurement of human eyes, Carolyn Gendron (OHSU Cancer Center) for paraffin sectioning and histology, Aurelie Snyder (OHSU Molecular Microbiology and Immunology Core) for confocal microscopy, and Genevieve Long, PhD, for editorial assistance.
Supported by National Eye Institute Grants EY003279, EY008247, and EY010572 and by a grant from Research to Prevent Blindness (New York, NY).
Footnotes
Disclosure: M. Aga, None; J.M. Bradley, None; K.E. Keller, None; M.J. Kelley, None; T.S. Acott, None
References
- 1.Quigley HA. Open-angle glaucoma. N Engl J Med. 1993;328:1097–1106. doi: 10.1056/NEJM199304153281507. [DOI] [PubMed] [Google Scholar]
- 2.Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996;80:389–393. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shields MB. Textbook of Glaucoma. 4th. Baltimore: Williams & Wilkins; 1998. [Google Scholar]
- 4.Boland MV, Quigley HA. Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007;16:406–418. doi: 10.1097/IJG.0b013e31806540a1. [DOI] [PubMed] [Google Scholar]
- 5.Bill A, Maepea O. Mechanisms and routes of aqueous humor drainage. In: Albert DM, Jakotiec FM, editors. Principles and Practices of Ophthalmology. Philadelphia: WB Saunders; 1994. pp. 206–226. [Google Scholar]
- 6.Maepea O, Bill A. Pressures in the juxtacanalicular tissue and Schlemm's canal in monkeys. Exp Eye Res. 1992;54:879–883. doi: 10.1016/0014-4835(92)90151-h. [DOI] [PubMed] [Google Scholar]
- 7.Grant WM. Experimental aqueous perfusion in enucleated human eyes. Arch Ophthalmol. 1963;69:783–801. doi: 10.1001/archopht.1963.00960040789022. [DOI] [PubMed] [Google Scholar]
- 8.Johnson M. What controls aqueous humour outflow resistance? Exp Eye Res. 2006;82:545–557. doi: 10.1016/j.exer.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fautsch MP, Johnson DH. Aqueous humor outflow: what do we know? Where will it lead us? Invest Ophthalmol Vis Sci. 2006;47:4181–4187. doi: 10.1167/iovs.06-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ethier CR. The inner wall of Schlemm's canal. Exp Eye Res. 2002;74:161–172. doi: 10.1006/exer.2002.1144. [DOI] [PubMed] [Google Scholar]
- 11.Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork (review) Exp Eye Res. 2008;86:543–561. doi: 10.1016/j.exer.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman PL, Erickson KA. Cytochalasin B and D dose-outflow facility response relationships in the cynomolgus monkey. Invest Ophthalmol Vis Sci. 1982;23:646–650. [PubMed] [Google Scholar]
- 13.Sabanay I, Gabelt B, Tian B, Kaufman P, Geiger B. H-7 effects on the structure and fluid conductance of monkey trabecular meshwork. Arch Ophthalmol. 2000;118:955–962. [PubMed] [Google Scholar]
- 14.Bradley JMB, Vranka JA, Colvis CM, et al. Effects of matrix metalloproteinase activity on outflow in perfused human organ culture. Invest Ophthalmol Vis Sci. 1998;39:2649–2658. [PubMed] [Google Scholar]
- 15.Bárány EH, Scotchbrook S. Influence of testicular hyaluronidase on the resistance to flow through the angle of the anterior chamber. Acta Physiol Scand. 1954;30:240–248. doi: 10.1111/j.1748-1716.1954.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 16.Johnson D. The effect of cytochalasin D on outflow facility and the trabecular meshwork of the human eye in perfusion organ culture. Invest Ophthalmol Vis Sci. 1997;38:2790–2799. [PubMed] [Google Scholar]
- 17.Knepper PA, Farbman AI, Telser AG. Exogenous hyaluronidases and degradation of hyaluronic acid in the rabbit eye. Invest Ophthalmol Vis Sci. 1984;25:286–293. [PubMed] [Google Scholar]
- 18.Epstein D, Rowlette LL, Roberts B. Acto-myosin drug effects and aqueous outflow function. Invest Ophthalmol Vis Sci. 1999;40:74–81. [PubMed] [Google Scholar]
