

Abstract
The active form of vitamin D, 1,25-dihydroxyvitamin D (1,25(OH)2D) enhances innate immunity by inducing the cathelicidin antimicrobial peptide (hCAP). In monocytes/macrophages, this occurs primarily in response to activation of toll-like receptors (TLR), that induce expression of the vitamin D receptor and localized synthesis of 1,25(OH)2D from precursor 25-hydroxyvitamin D3 (25OHD). To clarify the relationship between vitamin D and innate immunity, we assessed changes in hCAP expression in vivo and ex vivo in human subjects attending a bone clinic (n=50). Of these, 38% were vitamin D-insufficient (<75 nM 25OHD) and received supplementation with vitamin D (50,000 IU vitamin D2 twice weekly for 5 weeks). Baseline 25OHD status or vitamin D supplementation had no effect on circulating levels of hCAP. Therefore, ex vivo changes in hCAP for each subject were assessed using peripheral blood monocytes cultured with 10% autologous serum (n=28). Under these vitamin D ‘insufficient’ conditions the TLR2/1 ligand 19 kDa lipopeptide (19 kDa) or the TLR4 ligand lipopolysaccharide, monocytes showed increased expression of the vitamin D-activating enzyme CYP27b1 (5- and 5.5-fold respectively, both p<0.01) but decreased expression of hCAP mRNA (10-fold and 30-fold, both p<0.001). Following treatment with 19 kDa, expression of hCAP: 1) correlated with 25OHD levels in serum culture supplements (R=0.649, p<0.001); 2) was significantly enhanced by exogenous 25OHD (5 nM); 3) was significantly enhanced with serum from vivo vitamin D-supplemented patients. These data suggest that a key role of vitamin D in innate immunity is to maintain localized production of antibacterial hCAP following TLR activation of monocytes.
Keywords: vitamin D, CYP27b1, toll-like receptor, macrophage, cathelicidin
Introduction
The active, hormonal form of vitamin D, 1,25-dihydroxyvitamin D (1,25(OH)2D) exerts biological effects that extend far beyond its established role in calcium and bone homeostasis. In particular, recent studies have shown that 1,25(OH)2D is potent modulator of both innate and adaptive immunity (1–3). A crucial facet of these reports has been the recognition that immune responses to vitamin D are more likely to be due to localized synthesis of 1,25(OH)2D rather than systemic production of the hormone (4). Experiments using monocytes (5), dendritic cells (6–8) and lymphocytes (8, 9) have demonstrated expression of both the intracellular receptor for 1,25(OH)2D (vitamin D receptor, VDR), and the enzyme that catalyzes conversion of pro-hormone 25-hydroxyvitamin D (25OHD) to 1,25(OH)2D, 25-hydroxyvitamin D3-1α-hydroxylase (CYP27b1). Previous studies from our group have shown that coincident expression of these proteins by cells from the immune system facilitates immune responses to 25OHD via intracrine conversion to 1,25(OH)2D (5, 6).
In recent experiments we have shown that activation of the toll-like receptor (TLR)-2,1 signaling cascade by a 19 kDa lipopeptide from Mycobacterium tuberculosis (M tb.) increases expression of both CYP27b1 and VDR in monocyte/macrophages from normal human hosts (5). The resulting 1,25(OH)2D-VDR transcription complex is then able to stimulate expression of the cathelicidin antimicrobial protein (hCAP) with concomitant killing of phagocytosed mycobacteria. This induction of innate immune responses was hindered by incubation of TLR-stimulated human monocyte/macrophages with serum from vitamin D (25OHD)-insufficient subjects and rescued in vitro by the simple addition of supplemental 25OHD to medium conditioning those cells.
Serum levels of 25OHD are a direct reflection of vitamin D ‘status’, and several reports have highlighted the worldwide prevalence of 25OHD-insufficiency as a consequence of inadequate oral consumption or cutaneous biosynthesis of vitamin D (10, 11). In view of the link between 25OHD and macrophage function outlined above, we have proposed that populations with known susceptibility to vitamin D insufficiency may also exhibit impaired innate immunity. To test this hypothesis, we used cells and serum from a cohort of human subjects pre- and post- vitamin D supplementation to assess various parameters associated with responses to TLR activation. Data reveal a direct correlation between serum concentration of 25OHD and monocyte expression of hCAP following treatment with ligands to pathogen-responsive toll-like receptors (TLRs). Furthermore, we show that in vivo supplementation of vitamin D insufficient patients significantly enhanced ex vivo innate immune responses by rescuing TLR-mediated suppression of hCAP expression.
Materials and Methods
Human subjects
A cohort of fifty human subjects (40 female, 10 male, mean age 63.3 ± 14.9) was recruited from patients visiting the Cedars-Sinai Medical Center Bone Clinic for treatment of low bone mineral density, but who were otherwise healthy. The clinical parameters for inclusion in the study were established as part of an IRB-approved protocol (CSMC I.D. 7995) to: 1] screen serum and urine for diagnosis of vitamin D insufficiency (and other causes of secondary osteoporosis) by measuring serum calcium, phosphate, magnesium, creatinine, parathyroid hormone, 25OHD, 1,25(OH)2D and urine calcium: creatinine excretion ratio); 2] collect serum and peripheral blood mononuclear cells (PBMCs) from patients; 3] recollect serum and PBMCs from those patients with low vitamin D status after a course of vitamin D treatment to restore 25OHD levels to normal. Patients classified as vitamin D insufficient (serum 25OHD < 75 nM [30 ng/ml]) following the initial visit to the clinic were placed on a course of oral vitamin D therapy to restore vitamin D sufficiency (50,000 IU vitamin D2 twice per week for 5 weeks). All 50 patients recruited to the trial, provided serum for biochemical analyses and 28 provided PBMCs for ex vivo analyses.
Culture of monocytic cell lines
Human THP-1 and mouse J774A macrophage cell lines were cultured in RPMI 1640 growth medium supplemented with 10% fetal calf serum (FCS) at 37°C and 5% carbon dioxide. In vitro treatments included: the toll-like receptor (TLR) 2/1 ligand 19 kDa lipopeptide (19 kDa, 1 ng/ml); the TLR4 ligand lipopolysaccharide (LPS, 100 ng/ml) and 25OHD (5–100 nM).
Patient blood collection and isolation of serum and PBMCs
All patients entering the trial donated 10 ml of blood for separation of serum. Serum samples were: 1) stored at −80°C for subsequent use in ex vivo culture experiments; 2) used for analysis of serum vitamin D metabolites; 3) used for ELISA analysis of serum hCAP protein (as described in (12)).
