

Abstract
Nitric oxide (NO) produced by NO synthase type 3 (NOS3) in medullary thick ascending limbs (mTHALs) inhibits Cl− reabsorption. Acutely, angiotensin II stimulates THAL NO production. In endothelial cells NO inhibits NOS3 expression. Therefore, we hypothesized that angiotensin II decreases NOS3 expression via NO in mTHALs. After 24 hrs, 10 and 100 nmol/L angiotensin II decreased NOS3 expression by 23 ± 9% (n=6, p<0.05) and 50 ± 5% (n=7, p<0.001) respectively, in primary cultures of rat mTHALs. NOS inhibition by 4 mmol/L NG-nitro-L-arginine methylester hydrochloride (L-NAME) prevented angiotensin II from decreasing NOS3 expression (Δ= −5 ± 8%, n=5). In the presence of L-NAME, addition of exogenous NO (1 μmol/L spermine NONOate) restored the angiotensin II-induced decreases in NOS3 expression (−22 ± 6%; n=7, p<0.013). In addition, NO scavenging with 10 μmol/L carboxy -PTIO abolished the effect of angiotensin II in NOS3 expression (Δ= −1 ± 8% vs carboxy-PTIO alone, n=6). Angiotensin II increases superoxide, and superoxide scavenges NO. Thus, we tested whether scavenging superoxide enhances the angiotensin II-induced reduction in NOS3 expression. Surprisingly, treatment with 100 μmol/L tempol, a superoxide dismutase mimetic, blocked the angiotensin II-induced decrease in NOS3 expression (Δ= −3 ± 7%, n=6). This effect was not due to increased hydrogen peroxide. We conclude that angiotensin II-induced decreases in NOS3 expression in mTHALs requires both NO and superoxide. Decreased NOS3 expression by angiotensin II in mTHALs could contribute to increased salt retention observed in angiotensin II-induced hypertension.
Keywords: reactive oxygen species, oxidative stress, hypertension, peroxynitrite, endothelial nitric oxide synthase
Introduction
Thick ascending limbs (THALs) reabsorb 20 to 30% of the filtered NaCl load.1 The importance of THALs in salt-volume regulation is evidenced by the efficiency of loop diuretics, which inhibit transport in this segment, to induce natriuresis.2 Nitric oxide (NO) produced by NO synthase type 3 (NOS3 or eNOS) acts as an autacoid to inhibit transport in this segment.3–5 NOS3 activity is regulated by both changes in expression6 and allosteric modulation.7,8 Thus, studying the factors that regulate NOS3 expression in THALs is of physiological relevance.
Angiotensin II (Ang II) regulates NOS3 expression in the kidney. Chronic Ang II infusion increases NO-dependent renal blood flow in the cortex9 as well as NOS3 expression.10–12 Conversely, in the medulla, Ang II decreases NO-dependent renal blood flow,9 but the effect on NOS3 expression is controversial. In 2-kidney, 1-clip (2K1C) hypertension, a model of elevated Ang II, NOS3 expression is reduced in the outer medulla.11 In contrast, Ang II infusion has been reported to increase10 and not to change11,12 NOS3 expression in the whole medulla. This discrepancy raises the likelihood that either: a) Ang II reduces NOS3 expression in some medullary structures while increasing it in others; or b) Ang II stimulates multiple pathways that have opposing effects on NOS3 expression, and therefore the final result changes depending on experimental conditions.
Acutely, Ang II enhances NO and superoxide (O2−) production in the kidney in general,13,14 and in the THAL specifically.15–17 In endothelial cells, NO reduces NOS3 activity18 and expression.19 In contrast, O2− not only decreases NO bioavailability in THALs,20 but also enhances NOS3 expression in endothelial cells.21 Since the renal medulla has the highest capacity for NO production in the kidney,22 we hypothesized that Ang II decreases NOS3 expression in medullary THALs (mTHALs) via NO, and that this reduction is partially mitigated by the Ang II-induced increase in O2−.