- 19.Rao PV, Deng P, Maddala R, Epstein DL, Li CY, Shimokawa H. Expression of dominant negative Rho-binding domain of Rho-kinase in organ cultured human eye anterior segments increases aqueous humor outflow. Mol Vis. 2005;11:288–297. [PubMed] [Google Scholar]
- 20.Tian B, Kaufman PL, Volberg T, Gabelt BT, Geiger B. H-7 disrupts the actin cytoskeleton and increases outflow facility. Arch Ophthalmol. 1998;116:633–643. doi: 10.1001/archopht.116.5.633. [DOI] [PubMed] [Google Scholar]
- 21.Bradley JMB, Kelley MJ, Zhu XH, Anderssohn AM, Alexander JP, Acott TS. Effects of mechanical stretching on trabecular matrix metalloproteinases. Invest Ophthalmol Vis Sci. 2001;42:1505–1513. [PubMed] [Google Scholar]
- 22.Bradley JM, Kelley MJ, Rose A, Acott TS. Signaling pathways used in trabecular matrix metalloproteinase response to mechanical stretch. Invest Ophthalmol Vis Sci. 2003;44:5174–5181. doi: 10.1167/iovs.03-0213. [DOI] [PubMed] [Google Scholar]
- 23.Vittitow J, Borras T. Genes expressed in the human trabecular meshwork during pressure-induced homeostatic response. J Cell Physiol. 2004;201:126–137. doi: 10.1002/jcp.20030. [DOI] [PubMed] [Google Scholar]
- 24.Tumminia SJ, Mitton KP, Arora J, Zelenka P, Epstein PL, Russell P. Mechanical stretch alters the actin cytoskeletal network and signal transduction in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1998;39:1361–1371. [PubMed] [Google Scholar]
- 25.Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2005;46:2857–2868. doi: 10.1167/iovs.05-0075. [DOI] [PubMed] [Google Scholar]
- 26.Keller KE, Kelley MJ, Acott TS. Extracellular matrix gene alternative splicing by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2007;48:1164–1172. doi: 10.1167/iovs.06-0875. [DOI] [PubMed] [Google Scholar]
- 27.WuDunn D. The effect of mechanical strain on matrix metalloproteinase production by bovine trabecular meshwork cells. Curr Eye Res. 2001;22:394–397. doi: 10.1076/ceyr.22.5.394.5500. [DOI] [PubMed] [Google Scholar]
- 28.Okada Y, Matsuo T, Ohtsuki H. Bovine trabecular cells produce TIMP-1 and MMP-2 in response to mechanical stretching. Jpn J Ophthalmol. 1998;42:90–94. doi: 10.1016/s0021-5155(97)00129-9. [DOI] [PubMed] [Google Scholar]
- 29.Sato Y, Matouo T, Ohtsuki H. A novel gene (oculomedin) induced by mechanical stretching of human trabecular cells of the eye. Biochem Biophys Res Commun. 1999;259:349–351. doi: 10.1006/bbrc.1999.0797. [DOI] [PubMed] [Google Scholar]
- 30.Stamer WD, Roberts BC, Epstein DL. Hydraulic pressure stimulates adenosine 3′,5′-cyclic monophosphate accumulation in endothelial cells from Schlemm's canal. Invest Ophthalmol Vis Sci. 1999;40:1983–1988. [PubMed] [Google Scholar]
- 31.Mitton KP, Tumminia SJ, Arora J, Zelenka P, Epstein DL, Russell P. Transient loss of αB-crystallin: an early cellular response to mechanical stretch. Biochem Biophys Res Commun. 1997;235:69–73. doi: 10.1006/bbrc.1997.6737. [DOI] [PubMed] [Google Scholar]
- 32.Tamm ER, Russell P, Epstein DL, Johnson DH, Piatigorsky J. Modulation of myocillin: TIGR expression in human trabecular meshwork. Invest Ophthalmol Vis Sci. 1999;40:2577–2582. [PubMed] [Google Scholar]
- 33.Basbaum CB, Werb Z. Focalized proteolysis: spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr Opin Cell Biol. 1996;8:731–738. doi: 10.1016/s0955-0674(96)80116-5. [DOI] [PubMed] [Google Scholar]