Patients also donated 20ml of heparinized blood for isolation of PBMCs. Briefly heparinized whole blood was diluted 1:2 in PBS and layered on 5ml of Lymphoprep™ (Greiner Bio-One, Munroe, NC) and centrifuged at 500 × g for 30 mins. PBMCs were isolated from the interface and washed twice with PBS to remove residual Lymphoprep™. The resulting cell pellet was then resuspended in PBS and the sample divided into two. The first aliquot was lysed immediately with RNAzol to provide PBMC total RNA for subsequent gene analysis. The second aliquot was re-centrifuged, resuspended in serum-free RMPI and aliquots of this suspension plated in 24 well plastic cell culture plates for 2 hrs. Non-adherent cells were then removed and lysed in RNAzol to provide total RNA from a lymphocyte-enriched population of cells. The remaining adherent monocytes were separated into two populations. The first was lysed with RNAzol to provide monocyte total RNA for subsequent gene analysis. The second population of monocytes was retained for ex vivo culture experiments (as described in the following section).
Ex vivo analysis of patient monocytes using autologous serum cultures
Monocytes isolated from patient PBMCs were cultured for 24 hrs in RPMI 1640 medium supplemented with 10% autologous serum. Cells cultured under these conditions underwent four main treatments: 1] monocytes + autologous serum (control); 2] monocytes + autologous serum + TLR2 ligand (19 kDa bacterial lipopeptide [19 kDa], 1 μg/ml); 3] monocytes + autologous serum + ex vivo added 25OHD (5 or 100 nM); 4] monocytes + autologous serum + TLR2 ligand (19 kDa) + ex vivo added 25OHD (5 or 100 nM). Following treatment of monocytes for 24 hrs, culture supernatants were removed and stored at −80°C for subsequent analysis of 1,25(OH)2D production using an IRA kit (Diasorin, Stillwater, MN) and previously described methods (13). The remaining adherent cells were then lysed with RNAzol to generate total RNA for subsequent gene analysis (see below).
Extraction of RNA and reverse transcription
RNA was extracted from mouse tissues using the RNeasy™ Total RNA extraction kit as detailed by the manufacturer (Qiagen, Valencia, CA). RNA was eluted in RNase-free elution solution and aliquots (1.5 μg) were reverse-transcribed using Powerscript™ MMLV reverse transcriptase as described by the manufacturer (ABI, Foster City, CA).
Quantitative real time RT-PCR amplification of cDNAs
Expression of mRNAs for VDR, CYP27b1, 24-hydroxylase (CYP24), hCAP, β-defensin-4 and other specified genes was quantified using an ABI 7700 sequence detection system (ABI) as described previously (14). Approximately 50 ng of cDNA was used per reaction. All reactions were multiplexed with the housekeeping 18S rRNA gene (Assays-on-Demand™ (ABI) primer and probe mix Hs99999901_s1), enabling data to be expressed in relation to an internal reference to allow for differences in sampling. Data were obtained as Ct values (the cycle number at which logarithmic PCR plots cross a calculated threshold line), and used to determine ΔCt values (Ct of target gene–Ct of housekeeping 18S rRNA gene). PCR amplification of target gene cDNA was carried out using the following Taqman human gene expression assays: CYP27b1, forward primer 5′-TTGGCAAGCGCAGCTGTAT-3′, reverse primer 5′-TGTGTTAGGATCTGGGCCAAA-3′, TaqMan probe 5′-TTGCAATTCAAGCTCTGCCAGGCG-3′; VDR, forward primer 5′-CTTCAGGCGAAGCATGAAGC-3′, reverse primer 5′-CCTTCATCATGCCGATGTCC-3′, Taqman probe 5′-AAGGCACTATTCACCTGCCCCTTCAA-3′; CYP24, Forward primer, 5′-CAAACCGTGGAAGGCCTATC-3′; reverse primer 5′-AGTCTTCCCCTTCCAGGATCA-3′;TaqMan probe 5′-ACTACCGCAAAGAAGGCTACGGGCTG-3′; hCAP, Assays-on-Demand™ (ABI) primer and probe mix Hs00189038_m1; DEFB4 Hs00175474_m1; interleukin-1 (IL-1β). In addition, for studies using J774A mouse macrophages, real time RT-PCR was carried out for the following mouse genes: cathelicidin-related antimicrobial peptide (CRAMP), ABI primer and probe mix Mm00438285_m1; CYP27b1 (Mm01165922); VDR (Mn004337297); interleukin 1β (IL-1) (00439620). All cDNAs were amplified under the following conditions: 50 °C for 2 min; 95 °C for 10 min followed by 40 cycles of 95 °C for 15 sec and 60 °C for 1 min. All reactions were performed in triplicate and initially expressed as mean ± SD ΔCt values which were employed in statistical comparisons. Visual representation of data was carried out by converting ΔCt values to fold-change data relative to ΔCt values for control (vehicle-treated) cells using the equation 2−ΔΔCt.
Statistical analyses
Data were expressed as mean ±SD unless otherwise stated. Statistical evaluation of correlations for circulating vitamin D metabolites was by linear regression. Statistical analysis of ex vivo treatment studies was carried out by one way analysis of variance (ANOVA) with the Holm-Sidak method as a post hoc multiple comparison procedure applied to raw ΔCt values from RT-PCR assays. Statistical analysis of pre- and post-vitamin D supplementation data was carried out using a students t-test. All statistical values were defined using Sigmaplot 9.0 software (Systat Inc., San Jose, CA, USA).
Results
Lack of correlation between serum vitamin D metabolites and circulating levels of hCAP
Previous studies ex vivo have shown that serum levels of 25OHD are a key determinant of antimicrobial hCAP expression by monocytes following immune challenge by pathogens such as M. tb (5). In a similar fashion, studies in vivo have shown that supplementation with vitamin D suppresses the growth of M. tb in samples of whole blood from healthy adults (15), and reduces the time for sputum smear conversion from acid fast bacteria (AFB) positive to AFB-negative status in patients with tuberculosis (TB) (16). We have shown previously that monocyte responses to vitamin D involve localized autocrine induction of hCAP (5). What is less clear is whether this antimicrobial activity also extends to systemic hCAP. To investigate further the extent to which innate immunity is influenced by vitamin D status in the host, and to ascertain whether antibacterial effects are due to a predictable change in circulating concentrations of hCAP, we assessed the relationship between serum concentrations of 25OHD, 1,25(OH)2D and hCAP in a cohort of human subjects.
Data in Figure 1 show that although serum hCAP levels varied considerably, there was no significant correlation with serum 25OHD or 1,25(OH)2D concentrations. Likewise, no significant correlation was observed between serum vitamin D metabolites and mRNA levels for hCAP in PBMCs isolated from the same blood sample (Figure 2A and 2B). A similar lack of correlation between serum vitamin D metabolites and hCAP mRNA was also observed for macrophages and lymphocytes isolated from the PBMCs (data not shown). It was interesting to note that circulating concentrations of protein for hCAP did not correlate with levels of hCAP mRNA isolated from PBMCs. This is consistent with reports indicating that plasma hCAP levels are primarily generated by the bone marrow rather than cells in the circulation (12).
Figure 1.