Methods
Primary cultures of medullary THALs
All protocols involving animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Henry Ford Hospital. The composition of physiological saline used was (in mmol/L) 130 NaCl, 2.5 NaH2PO4, 4 KCl, 1.2 MgSO4, 6 D/L-alanine, 1 trisodium citrate, 5.5 glucose, 2 calcium dilactate, and 10 HEPES. The solution was adjusted to 320 ± 3 mosmol/kgH2O with mannitol and was pH 7.4 at room temperature. Male Sprague-Dawley rats (Charles River Breeding Laboratories, Wilmington, MA) were maintained on a diet containing 0.4 % sodium and 1% potassium (Teklad Rodent Diet #8640, Harland-Teklad, Madison, WI). Rats weighing 200–250 g were anesthetized with ketamine and xylazine (100 and 20 mg/kg body wt ip, respectively). mTHALs primary culture were generated as previously described.6 Briefly, the abdominal cavity was opened, and the kidneys were flushed with 40 mL of ice-cold 0.1% collagenase (Sigma; St. Louis, MO) and 100 U heparin in physiological saline via retrograde perfusion of the aorta. Kidneys were removed, and coronal slices were cut. The inner stripe of the outer medulla was minced and digested in 0.1% collagenase at 37°C for 30 min. During each 5 min period, the tissue was gently agitated and gassed with 100% oxygen. Tissue was centrifuged at 60×g for 2 min; the pellet was re-suspended in cold physiological saline and stirred on ice for 30 min. The suspension was filtered through a 250 μm nylon mesh and centrifuged at 60×g for 2 min. The pellet was washed, centrifuged again, and re-suspended in 1 mL of DMEM/F-12 (Invitrogen; Eugene, Oregon) supplemented with 5% heat-inactivated fetal bovine serum (HyClone; Logan, UT), 100 U/mL penicillin and 100 μg/mL streptomycin (HyClone) and 20 ng/mL EGF (Invitrogen; Eugene, Oregon). Cells were plated on collagen-coated inserts (0.4-μm pore size, 4.7-cm2 area, Corning Costar, Cambridge, MA) at a concentration of 80 μg protein/insert and placed in anincubator at 37°C and 95% O2,5% CO2. Previously we found that 92 % of cells in primary cultures were THALs as evidenced by positive Tamm-Horsfall staining.6 After 40 hrs of seeding, cells were treated with either vehicle (DMEM/F-12 medium) or Ang II 0.1, 1, 10 or 100 nmol/L (Calbiochem; EMD, La Jolla, CA) for 24 hrs. In experiments involving NG-nitro-L-arginine methylester hydrochloride (L-NAME, a NOS inhibitor), tempol (a superoxide dismutase mimetic; Sigma St. Louis, MO) or Carboxy-PTIO (c-PTIO, a NO scavenger; Cayman Chemical Ann Arbor, Michigan), cells were preincubated for 1 hr with the reagent and then the medium was changed to one containing 100 nmol/L Ang II plus; 1) L-NAME, 2) tempol, 3) c-PTIO or 4) L-NAME + spermine NONOate (NO donor, Cayman Chemical Ann Arbor, Michigan) for 24 hrs. All treatments were done in the presence of 5% fetal bovine serum.
Western blot analysis
Cells were washed once with cold phosphate buffered saline pH 7.4 (EMD, La Jolla, CA) then scraped and lysed in 100 μL of buffer containing 20 mmol/L HEPES (pH 7.5), 2 mmol/L EDTA, 0.3 mol/L sucrose, 1.0% Igepal CA-630, 0.1% sodium dodecyl sulfate, 5 μg/mL antipain, 10 μg/mL aprotinin, 5 μg/mL leupeptin, 4 mmol/L benzamidine, 5 μg/mL chymostatin, 5 μg/mL pepstatin A, and 0.116 mol/L pf-block (Sigma; St Louis, MO). Debris was removed by centrifugation for 5 min at 5,600×g. Protein concentration was determined by a colorimetric assay (Coomassie Plus protein assay, Pierce, Rockford, IL). For each experiment, 30 μg of total fresh protein was loaded into each lane of an 8% SDS-polyacrylamide gel, separated by electrophoresis, and transferred to a PVDF membrane (Millipore, Bedford, MA). The membrane was incubated in blocking buffer containing 50 mmol/L Tris, 500 mmol/L NaCl, 0.1% Tween 20(TBS-T) and 5% nonfat dry milk for 60 min and then with a 1:1,000 dilution of an NOS3-specific monoclonal antibody (BD Transduction Laboratories, San Diego, CA) in blocking buffer for 60 min at room temperature. The membrane was washed with TBS-T and incubated with a 1:1,000 dilution of secondary antibody against mouse IgG conjugated to horseradish peroxidase (Amersham Pharmacia Biotech, Arlington Heights, IL). Reaction products were detected with a chemiluminescence’s kit (Pierce, Rockford, IL). The signal was detected by exposure to Fuji Super RX film which was scanned (EPSON expression 1680 scanner) and densitometry was performed with a custom program. The exposure times for the film were standardized to get bands of mean optical density between 0.40 and 1.