- 34.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 35.Ayala I, Baldassarre M, Caldieri G, Buccione R. Invadopodia: a guided tour. Eur J Cell Biol. 2006;85:159–164. doi: 10.1016/j.ejcb.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–117. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 39.Jurdic P, Saltel F, Chabadel A, Destaing O. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85:195–202. doi: 10.1016/j.ejcb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Spinardi L, Marchisio PC. Podosomes as smart regulators of cellular adhesion. Eur J Cell Biol. 2006;85:191–194. doi: 10.1016/j.ejcb.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Linder S, Kopp P. Podosomes at a glance. J Cell Sci. 2005;118:2079–2082. doi: 10.1242/jcs.02390. [DOI] [PubMed] [Google Scholar]
- 42.Webb BA, Eves R, Mak AS. Cortactin regulates podosome formation: roles of the protein interaction domains. Exp Cell Res. 2006;312:760–769. doi: 10.1016/j.yexcr.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 43.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 44.Murphy G, Willenbrock F, Crabbe T, et al. Regulation of matrix metalloproteinase activity (review) Ann N Y Acad Sci. 1994;732:31–41. doi: 10.1111/j.1749-6632.1994.tb24722.x. [DOI] [PubMed] [Google Scholar]
- 45.Murphy G, Willenbrock F, Ward RV, Cockett MI, Eaton D, Docherty AJ. The C-terminal domain of 72 kDa gelatinase A is not required for catalysis, but is essential for membrane activation and modulates interactions with tissue inhibitors of metalloproteinases. Biochem J. 1992;283:637–641. doi: 10.1042/bj2830637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagase H, Woessner J. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 47.Parks W, Mecham R, editors. Matrix Metalloproteinases. San Diego, CA: Academic Press; 1998. [Google Scholar]
- 48.Fu X, Parks WC, Heinecke JW. Activation and silencing of matrix metalloproteinases. Semin Cell Dev Biol. 2008;19:2–13. doi: 10.1016/j.semcdb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 50.Ochoa GC, Slepnev VI, Neff L, et al. A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol. 2000;150:377–389. doi: 10.1083/jcb.150.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Asawa T, Takato T, Sakai R. Cooperative roles of Fyn and cortactin in cell migration of metastatic murine melanoma. J Biol Chem. 2003;278:48367–48376. doi: 10.1074/jbc.M308213200. [DOI] [PubMed] [Google Scholar]
- 52.Gu Z, Kordowska J, Williams GL, Wang CL, Hai CM. Erk1/2 MAPK and caldesmon differentially regulate podosome dynamics in A7r5 vascular smooth muscle cells. Exp Cell Res. 2007;313:849–866. doi: 10.1016/j.yexcr.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW. Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol. 2000;151:187–198. doi: 10.1083/jcb.151.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alexander JP, Acott TS. Involvement of protein kinase C in TNFα regulation of trabecular matrix metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci. 2001;42:2831–2838. [PubMed] [Google Scholar]
- 55.Alexander JP, Acott TS. Involvement of Erk-MAP kinase pathway in TNFα regulation of trabecular metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci. 2003;44:164–169. doi: 10.1167/iovs.01-1201. [DOI] [PubMed] [Google Scholar]
- 56.Alexander JP, Samples JR, Van Buskirk EM, Acott TS. Expression of matrix metalloproteinases and inhibitor by human trabecular meshwork. Invest Ophthalmol Vis Sci. 1991;32:172–180. [PubMed] [Google Scholar]
- 57.Polansky JR, Weinreb R, Alvarado JA. Studies on human trabecular cells propagated in vitro. Vision Res. 1981;21:155–160. doi: 10.1016/0042-6989(81)90151-6. [DOI] [PubMed] [Google Scholar]
- 58.Polansky JR, Weinreb RN, Baxter JD, Alvarado J. Human trabecular cells. I. Establishment in tissue culture and growth characteristics. Invest Ophthalmol Vis Sci. 1979;18:1043–1049. [PubMed] [Google Scholar]