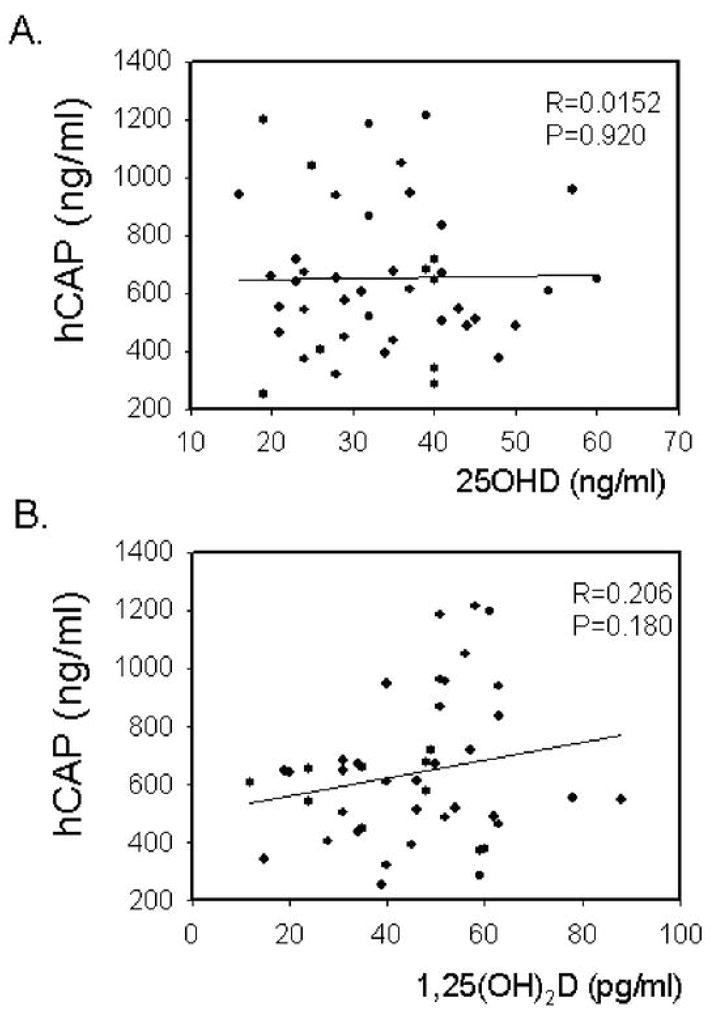
Serum hCAP levels do not correlate with serum vitamin D metabolites in normal human donors. A) Effect of donor serum 25OHD concentration (ng/ml) on serum concentration of hCAP (ng/ml). B) Effect of donor serum 1,25(OH)2D concentration (pg/ml) on serum concentration of hCAP (ng/ml). Correlation coefficient (R) and statistical significance (p value) are shown for each plot.
Figure 2.
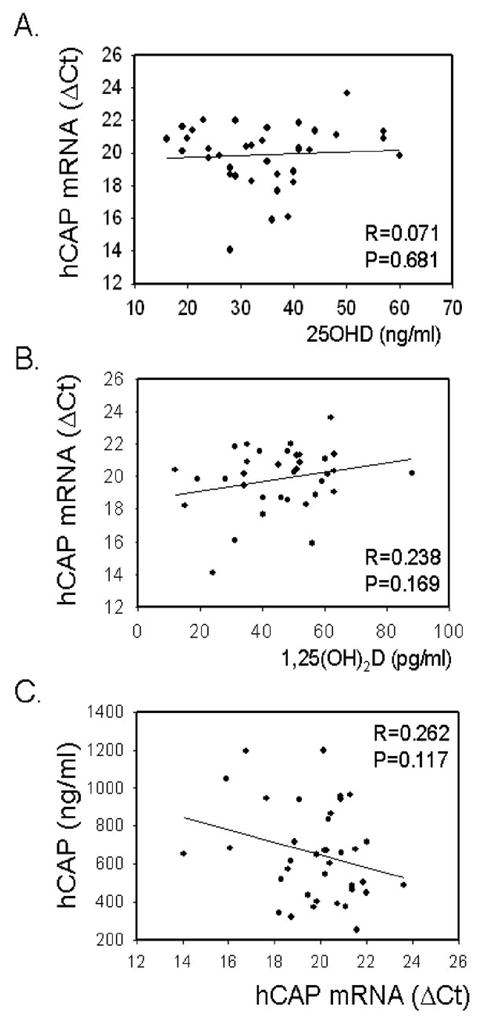
Leukocyte hCAP mRNA expression does not correlate with serum vitamin D metabolites or serum hCAP levels. A) Effect of donor serum 25OHD concentration (ng/ml) on levels of hCAP mRNA (dCt) in donor leukocytes. B) Effect of donor serum 1,25(OH)2D concentration (pg/ml) on levels of hCAP mRNA (dCt) in donor leukocytes. C) Relationship between donor leukocyte hCAP mRNA expression (dCt) and serum hCAP levels (ng/ml). Correlation coefficient (R) and statistical significance (p value) are shown for each plot.
Expression of CYP27b1 and VDR in ex vivo monocyte cultures
The absence of any significant relationship between serum vitamin D metabolites and circulating levels of hCAP supported our long-held hypothesis that immune responses to vitamin D occur primarily at a localized, cell-specific level. As such, further experiments were carried out using ex vivo culture of monocytes isolated from patient PBMCs and cultured for 24 hrs in medium supplemented with autologous serum (n=28). Under these conditions monocytes treated with the TLR2 ligand 19 kDa, showed significantly enhanced expression of CYP27b1 and VDR (Figure 3A and 3B). Similar induction of CYP27b1 was also observed following treatment with the TLR4 ligand LPS, indicating that at least one other TLR pathway is capable of activating vitamin D metabolism and function (Figure 3A). Expression of CYP27b1 and VDR was unaffected in monocytes cultured with supplementary 25OHD (Figure 3A and 3B). Analysis of 1,25(OH)2D levels in conditioned medium from cells treated with or without supplementary 25OHD showed that even in the absence of TLR induction, there was significant conversion of the pro-hormone to 1,25(OH)2D when 25OHD at a concentration of 100 nM was added to the macrophages (Figure 3C). These results indicate that there is a basal level of monocyte CYP27b1 activity that is dependent on the availability of substrate 25OHD alone, and which does not require TLR-mediated upregulation of the enzyme.
Figure 3.