Statistical analysis
Results are expressed as percentage of control ± standard error. Data was analyzed by the Biostatistics and Research Epidemiology Department from Henry Ford Hospital with an analysis of slope for concentration dependent responses and paired t test. In some experiments ANOVA was used with post hoc testing. When multiple pair-wise comparisons were done, a procedure for multiple tests of significance was applied using Hochberg’s significance limits.23
Results
To test our hypothesis we first studied the effect of Ang II on NOS3 expression in primary cultures of mTHALs. Treatment of mTHALs with 10 and 100 nmol/L Ang II for 24 hrs decreased NOS3 expression by 23 ± 9% (n=6, p<0.05 vs control) and 50 ± 5% (n=7, p<0.001 vs control) respectively (Figure 1). Lower Ang II concentrations (0.1 and 1 nmol/L) did not significantly affect NOS3 expression (Δ= −7 ± 10 and Δ= −14 ± 22 vs control, respectively).
Figure 1. Effect of Ang II in NOS3 expression in mTHALs.

mTHAL primary cultures were incubated with 10 and 100 nmol/L Ang II for 24 hrs and NOS3 expression was assessed by Western blot. Top: Representative Western blot. Bottom: Cumulative data from 6 and 7 independent measurements for 10 and 100nmol/L, *=p<0.05, **=p<0.001 vs 0 nmol/L respectively.
Acutely, Ang II activates NOS in THALs 15 and NO negatively regulates NOS3 expression in endothelial cells19. Therefore, we next studied whether Ang II decreases NOS3 expression via NOS activation and NO production. First we tested whether NOS activation is required for the Ang II-induced reduction of NOS3 expression. In these experiments, 100 nmol/L Ang II alone reduced NOS3 expression by 27 ± 5% (n=5). In contrast, in the presence of L-NAME, a NOS inhibitor, Ang II had no significant effect on NOS3 expression (Δ= −5 ± 8% vs control, n=5; p<0.007 vs Ang II alone; Figure 2). L-NAME alone had no effect on basal NOS3 expression (Δ= −2 ± 8% vs control, n=5). These data indicate that NOS activity is required for Ang II to reduce NOS3 expression.
Figure 2. Effect of NOS inhibition on Ang II-induced decrease in NOS3 expression in mTHALs.
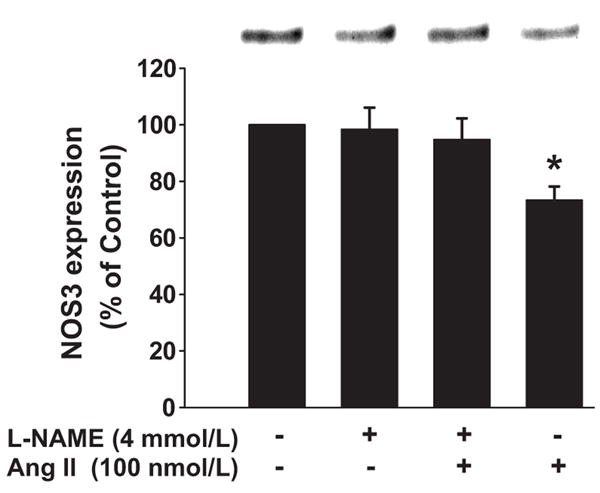
Top: Representative Western blot. Bottom: Cumulative data from 5 independent measurements; *=p<0.007 vs L-NAME+ Ang II.