- 59.Stamer WD, Seftor RE, Williams SK, Samaha HA, Snyder RW. Isolation and culture of human trabecular meshwork cells by extracellular matrix digestion. Curr Eye Res. 1995;14:611–617. doi: 10.3109/02713689508998409. [DOI] [PubMed] [Google Scholar]
- 60.Bradley JMB, Anderssohn AM, Colvis CM, et al. Mediation of laser trabeculoplasty-induced matrix metalloproteinase expression by IL-1β and TNFα. Invest Ophthalmol Vis Sci. 2000;41:422–430. [PubMed] [Google Scholar]
- 61.Johnson DH, Tschumper RC. The effect of organ culture on human trabecular meshwork. Exp Eye Res. 1989;49:113–127. doi: 10.1016/0014-4835(89)90080-8. [DOI] [PubMed] [Google Scholar]
- 62.Johnson DH, Tschumper RC. Human trabecular meshwork organ culture. Invest Ophthalmol Vis Sci. 1987;28:945–953. [PubMed] [Google Scholar]
- 63.Bachmann B, Birke M, Kook D, Eichhorn M, Lütjen-Drecoll E. Ultrastructural and biochemical evaluation of the porcine anterior chamber perfusion model. Invest Ophthalmol Vis Sci. 2006;47:2011–2020. doi: 10.1167/iovs.05-1393. [DOI] [PubMed] [Google Scholar]
- 64.Acott TS, Westcott M, Passo MS, Van Buskirk EM. Trabecular meshwork glycosaminoglycans in human and cynomolgus monkey eye. Invest Ophthalmol Vis Sci. 1985;26:1320–1329. [PubMed] [Google Scholar]
- 65.Keller KE, Bradley JM, Kelley MJ, Acott TS. Effects of modifiers of glycosaminoglycan biosynthesis on outflow facility in perfusion culture. Invest Ophthalmol Vis Sci. 2008;49:2495–2505. doi: 10.1167/iovs.07-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt R, Wurm CA, Jakobs S, Engelhardt J, Egner A, Hell SW. Spherical nanosized focal spot unravels the interior of cells. Nat Methods. 2008;5:539–544. doi: 10.1038/nmeth.1214. [DOI] [PubMed] [Google Scholar]
- 67.Daly RJ. Cortactin signalling and dynamic actin networks. Biochem J. 2004;382:13–25. doi: 10.1042/BJ20040737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varon C, Tatin F, Moreau V, et al. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Mol Cell Biol. 2006;26:3582–3594. doi: 10.1128/MCB.26.9.3582-3594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Machesky L, Jurdic P, Hinz B. Grab, stick, pull and digest: the functional diversity of actin-associated matrix-adhesion structures: workshop on invadopodia, podosomes and focal adhesions in tissue invasion. EMBO Rep. 2008;9:139–143. doi: 10.1038/sj.embor.7401162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mao Y, Schwarzbauer JE. Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J Cell Sci. 2005;118:4427–4436. doi: 10.1242/jcs.02566. [DOI] [PubMed] [Google Scholar]
- 71.Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24:389–399. doi: 10.1016/j.matbio.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 72.Johnson DH, Richardson TM, Epstein DL. Trabecular meshwork recovery after phagocytic challenge. Curr Eye Res. 1989;8:1121–1130. doi: 10.3109/02713688909000037. [DOI] [PubMed] [Google Scholar]
- 73.Acott TS, Samples JR, Bradley JMB, Bacon DR, Bylsma SS, Van Buskirk EM. Trabecular repopulation by anterior trabecular meshwork cells after laser trabeculoplasty. Am J Ophthalmol. 1989;107:1–6. doi: 10.1016/0002-9394(89)90805-2. [DOI] [PubMed] [Google Scholar]
- 74.Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M, Howe W. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994;35:281–294. [PubMed] [Google Scholar]
- 75.Clark AF, Brotchie D, Read AT, et al. Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil Cytoskeleton. 2005;60:83–95. doi: 10.1002/cm.20049. [DOI] [PubMed] [Google Scholar]