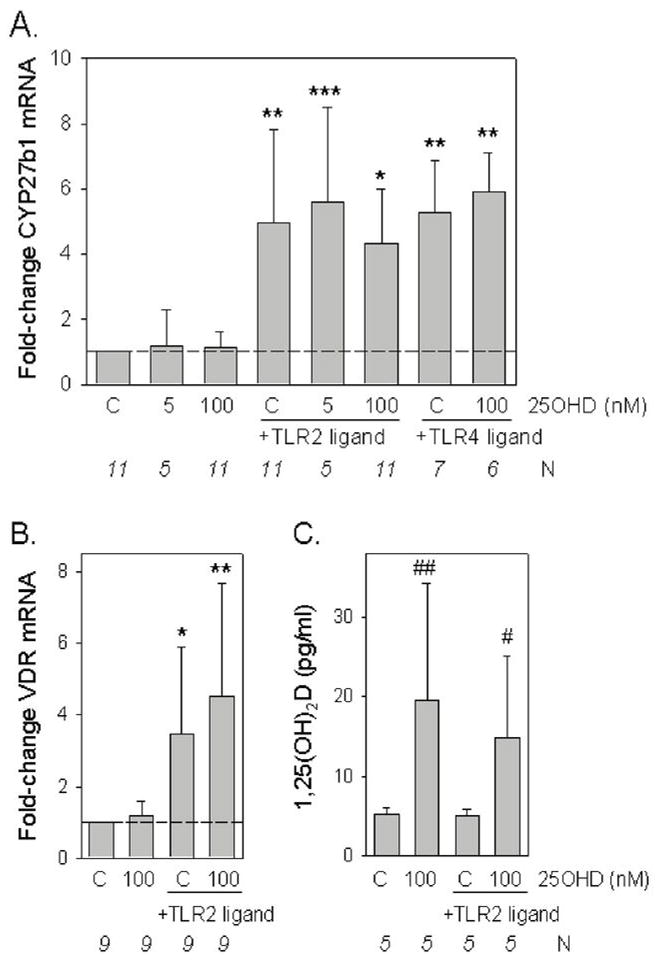
Induction of CYP27b1 expression and activity following TLR activation of monocytes cultured in autologous serum conditions. A. Changes in CYP27b1 expression following treatment with ligands to TLR2/1 (19 kDa lipopeptide, 1 ng/ml) or TLR4 (lipopolysaccharide, 100 ng/ml), in monocytes cultured in medium supplemented with autologous serum with or without added 25OHD. Data shown are the mean fold-change in CYP27b1 mRNA expression relative to vehicle-treated cells based on mean ± SD ΔCt values for each different. B) Changes in VDR expression following treatment with ligands to TLR2 in monocytes cultured in medium supplemented with autologous serum with or without added 25OHD. Data shown are the mean fold-change in VDR mRNA expression relative to vehicle-treated cells based on mean ± SD ΔCt values for each different treatment. C) Changes in monocytes synthesis of 1,25(OH)2D following treatment with ligand to TLR2, in the presence or absence of added 25OHD. * = mean ΔCT values statistically different to ΔCT value for vehicle-treated control monocytes, p<0.05. ** = mean ΔCT values statistically different to ΔCT value for vehicle-treated control monocytes, p<0.01. *** = mean dCT values statistically different to ΔCT value for vehicle-treated control monocytes, p<0.001. # = level of 1,25(OH)2D statistically different from TLR-treated control p<0.05. # = level of 1,25(OH)2D statistically different from vehicle-treated control p<0.01.
Vitamin D and ex vivo regulation of hCAP expression
Analysis of hCAP expression in the ex vivo monocyte cultures highlighted two key responses (Figure 4). Firstly, treatment with 25OHD enhanced expression of hCAP. At a concentration of 100 nM 25OHD stimulated hCAP expression in both the absence (12-fold, p<0.001 compared to vehicle) or presence of 19 kDa (8.4-fold, p<0.001 compared to vehicle) (Figure 4A). Similar observations were also made following combined treatment of monocytes with LPS and 100 nM 25OHD (Figure 4B). In the absence of any TLR-activation of CYP27b1, treatment with 5 nM 25OHD had no effect on monocyte hCAP expression. However, when used in combination with 19 kDa, 5 nM 25OHD significantly enhanced hCAP expression relative to cells treated with 19 kDa only (p<0.05) (Figure 4A). Similar induction of the VDR-target gene CYP24 was also observed in monocytes treated with 19 kDa and 5 nM 25OHD (see Supplement 1), suggesting an autocrine mechanism of action via localized induction of CYP27b1 expression and concomitant endogenous synthesis of 1,25(OH)2D. This was further endorsed by studies using intraconazole (ITRA, 5 μM) to inhibit CYP27b1 activity. In heterologous culture experiments using n=3 of the serum samples employed for autologous culture, but with a single population of donor monocytes, cells exposed to ITRA showed lower levels of hCAP expression when treated in combination with 19 kDa (4.3-fold, P<0.05 compared to 19 kDa only) or 19 kDa + 5 nM 25OHD (2.3-fold, p<0.05 compared to 19 kDa + 5 nM 25OHD only).
Figure 4.

Regulation of monocyte cathelicidin (hCAP) by TLR ligands and 25OHD. Changes in hCAP expression following treatment with ligands to: A) TLR2/1 (19 kDa lipopeptide, 19 kDa, 1 ng/ml) or B) TLR4 (lipopolysaccharide, 100 ng/ml) in monocytes cultured in medium supplemented with autologous serum with or without added 25OHD (5 or 100 nM). Data shown are the mean fold-change in hCAP mRNA expression relative to vehicle-treated cells based on mean ± SD ΔCt values for each different. *** = mean ΔCT values statistically different to ΔCT value for vehicle-treated control monocytes, p<0.001. # = mean ΔCT values statistically different to ΔCT value for TLR-treated control monocytes, p<0.05. ## = mean ΔCT values statistically different to ΔCT value for TLR-treated control macrophages, p<0.01. ### = mean dCT values statistically different to ΔCT value for TLR-treated control monocytes, p<0.001. Numbers shown in bars indicate mean fold-change values compared to vehicle-treated controls (C).
In previous studies we observed that monocytes cultured in 10% heterologous serum without added 25OHD exhibit decreased levels of mRNA for hCAP when treated with 19 kDa (5). It was therefore interesting to note that 19 kDa or LPS induced a similar suppression of hCAP mRNA in monocytes cultured in 10% autologous serum (Figure 4). This was in stark contrast to the antimicrobial defensin DEFB4 which was potently upregulated following activation of TLR2 or TLR4, but showed no response to treatment with 25OHD, even at concentrations of 100 nM (Figure 5). In view of the fact that the suppression of macrophage hCAP expression by TLR ligands was rescued in vitro by the addition of a relatively small amount of supplementary 25OHD (5 nM) (see Figure 4A), we reasoned that naturally occurring variations in the serum levels of this vitamin D metabolite may be a key determinant of macrophage production of hCAP following pathogen-sensing by TLRs. To test this postulate, we determined the magnitude of TRL2/1-regulated hCAP expression for each patient following treatment with 19 kDa (Figure 6). The resulting change in hCAP expression was then compared to the levels of 25OHD and 1,25(OH)2D in each 10% serum sample used for autologous culture of macrophages. Data in Figure 6A revealed a statistically significant correlation between serum 25OHD levels in 10% donor serum and hCAP mRNA in ex vivo cultures of donor monocytes following activation of TLR2. Although a similar trend was observed for serum 1,25(OH)2D, this was not statistically significant (Figure 6B).