Next, we tested whether NO per se was involved in the Ang II-induced inhibition of NOS3 expression, first by using a NO donor. When cells were treated with Ang II in the presence of L-NAME plus the NO donor spermine NONOate (1 μmol/L), NOS3 expression decreased by 22 ± 6% (n=7; p<0.013 vs L-NAME + NO donor; Figure 3). Similarly Ang II alone decreased expression by 28 ± 7% (n=7). L-NAME plus spermine NONOate did not have any effect on NOS3 expression in the absence of Ang II (Δ= 2 ± 10 %, n=7).
Figure 3. Effect of NOS inhibition and addition of exogenous NO on Ang II-induced decrease in NOS3 expression in mTHALs.
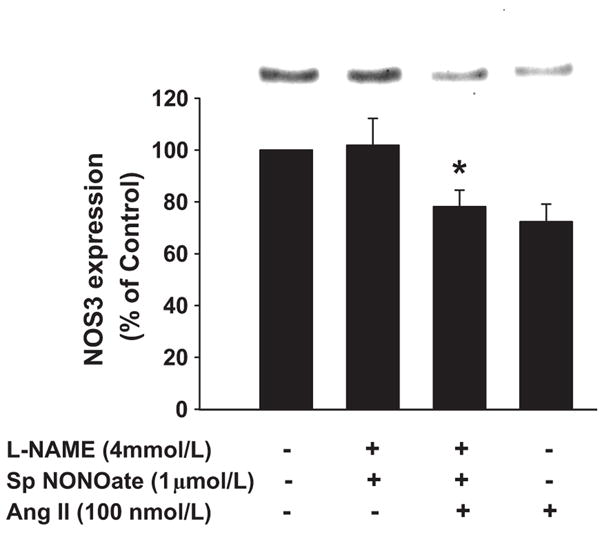
Top: Representative Western blot. Bottom: Cumulative data from 7 independent measurements; *=p<0.013 vs L-NAME + spermine NONOate.
Next, we used c-PTIO, a NO scavenger.24 In these experiments Ang II alone reduced NOS3 expression by 33 ± 4% (n=6). In contrast, in the presence of 10 μmol/L c-PTIO, Ang II had no significant effect on NOS3 expression (Δ= −1 ± 8%, n=6 vs c-PTIO alone; Figure 4). c-PTIO alone did not significantly change NOS3 expression (n=6). Taken together, these data indicate that NO is required for Ang II to reduce NOS3 expression.
Figure 4. Effect of NO scavenging on Ang II-induced decrease in NOS3 expression in mTHALs.
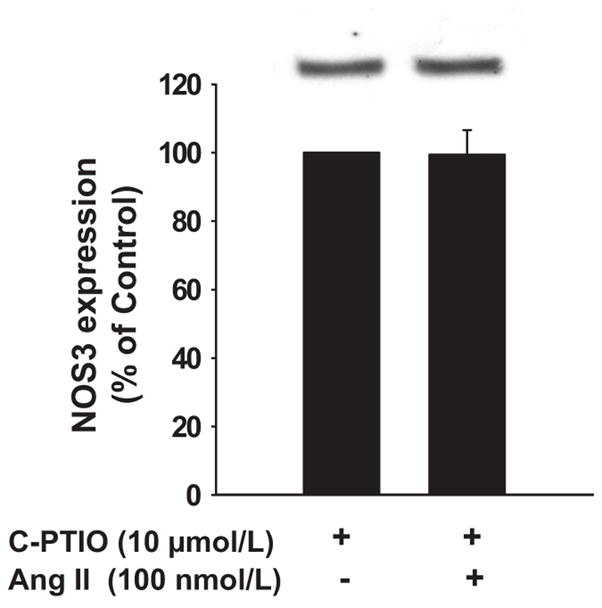
Top: Representative Western blot. Bottom: Cumulative data from 6 independent measurements, no significant difference.
Ang II not only increases THAL NO, but also O2− production,16,17 which scavenges NO.20 Consequently, we studied whether scavenging O2− with tempol25 exacerbated the effect of Ang II on NOS3 expression. Ang II alone reduced NOS3 expression by 29 ± 3% (n=6). Surprisingly, Ang II failed to decrease NOS3 expression in the presence of tempol (Δ= −3 ± 7%, n=6; p<0.015 vs Ang II). Tempol alone did not affect NOS3 expression (Δ= 6 ± 12%; n=6; Figure 5).