- 76.Filla MS, Woods A, Kaufman PL, Peters DM. Beta1 and beta3 integrins cooperate to induce syndecan-4-containing cross-linked actin networks in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2006;47:1956–1967. doi: 10.1167/iovs.05-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gabelt BT, Hu Y, Vittitow JL, et al. Caldesmon transgene expression disrupts focal adhesions in HTM cells and increases outflow facility in organ-cultured human and monkey anterior segments. Exp Eye Res. 2006;82:935–944. doi: 10.1016/j.exer.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 78.Grosheva I, Vittitow JL, Goichberg P, et al. Caldesmon effects on the actin cytoskeleton and cell adhesion in cultured HTM cells. Exp Eye Res. 2006;82:945–958. doi: 10.1016/j.exer.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 79.Hai CM, Gu Z. Caldesmon phosphorylation in actin cytoskeletal remodeling. Eur J Cell Biol. 2006;85:305–309. doi: 10.1016/j.ejcb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 80.Wang CL. Caldesmon and smooth-muscle regulation. Cell Biochem Biophys. 2001;35:275–288. doi: 10.1385/cbb:35:3:275. [DOI] [PubMed] [Google Scholar]
- 81.Dabrowska R, Kulikova N, Gagola M. Nonmuscle caldesmon: its distribution and involvement in various cellular processes (review) Protoplasma. 2004;224:1–13. doi: 10.1007/s00709-004-0057-3. [DOI] [PubMed] [Google Scholar]
- 82.Eves R, Webb BA, Zhou S, Mak AS. Caldesmon is an integral component of podosomes in smooth muscle cells. J Cell Sci. 2006;119:1691–1702. doi: 10.1242/jcs.02881. [DOI] [PubMed] [Google Scholar]
- 83.Mizutani K, Miki H, He H, Maruta H, Takenawa T. Essential role of neural Wiskott-Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src-transformed fibroblasts. Cancer Res. 2002;62:669–674. [PubMed] [Google Scholar]
- 84.Yamaguchi H, Lorenz M, Kempiak S, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 86.Rohatgi R, Ma L, Miki H, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 87.Weaver AM, Heuser JE, Karginov AV, Lee WL, Parsons JT, Cooper JA. Interaction of cortactin and N-WASp with Arp2/3 complex. Curr Biol. 2002;12:1270–1278. doi: 10.1016/s0960-9822(02)01035-7. [DOI] [PubMed] [Google Scholar]
- 88.Caron E. Regulation of Wiskott-Aldrich syndrome protein and related molecules. Curr Opin Cell Biol. 2002;14:82–87. doi: 10.1016/s0955-0674(01)00298-8. [DOI] [PubMed] [Google Scholar]
- 89.Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–311. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 90.Guerriero CJ, Weisz OA. N-WASP inhibitor wiskostatin nonselectively perturbs membrane transport by decreasing cellular ATP levels. Am J Physiol Cell Physiol. 2007;292:C1562–C1566. doi: 10.1152/ajpcell.00426.2006. [DOI] [PubMed] [Google Scholar]
- 91.Leung DW, Morgan DM, Rosen MK. Biochemical properties and inhibitors of (N-)WASP. Methods Enzymol. 2006;406:281–296. doi: 10.1016/S0076-6879(06)06021-6. [DOI] [PubMed] [Google Scholar]
- 92.Inomata H, Bill A, Smelser GK. Aqueous humor pathways through the trabecular meshwork and into Schlemm's canal in the cynomolgus monkey (Macaca irus): an electron microscopic study. Am J Ophthalmol. 1972;73:760. doi: 10.1016/0002-9394(72)90394-7. [DOI] [PubMed] [Google Scholar]
- 93.Grierson I, Lee WR, Abraham S, Howes RC. Associations between the cells of the walls of Schlemm's canal. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1978;208:33–47. doi: 10.1007/BF00406980. [DOI] [PubMed] [Google Scholar]
- 94.Carr DW, Acott TS. Intracellular pH regulates bovine sperm motility and protein phosphorylation. Biol Reprod. 1989;41:907–920. doi: 10.1095/biolreprod41.5.907. [DOI] [PubMed] [Google Scholar]