Figure 5.
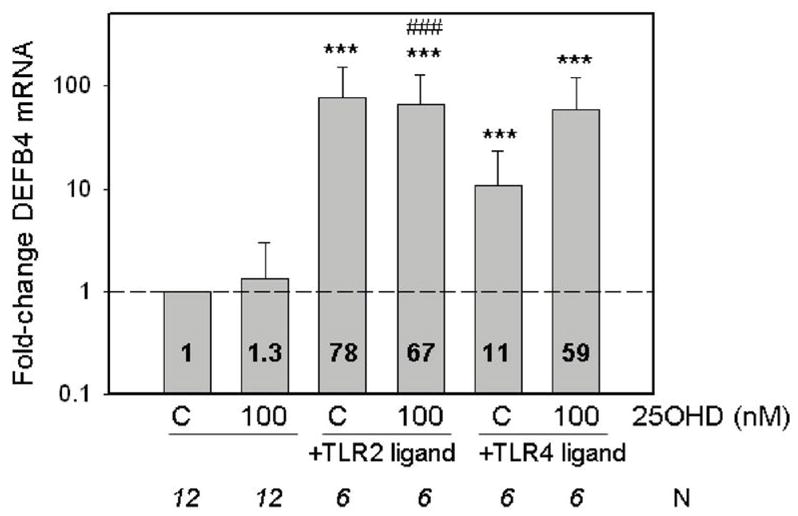
Monocyte DEFB is induced by TLR ligands but not 25OHD. Changes in DEFB4 expression following treatment with ligands to: TLR2/1 (19 kDa lipopeptide, 19 kDa, 1 ng/ml) or TLR4 (lipopolysaccharide, 100 ng/ml) in monocytes cultured in medium supplemented with autologous serum with or without added 25OHD. Data shown are the mean fold-change in DEFB mRNA expression relative to vehicle-treated cells based on mean ± SD ΔCt values for each different treatment. *** = mean ΔCT values statistically different to ΔCT value for vehicle-treated control macrophages, p<0.001. ### = mean ΔCt value statistically different from monocytes treated with 25OHD alone, p<0.001. Numbers shown in bars indicate mean fold-change values compared to vehicle-treated controls (C).
Figure 6.
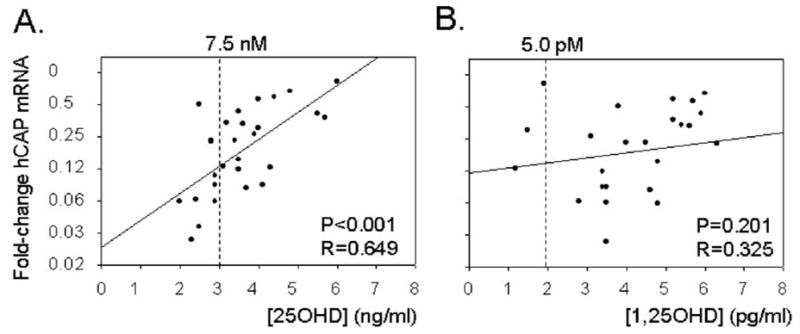
TLR2/1-induced cathelicidin (hCAP) in monocytes correlates with levels of serum 25OHD in ex vivo culture supplements. Change in levels of monocyte hCAP mRNA following activation of TLR2/1 by 19 kDa lipopeptide (19 kDa, 1 ng/ml) related to the concentration of: A) 25OHD; B) 1,25(OH)2D in 10% human serum used for autologous monocyte cultures. Concentrations of 25OHD [25OHD] and 1,25(OH)2D [1,25(OH)2D] are shown as ng/ml and pg/ml respectively on the lower x-axis, with a corresponding nM and pM reference point shown on the upper x-axis.
In vivo supplementation with vitamin D enhances monocyte production of hCAP ex vivo
As indicated in Figure 6A, approximately 40% of the patients recruited to the study presented with serum levels of 25OHD that were classified as vitamin D insufficient (< 75 nM or 30 ng/ml). When compared to the patients classified as vitamin D sufficient (> than 75 nM or 30 ng/ml), the deficient patients showed greater suppression of hCAP following TLR2/1-activation (12.5-fold decrease versus 3.5-fold decrease, p<0.05) (Figure 7A). As part of the study, these patients underwent a course of oral vitamin D therapy (50,000 IU vitamin D2 twice per week for 5 weeks), which significantly increased their serum 25OHD concentrations (24.7 ± 4.1 ng/ml to 40.3 ± 9.7 ng/ml, p<0.01) (Figure 7B). Notably, there was no change in the serum levels of 1,25(OH)2D in these patients following the course of vitamin D therapy (Figure 7B). After completion of the course of vitamin D supplementation, patient blood was again used to isolate monocytes for autologous serum cultures (n=7). Repeat analysis of mRNA expression in these cells following treatment showed that expression of hCAP was significantly higher less than when compared to mean data for this group of patients pre-vitamin D supplementation (p=0.033)(Figure 7C).
Figure 7.
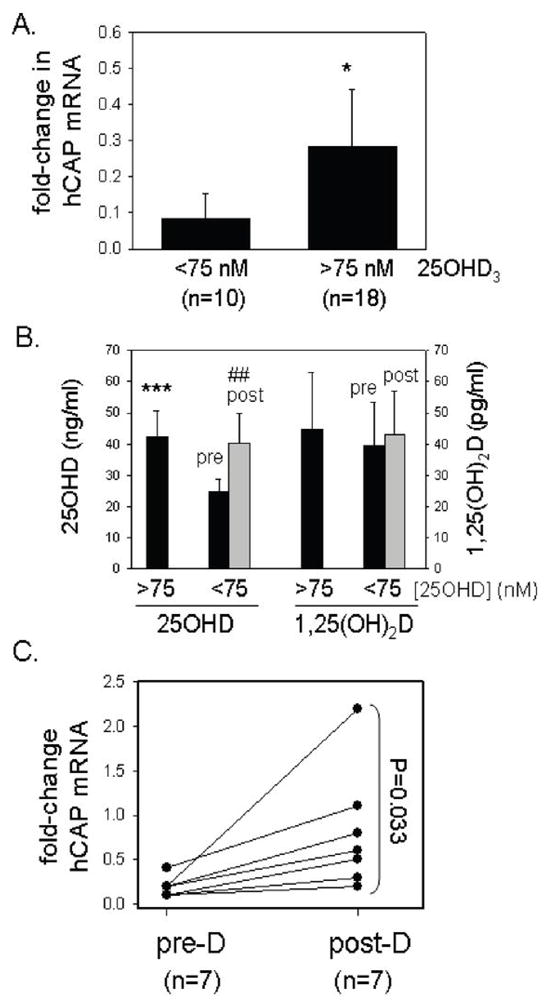
Vitamin D deficiency is associated with decreased expression of monocyte cathelicidin (hCAP) following TLR2/1 challenge. A) Expression of hCAP in ex vivo cultures of human monocytes following 24 hr treatment with the TLR2/1 ligand 19 kDa lipopeptide (1 ng/ml) under autologous serum culture conditions. Data are shown as the mean fold-change in hCAP expression (± SD) of TLR-activated monocytes compared to vehicle treated cells for subjects categorized as vitamin D-sufficient (serum 25OHD greater than 75 nM, n=18) or vitamin D-deficient (serum 25OHD less than 75 nM, n=10). B) Effect of supplementation with vitamin D on serum 25OHD and 1,25(OH)2D levels in the patients initially categorized as vitamin D-deficient (serum 25OHD less than 75 nM, n=10). C) Effect of vitamin D supplementation of patients (Post-D) initially categorized as vitamin D-deficient (Pre-D) on expression of hCAP in ex vivo cultures of human monocytes treated for 24 hrs with the TLR2/1 ligand 19 kDa lipopeptide under autologous serum culture conditions. Data are shown as the mean fold-change in hCAP expression (± SD) of TLR-activated monocytes compared to vehicle treated cells for vitamin D-insufficient subjects before (Pre-D) and after (Post-D) supplementation with vitamin D. *** = statistically different from values for the <75 nM 25OHD group, p<0.001. * = statistically different from values for the <75 nM 25OHD group, p<0.05. ## = statistically different from values for the pre-treatment <75 nM 25OHD group, p<0.01.