Figure 5. Effect of O2− dismutation on Ang II-induced decrease in NOS3 expression in mTHALs.

Top: Representative Western blot. Bottom: Cumulative data from 6 independent measurements; *=p<0.015 vs tempol + Ang II.
The tempol results could be due to either: 1) a reduction in O2− which is required for Ang II to decrease NOS3; or 2) the generation of hydrogen peroxide (H2O2) which has been reported to enhance NOS3 expression in endothelial cells.21 We reasoned that if H2O2 was masking the effect of NO in NOS3 expression then in the presence of tempol Ang II should increase NOS3 expression when NOS is inhibited. Thus, we incubated cells with or without Ang II in the presence and absence of both L-NAME and tempol. Ang II alone reduced NOS3 expression by 40 ± 6%. In contrast, Ang II had no effect on NOS3 expression in the presence of L-NAME and tempol, (Δ= 2 ± 7% vs L-NAME+ tempol). L-NAME and tempol did not affect basal NOS3 expression in the absence of Ang II. Taken together these data indicate that the ability of tempol to block Ang II-induced inhibition of NOS3 expression is due to a reduction in O2− rather than an increase in H2O2.
Discussion
Our hypothesis was that Ang II decreases NOS3 expression in mTHALs via enhanced NO production. We used primary cultures of mTHALs to avoid the confounding influences that changes in blood pressure,26 renal blood flow,27 tubuloglomerular feedback28,29 and transport in the proximal tubule30,31 caused by Ang II infusion may have on mTHAL NOS3 expression. We found that treatment of mTHAL primary cultures with 10 and 100 nmol/L Ang II for 24 hrs decreased NOS3 expression in a concentration dependent manner. To our knowledge this is the first time that the chronic effect of Ang II on NOS3 expression in mTHALs has been studied.
The literature concerning the effect of Ang II on renal medullary NOS3 expression is controversial. Decreases, no effect and increases have been reported. Wickman et al. found that in 2K1C-hypertension, a model of high circulating Ang II, NOS3 expression is decreased in the outer medulla, the location of mTHALs. This result was observed in both clipped and un-clipped kidneys after 42 days of clipping, suggesting that the effect was due to Ang II and not increased renal perfusion pressure.11 In contrast, 7 days of Ang II infusion at 600 ng.Kg−1.min−1 did not affect expression in the outer medulla.11 Infusion for 14 days at about 280 ng.Kg−1.min−1 produced similar results when whole medulla was studied.12 Finally, when infused at 200 ng.Kg−1.min−1 for 3 days Ang II increased NOS3 expression in the whole medulla.10
The apparent discrepancies may be explained by differences in experimental design including tissue studied, dose of Ang II used and time of treatment. The medulla is composed of THALs, thin descending and ascending limbs, outer and inner medullary collecting ducts, vasa recta and interstitial cells. Thus, the disparate results in these studies may simply be due to Ang II having different effects on the various cell types. Support for such a hypothesis comes from data showing that Ang II enhances NOS3 expression in the cortex,11,12 which is 80–90% proximal tubule cells, whereas our data show that it decreases NOS3 expression in mTHALs.
Differences in the dose of Ang II used may also account for the contrasting results. The concentration-response relationship between Ang II and NOS3 expression in the outer medulla may be complex. The effect of Ang II on ion transport32,33 and regulation of NOS3 expression by endothelin-16 in the THAL is biphasic. We found that 10 and 100 nmol/L Ang II, similar to concentrations measured in the kidney in vivo,34–36 inhibit NOS3 expression. We also found that 0.1 and 1 nmol/L had no effect, but did not test lower concentrations. It is possible that concentrations of Ang II lower than 0.1 nmol/L stimulate expression as it does transport.33
Finally, the effect of Ang II on NOS3 expression may be time dependent. In mTHALs, high salt diet increases NOS3 expression in a biphasic manner, peaking at 3 days and returning to basal levels at 14 and 28 days.37
In endothelial cells NO donors decrease NOS3 expression19 and acutely, Ang II increases NO production in the THAL.15 Thus, we next tested whether Ang II reduces NOS3 expression via an increase in NO through negative feedback in mTHALs. We found that the effect of Ang II on NOS3 expression was blocked by L-NAME. Thus, we studied whether NO per se was involved by using two additional approaches: 1) blocking NOS with L-NAME and adding NO back to see whether it restores the effect; and 2) scavenging NO with c-PTIO. Addition of the NO donor spermine NONOate to incubation media in the presence of L-NAME restored the effect of Ang II on NOS3 expression; furthermore, scavenging NO with c-PTIO blocked Ang II-induced decreases in NOS3 expression. Together these data indicate that NO is required for Ang II to inhibit NOS3 expression in mTHALs.