Discussion
In recent years, our perception of what constitutes normal vitamin D status has changed dramatically. Vitamin D sufficiency was originally defined as the minimum serum concentration of 25OHD that would prevent rickets in children and osteomalacia in adults, approximately 20 nM (8 ng/ml) (11). However, the observation that 25OHD concentrations as high as 80–100 nM (32–40 pg/ml) are inversely correlated with serum parathyroid hormone (17–19), and continue to enhance gastrointestinal calcium uptake (20), have led to a revision of these parameters. Specifically, current data suggest that serum 25OHD is biologically optimal above 75 nM (30 ng/ml), with concentrations between 20 and 75 nM constituting sub-optimal levels, or vitamin D ‘insufficiency (21). As a consequence of these new definitions, it has been estimated that as many as 1 billion people worldwide may be classified as vitamin D insufficient (11), with specific groups such as the elderly, children and pregnant mothers being particularly vulnerable. Individuals with dark skin pigmentation who are less able to generate vitamin D in the epidermis via photolytic modulation of 7-dehydrocholesterol also have a greater risk of vitamin D insufficiency, particularly those living in geographically less sunny areas (22).
The re-classification of optimal vitamin D status raises a key clinical question, namely what are the health consequences of vitamin insufficiency? In common with the rickets/osteomalacia that is characteristic of vitamin D deficiency, sub-optimal levels of 25OHD may also compromise skeletal homeostasis, with osteopenia or osteoporosis being the principal problem (21, 23). However, the well-documented non-classical effects of active 1,25(OH)2D suggest that vitamin D insufficiency will have a much broader clinical impact. In particular, expression of CYP27b1 and VDR by macrophages and dendritic cells has highlighted a potential role for intracrine synthesis of 1,25(OH)2D as a modulator of both innate and adaptive immunity (1–3). We have shown recently that intracrine conversion of 25OHD to 1,25(OH)2D by macrophages is potently stimulated by the TLR2/1 ligand 19 kDa, and this in turn induces expression of the antimicrobial peptide hCAP (5). Further studies have confirmed that this is a key requirement for host defense against pathogens such as Mycobacterium tuberculosis (M. tb) (24). Perhaps most significantly we also demonstrated that the efficacy of this innate immune mechanism is dependent on the availability of substrate for macrophage CYP27b1, namely the pro-hormone 25OHD. Specifically, ex vivo culture of monocytes using serum from 25OHD-sufficient (Caucasian) subjects supported levels of TLR-induced hCAP production that were much higher than observed with serum from 25OHD-deficient (African-American) populations (5). In data presented here we have expanded these observations to show that the ability of human macrophages to induce antimicrobial hCAP in response to TLR-activation is directly proportional to serum vitamin D status, with this being enhanced in vitamin D-insufficient patients treated with supplementary vitamin D.
Although 1,25(OH)2D is known to be a direct regulator of hCAP gene expression (25, 26), to our knowledge data in Figure 1 represent the first assessment of the impact of vitamin D metabolites on circulating levels of the antimicrobial peptide. The lack of association between circulating 25OHD, 1,25(OH)2D and hCAP levels emphasizes the importance of a local, intracrine mechanism of action. However, it should also be recognized that the patients studied in this trial were relatively vitamin D-replete, with 70% of those used in the ex vivo studies being classified as vitamin D sufficient and no patients technically vitamin D ‘deficient’. Thus, it is possible that circulating levels of hCAP will be more dependent on vitamin D status at very low levels of serum 25OHD. It was also interesting to note that serum levels of hCAP varied considerably independent of either vitamin D metabolite, suggesting alternative determinants of its background expression. The effect of vitamin D on serum hCAP may also be tissue specific: no association was found between serum hCAP protein and its mRNA from PBMCs (see Figure 2C), suggesting that other cell types contribute to the peripheral reservoir of this antimicrobial peptide (12).
In previous studies we showed that activation of TLR2 stimulated expression of CYP27b1 and VDR (5). Here we show that similar effects are also evident with the TLR4 ligand LPS indicating that induction of localized production of 1,25(OH)2D may occur in response to gram+VE or gram−VE bacteria (Figure 3). ELISA data suggest that, under these conditions, relatively low levels of 1,25(OH)2D are generated by human macrophages. Following the addition of 100 nM 25OHD, less than 50 pM 1,25(OH)2D was synthesized (see Figure 3C). Due to the detection limits of the assay kit employed, we were unable to quantify any significant changes in 1,25(OH)2D production using 5 nM 25OHD as substrate for CYP27b1, even when the enzyme was induced by TLR-activation. This underlines the relatively low levels of 1,25(OH)2D produced by this intracrine system but is also consistent with previous reports in which we demonstrated the relative efficiency of intracrine versus endocrine delivery of 1,25(OH)2D in regulating immune cell function (4). It was also interesting to note that 100 nM 25OHD facilitated the synthesis of 1,25(OH)2D without any need for TLR-mediate stimulus. This suggests that there is a low baseline expression of CYP27b1 in macrophages which is capable of synthesizing 1,25(OH)2D in the presence of high levels of substrate 25OHD, but which requires transcriptional activation via TLRs to effectively metabolize low levels of 25OHD. This proposal was endorsed by hCAP expression studies in Figure 4 which showed that in the absence of TLR ligands, 100 nM 25OHD was able to significantly induce expression of mRNA for the antimicrobial peptide. By contrast, 5 nM 25OHD only increased hCAP expression in conjunction with 19 kDa or LPS.
A key observation from this study is that in the absence of any exogenous 25OHD, treatment of macrophages with ligands to TLR2/1 or TLR4 suppressed expression of hCAP (see Figure 4). This is consistent with similar reports of hCAP inhibition in macrophages infected with M. tb (27), and other pathogenic agents (28, 29). As a consequence of these studies it has been proposed that suppression of antibacterial peptides such as hCAP provides a mechanism by which pathogens escape innate immune surveillance and thereby survive in the host (28). To the best of our knowledge, the data we present here are the first to show specific suppression of hCAP by purified TLR ligands. However, it is important to stress that the 10% culture autologous serum culture conditions employed in this study are effectively equivalent to vitamin D “deficiency” in that the levels of 25OHD to which cells are exposed ex vivo are less than 10 nM (see Figure 6). We postulate that in vivo regulation of hCAP by TLR ligands will be different to that observed in vitro because of higher concentrations of serum and concomitantly higher levels of 25OHD. Specifically, in individuals with higher levels of serum 25OHD it is likely that TLR ligands will enhance hCAP expression, whilst those with low serum 25OHD will have less induction or possibly even suppression of hCAP expression. It is noteworthy that addition of 25OHD at concentrations as low as 5 nM was able to rescue TLR-mediated suppression of hCAP in the autologous serum experiments. In cultures with 10% supplementary serum, this represents an effective concentration of 50 nM in undiluted (100%) serum, enough to change all the vitamin D-insufficient sera into vitamin D-sufficient sera. Similar effects were also observed in vitamin D-insufficient patients whose serum 25OHD levels were elevated in vivo from 25 ng/ml (62 nM) to 40 ng/ml (100 nM) (Figure 7). Collectively, these data emphasize the fundamental importance of 25OHD as a determinant of hCAP expression following TLR-mediate immune challenge. However, it is also important to recognize that supplementation of any culture medium with 10% serum effectively represents an environment which is 25OHD ‘insufficient’, and this may have implications for in vitro experimentation per se.
Vitamin D-mediated induction of hCAP appears to have been a relatively recent evolutionary development, as the gene promoter vitamin D response element (VDRE) required for liganded VDR stimulation of this protein is only present in higher primates (26). Consistent with this, comparison of macrophage cell lines cultured under identical conditions of vitamin D insufficiency (i.e. 10% FCS-supplemented medium) showed that 25OHD induced hCAP in human cells but had no effect in mouse macrophages (see Appendix 2). A potential explanation for this is provided by the fact that under these culture conditions, TLR ligands suppressed hCAP in human THP-1 cells but conversely stimulated this protein in mouse J774A macrophages. Both cell types showed induction of IL-1 expression following treatment with 19 kDa or LPS, indicating similar levels of sensitivity to the TLR ligands. Moreover, in autologous 10% serum cultured human macrophages and THP-1, cells TLR activation stimulated the related antimicrobial defensin DEFB4. Although the gene for DEFB4 also has a promoter VDRE (26), it did not show the same 25OHD-mediated induction observed for hCAP (Figure 5 and Figure 8A). Thus, we can speculate that the sensitive regulation of hCAP by vitamin D may have arisen in as a mechanism that countered microbial subversion of innate immune responses. The efficacy of such a mechanism is illustrated by fact that sub-human primates have greatly elevated circulating levels of 25OHD when compared to humans (30) and thus monocyte synthesis of 1,25(OH)2D in these animals is likely to be an effective way of stimulating hCAP following infection. By contrast, the migration of Homo sapiens out of Africa and into Europe was accompanied by a significant fall in serum 25OHD, and this may have substantially compromised hCAP innate immunity.
Several other factors factors are also likely to influence the regulation of monocyte hCAP expression following TLR activation including the localized concentration of TLR ligands themselves, and potential modulation by other factors such as cytokines. Moreover, the cohort of donors used in the study were relatively senior (mean age 63.3 ± 14.9), and this may be a significant factor in the defining the interrelationship between TLR-activation, hCAP and vitamin D. In similar studies carried out using monocytes from a younger donor cohort (mean age 33.2 ± 8.2), we observed the same correlation between serum 25OHD concentrations and hCAP mRNA following activation of TLR2/1 (see Appendix 3). However, in this instance suppression of hCAP by 19 kDa was less pronounced despite the fact that donor serum 25OHD levels in this younger cohort (21.1 ± 13.0) were lower than those reported in the current study (35.8 ± 10.2, p<0.001). Thus, it is possible that the ability of monocytes to promote hCAP antibacterial responses via 25OHD metabolism decreases with age, further underlining the need for maintenance of vitamin D status in elderly individuals. This is emphasized by analysis of the serum levels of 25OHD required to support optimal innate immunity in the ex vivo culture model. In the cohort of older subjects (see Figure 6) a 2-fold increase in monocyte hCAP levels would require serum 25OHD levels of approximately 200 nM (80 ng/ml). By contrast, monocytes from the younger cohort of donors in Appendix 3 would require 25OHD levels of only 100 nM (40 ng/ml). Irrespective of the potential effects of donor age, what is clear is that vitamin D functions as fundamental rheostatic regulator of macrophage hCAP levels following TLR-activation, with the magnitude of this effect being entirely dependent on the 25OHD status of the individual.
In data presented here we have confirmed the close association between vitamin D (25OHD) status and innate immunity. Consistent with our previous studies (5), we have shown that ability of physiological levels of serum 25OHD to induce hCAP is dependent on enhanced expression of macrophage CYP27b1, although at relatively high concentrations 25OHD can stimulate hCAP without TLR-mediated activation of macrophages. The upregulation of hCAP by 25OHD appears to counteract TLR-mediated suppression of the antimicrobial peptide, suggesting that a key function of vitamin D in this setting is to prevent pathogen-induced evasion of innate immunity. We have also shown for the first time that vitamin D supplementation in vivo is capable of promoting improved innate immune responses. As such, these data provide further support for prospective clinical trials to assess the effects of vitamin D supplementation in preventing infectious diseases such as tuberculosis.
Supplementary Material
Acknowledgments
This work was supported by NIH grant RO1AR050626 to M.H. and a Winnick Family Clinical Scholarship to M.H. PTL is supported by the Microbial Pathogenesis Training Grant 2-T32-AI-07323.