Although mTHALs express all NOS isoforms,38 the source of Ang II-induced NO in this nephron segment is likely to be NOS3 itself. We have data showing that acutely, Ang II increases NOS3 phosphorylation by activating Akt resulting in increased NO production (Herrera M and Garvin J.L, unpublished data, 2008). In addition, we have previously shown that neither NOS1 nor NOS2 are involved in the NO-induced decrease THAL Cl− absorption,5 and that enhanced THAL NO production by flow,39,40 clonidine41 and high salt diet37 are a result of NOS3 activation.
In addition to stimulating NO synthesis, Ang II augments oxidative stress in the THAL by increasing O2− production;16,17 O2− in turn, decreases NO bioavailability.20 Thus, we investigated whether O2− dismutation enhanced the decrease in NOS3 expression caused by Ang II. Contrary to what we expected, tempol, a O2− dismutase mimetic,25 abolished the effect of Ang II on NOS3 expression. These data suggest that O2− is not counteracting the effect of NO on NOS3 expression, but is instead required for the Ang II-induced inhibition. However, since O2− dismutation results in increased H2O2,42 and H2O2 enhances NOS3 expression in endothelial cells,21 the ability of tempol to block Ang II’s effect on NOS3 could also be explained by increased H2O2. To show that the effect of tempol was due to O2− dismutation rather than a parallel increase in NOS3 expression induced by H2O2, we treated cells with both L-NAME and tempol. If Ang II increased NOS3 expression in the presence of L-NAME and tempol, this would mean that increased H2O2 is responsible for the apparent blocking effect of tempol. On the other hand, if Ang II had no effect on NOS3 expression in these experiments, it would indicate that O2− is required for the Ang II-induced reduction in NOS3 expression. Simultaneous treatment of mTHAL cells with L-NAME, tempol and Ang II did not change NOS3 expression. Thus we conclude that O2− participates in Ang II-induced decreases in NOS3 expression.
Our results show that both NO and O2− are required for Ang II to inhibit NOS3 expression in mTHALs. O2− can react with NO to form peroxynitrite (ONOO−)43 suggesting that ONOO− may mediate the actions of Ang II on NOS3. Ang II has been shown to increase tyrosine nitration, a marker of ONOO−,44,45 in endothelial cells,46 proximal tubule47 and renal outer medulla tissue.48. In bovine aortic endothelial cells, ONOO− decreases NOS3 expression,49 whereas in diabetes an inverse relationship between NOS3 expression and tyrosine nitration has been delineated.50 Moreover, it has been shown that treatment of endothelial cells with LDL and oxidized LDL results in increased ONOO− production and decreased NOS3 expression.51,52 Thus, ONOO− may mediate the effects of Ang II on mTHAL NOS3 expression. However, additional experiments are needed to confirm the involvement of ONOO−.
The source of O2− responsible for Ang II-induced decreases in mTHAL NOS3 expression needs further study. However, the most likely sources are NADPH oxidase and NOS itself. Ang II increases O2− production via NADPH oxidase in the THAL.17 Additionally, Ang II induces NOS uncoupling,46,53 thereby increasing O2− production. Thus, it is possible that NOS itself could become the source of both the NO and O2− required for Ang II to inhibit NOS3 expression. Although we showed that L-NAME blocks the effects of Ang II, and L-NAME could block both NO and O2− production by NOS,53–55 addition of exogenous NO was sufficient to revert the blockade of Ang II-induced decreases in NOS3 caused by L-NAME. Thus, these data appear to rule out the possibility that uncoupled NOS is the source of O2−.
Perspectives
We conclude that Ang II decreases NOS3 expression in mTHALs via both NO and O2−, and thus raises the possibility that ONOO− is involved. NO produced by THAL NOS3 inhibits Cl− reabsorption in this nephron segment.5 Conversely, Ang II is a pleiotropic hormone that produces vasoconstriction56 and enhances NaCl reabsorption by the kidney.57,58 Decreased NOS3 expression induced by Ang II may be one of the mechanisms by which this hormone enhances sodium retention. Reduced NOS3 may result in decreased NO production and bioavailability. This in turn, may increase NaCl transport and diminish NO diffusion to the vasa recta, leading to impaired medullary blood flow, all of which have been shown to induce hypertension.59,60 However, whether the same response is observed in vivo needs further study. Understanding the uniqueness of the effect of Ang II in each nephron segment and discerning the pathways that lead to an imbalance between pro and anti-oxidant species would allow design of better drugs for the treatment of hypertension.
Acknowledgments
Sources of Funding
This work was supported by grants from National Institutes of Health (HL 028982, HL 070985, DK080255) to J.L. Garvin.
Footnotes
Disclosures
None
References
- 1.Greger R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 2.Brater DC. Disposition and response to bumetanide and furosemide. Am J Cardiol. 1986;57:20A–25A. doi: 10.1016/0002-9149(86)91002-7. [DOI] [PubMed] [Google Scholar]
- 3.Plato CF, Stoos BA, Wang D, Garvin JL. Endogenous nitric oxide inhibits chloride transport in the thick ascending limb. Am J Physiol Renal Physiol. 1999;276:F159–F163. doi: 10.1152/ajprenal.1999.276.1.F159. [DOI] [PubMed] [Google Scholar]
- 4.Ortiz PA, Garvin JL. NO Inhibits NaCl Absorption by Rat Thick Ascending Limb Through Activation of cGMP-Stimulated Phosphodiesterase. Hypertension. 2001;37:467–471. doi: 10.1161/01.hyp.37.2.467. [DOI] [PubMed] [Google Scholar]
- 5.Plato CF, Shesely EG, Garvin JL. eNOS mediates L-arginine-induced inhibition of thick ascending limb chloride flux. Hypertension. 2000;35:319–323. doi: 10.1161/01.hyp.35.1.319. [DOI] [PubMed] [Google Scholar]
- 6.Herrera M, Garvin JL. Endothelin stimulates endothelial nitric oxide synthase expression in the thick ascending limb. Am J Physiol Renal Physiol. 2004;287:F231–F235. doi: 10.1152/ajprenal.00413.2003. [DOI] [PubMed] [Google Scholar]
- 7.Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- 8.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 9.Chin SY, Wang CT, Majid DS, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol. 1998;274:F876–F882. doi: 10.1152/ajprenal.1998.274.5.F876. [DOI] [PubMed] [Google Scholar]
- 10.Moreno C, Lopez A, Llinas MT, Rodriguez F, Lopez-Farre A, Nava E, Salazar FJ. Changes in NOS activity and protein expression during acute and prolonged ANG II administration. Am J Physiol Regul Integr Comp Physiol. 2002;282:R31–R37. doi: 10.1152/ajpregu.2002.282.1.R31. [DOI] [PubMed] [Google Scholar]
- 11.Wickman A, Andersson IJ, Jia J, Hedin L, Bergstrom G. Endothelial nitric oxide synthase protein is reduced in the renal medulla of two-kidney, one-clip hypertensive rats. J Hypertens. 2001;19:1665–1673. doi: 10.1097/00004872-200109000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Chin SY, Pandey KN, Shi SJ, Kobori H, Moreno C, Navar LG. Increased activity and expression of Ca(2+)-dependent NOS in renal cortex of ANG II-infused hypertensive rats. Am J Physiol. 1999;277:F797–F804. doi: 10.1152/ajprenal.1999.277.5.F797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baylis C, Harvey J, Engels K. Acute nitric oxide blockade amplifies the renal vasoconstrictor actions of angiotension II. J Am Soc Nephrol. 1994;5:211–214. doi: 10.1681/ASN.V52211. [DOI] [PubMed] [Google Scholar]