References
- 1.Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nature Clinical Practice Endocrinology and Metabolism. 2008;4:80–90. doi: 10.1038/ncpendmet0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams JS, Liu P, Chun R, Modlin RL, Hewison M. Vitamin D in Defense of the Human Immune Response. Ann N Y Acad Sci. 2007;1117:94–105. doi: 10.1196/annals.1402.036. [DOI] [PubMed] [Google Scholar]
- 3.Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med. 2007;13:117–124. doi: 10.1016/j.molmed.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Hewison M, Burke F, Evans KN, Lammas DA, Sansom DM, Liu P, Modlin RL, Adams JS. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Steroid Biochem Mol Biol. 2007;103:316–321. doi: 10.1016/j.jsbmb.2006.12.078. [DOI] [PubMed] [Google Scholar]
- 5.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 6.Hewison M, Freeman L, Hughes SV, Evans KN, Bland R, Eliopoulos AG, Kilby MD, Moss PA, Chakraverty R. Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol. 2003;170:5382–5390. doi: 10.4049/jimmunol.170.11.5382. [DOI] [PubMed] [Google Scholar]
- 7.Fritsche J, Mondal K, Ehrnsperger A, Andreesen R, Kreutz M. Regulation of 25-hydroxyvitamin D3-1 alpha-hydroxylase and production of 1 alpha,25-dihydroxyvitamin D3 by human dendritic cells. Blood. 2003;102:3314–3316. doi: 10.1182/blood-2002-11-3521. [DOI] [PubMed] [Google Scholar]
- 8.Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8:285–293. doi: 10.1038/ni1433. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin d3 on human B cell differentiation. J Immunol. 2007;179:1634–1647. doi: 10.4049/jimmunol.179.3.1634. [DOI] [PubMed] [Google Scholar]
- 10.Millen AE, Bodnar LM. Vitamin D assessment in population-based studies: a review of the issues. Am J Clin Nutr. 2008;87:1102S–1105S. doi: 10.1093/ajcn/87.4.1102S. [DOI] [PubMed] [Google Scholar]
- 11.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 12.Sorensen O, Cowland JB, Askaa J, Borregaard N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J Immunol Methods. 1997;206:53–59. doi: 10.1016/s0022-1759(97)00084-7. [DOI] [PubMed] [Google Scholar]
- 13.Wu S, Ren S, Nguyen L, Adams JS, Hewison M. Splice variants of the CYP27b1 gene and the regulation of 1,25-dihydroxyvitamin D3 production. Endocrinology. 2007;148:3410–3418. doi: 10.1210/en.2006-1388. [DOI] [PubMed] [Google Scholar]
- 14.Evans KN, Nguyen L, Chan J, Innes BA, Bulmer JN, Kilby MD, Hewison M. Effects of 25-Hydroxyvitamin D3 and 1,25-Dihydroxyvitamin D3 on Cytokine Production by Human Decidual Cells. Biol Reprod. 2006;6:816–822. doi: 10.1095/biolreprod.106.054056. [DOI] [PubMed] [Google Scholar]
- 15.Martineau AR, Wilkinson RJ, Wilkinson KA, Newton SM, Kampmann B, Hall BM, Packe GE, Davidson RN, Eldridge SM, Maunsell ZJ, Rainbow SJ, Berry JL, Griffiths CJ. A single dose of vitamin d enhances immunity to mycobacteria. Am J Respir Crit Care Med. 2007;176:208–213. doi: 10.1164/rccm.200701-007OC. [DOI] [PubMed] [Google Scholar]
- 16.Nursyam EW, Amin Z, Rumende CM. The effect of vitamin D as supplementary treatment in patients with moderately advanced pulmonary tuberculous lesion. Acta Med Indones. 2006;38:3–5. [PubMed] [Google Scholar]
- 17.Chapuy MC, Preziosi P, Maamer M, Arnaud S, Galan P, Hercberg S, Meunier PJ. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7:439–443. doi: 10.1007/s001980050030. [DOI] [PubMed] [Google Scholar]
- 18.Thomas MK, Lloyd-Jones DM, Thadhani RI, Shaw AC, Deraska DJ, Kitch BT, Vamvakas EC, Dick IM, Prince RL, Finkelstein JS. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–783. doi: 10.1056/NEJM199803193381201. [DOI] [PubMed] [Google Scholar]
- 19.Holick MF, Siris ES, Binkley N, Beard MK, Khan A, Katzer JT, Petruschke RA, Chen E, de Papp AE. Prevalence of Vitamin D inadequacy among postmenopausal North American women receiving osteoporosis therapy. J Clin Endocrinol Metab. 2005;90:3215–3224. doi: 10.1210/jc.2004-2364. [DOI] [PubMed] [Google Scholar]
- 20.Heaney RP, Dowell MS, Hale CA, Bendich A. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–146. doi: 10.1080/07315724.2003.10719287. [DOI] [PubMed] [Google Scholar]
- 21.Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int. 2005;16:713–716. doi: 10.1007/s00198-005-1867-7. [DOI] [PubMed] [Google Scholar]
- 22.Nesby-O’Dell S, Scanlon KS, Cogswell ME, Gillespie C, Hollis BW, Looker AC, Allen C, Doughertly C, Gunter EW, Bowman BA. Hypovitaminosis D prevalence and determinants among African American and white women of reproductive age: third National Health and Nutrition Examination Survey, 1988–1994. Am J Clin Nutr. 2002;76:187–192. doi: 10.1093/ajcn/76.1.187. [DOI] [PubMed] [Google Scholar]
- 23.Dawson-Hughes B, Bischoff-Ferrari HA. Therapy of osteoporosis with calcium and vitamin D. J Bone Miner Res. 2007;22(Suppl 2):V59–63. doi: 10.1359/jbmr.07s209. [DOI] [PubMed] [Google Scholar]
- 24.Liu PT, Stenger S, Tang DH, Modlin RL. Cutting Edge: Vitamin D-Mediated Human Antimicrobial Activity against Mycobacterium tuberculosis Is Dependent on the Induction of Cathelicidin. J Immunol. 2007;179:2060–2063. doi: 10.4049/jimmunol.179.4.2060. [DOI] [PubMed] [Google Scholar]
- 25.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173:2909–2912. doi: 10.4049/jimmunol.173.5.2909. [DOI] [PubMed] [Google Scholar]
- 26.Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. Faseb J. 2005;19:1067–1077. doi: 10.1096/fj.04-3284com. [DOI] [PubMed] [Google Scholar]
- 27.Martineau AR, Wilkinson KA, Newton SM, Floto RA, Norman AW, Skolimowska K, Davidson RN, Sorensen OE, Kampmann B, Griffiths CJ, Wilkinson RJ. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol. 2007;178:7190–7198. doi: 10.4049/jimmunol.178.11.7190. [DOI] [PubMed] [Google Scholar]
- 28.Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, Gudmundsson G. Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med. 2001;7:180–185. doi: 10.1038/84627. [DOI] [PubMed] [Google Scholar]
- 29.Chakraborty K, Ghosh S, Kole H, Mukhopadhyay AK, Ramamurthy T, Saha DR, Mukhopadhyay D, Roychowdhury S, Hamabata T, Takeda Y, Das S. Bacterial exotoxins downregulate cathelicidin (hCAP18/LL37) and human beta-defensin 1 (HBD-1) expression in the intestinal epithelial cells. Cell Microbiol. 2008;10:2520–2537. doi: 10.1111/j.1462-5822.2008.01227.x. [DOI] [PubMed] [Google Scholar]
- 30.Vieth R. What is the optimal vitamin D status for health? Prog Biophys Mol Biol. 2006;92:26–32. doi: 10.1016/j.pbiomolbio.2006.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.