- 14.Lopez B, Salom MG, Arregui B, Valero F, Fenoy FJ. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension. 2003;42:1150–1156. doi: 10.1161/01.HYP.0000101968.09376.79. [DOI] [PubMed] [Google Scholar]
- 15.Dickhout JG, Mori T, Cowley AW., Jr Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res. 2002;91:487–493. doi: 10.1161/01.res.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 16.Mori T, Cowley AW., Jr Angiotensin II-NAD(P)H oxidase-stimulated superoxide modifies tubulovascular nitric oxide cross-talk in renal outer medulla. Hypertension. 2003;42:588–593. doi: 10.1161/01.HYP.0000091821.39824.09. [DOI] [PubMed] [Google Scholar]
- 17.Li N, Yi FX, Spurrier JL, Bobrowitz CA, Zou AP. Production of superoxide through NADH oxidase in thick ascending limb of Henle’s loop in rat kidney. Am J Physiol Renal Physiol. 2002;282:F1111–F1119. doi: 10.1152/ajprenal.00218.2001. [DOI] [PubMed] [Google Scholar]
- 18.Buga GM, Griscavage JM, Rogers NE, Ignarro LJ. Negative feedback regulation of endothelial cell function by nitric oxide. Circ Res. 1993;73:808–812. doi: 10.1161/01.res.73.5.808. [DOI] [PubMed] [Google Scholar]
- 19.Vaziri ND, Wang XQ. cGMP-mediated negative-feedback regulation of endothelial nitric oxide synthase expression by nitric oxide. Hypertension. 1999;34:1237–1241. doi: 10.1161/01.hyp.34.6.1237. [DOI] [PubMed] [Google Scholar]
- 20.Ortiz PA, Garvin JL. Interaction of O(2)(−) and NO in the Thick Ascending Limb. Hypertension. 2002;39:591–596. doi: 10.1161/hy0202.103287. [DOI] [PubMed] [Google Scholar]
- 21.Zhen J, Lu H, Wang XQ, Vaziri ND, Zhou XJ. Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens. 2008;21:28–34. doi: 10.1038/ajh.2007.14. [DOI] [PubMed] [Google Scholar]
- 22.Moridani BA, Kline RL. Effect of endogenous L-arginine on the measurement of nitric oxide synthase activity in the rat kidney. Canadian Journal of Physiology & Pharmacology. 1996;74:1210–1214. [PubMed] [Google Scholar]
- 23.Hochberg Yosef. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- 24.Goldstein S, Russo A, Samuni A. Reactions of PTIO and carboxy-PTIO with *NO, *NO2, and O2-*. J Biol Chem. 2003;278:50949–50955. doi: 10.1074/jbc.M308317200. [DOI] [PubMed] [Google Scholar]
- 25.Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2-or as SOD mimics? J Biol Chem. 1996;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- 26.Young DB, Murray RH, Bengis RG, Markov AK. Experimental angiotensin II hypertension. Am J Physiol. 1980;239:H391–H398. doi: 10.1152/ajpheart.1980.239.3.H391. [DOI] [PubMed] [Google Scholar]
- 27.Mistry M, Muirhead EE, Yamaguchi Y, Nasjletti A. Renal function in rats with angiotensin II-salt-induced hypertension: effect of thromboxane synthesis inhibition and receptor blockade. J Hypertens. 1990;8:75–83. doi: 10.1097/00004872-199001000-00012. [DOI] [PubMed] [Google Scholar]
- 28.Braam B, Navar LG, Mitchell KD. Modulation of tubuloglomerular feedback by angiotensin II type 1 receptors during the development of Goldblatt hypertension. Hypertension. 1995;25:1232–1237. doi: 10.1161/01.hyp.25.6.1232. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Garvin JL, Carretero OA. Angiotensin II enhances tubuloglomerular feedback via luminal AT(1) receptors on the macula densa. Kidney Int. 2001;60:1851–1857. doi: 10.1046/j.1523-1755.2001.00999.x. [DOI] [PubMed] [Google Scholar]
- 30.Harris PJ, Young JA. Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Archiv -European Journal of Physiology. 1977;367:295–297. doi: 10.1007/BF00581370. [DOI] [PubMed] [Google Scholar]
- 31.Garvin JL. Angiotensin stimulates glucose and fluid absorption by rat proximal straight tubules. J Am Soc Nephrol. 1990;1:272–277. doi: 10.1681/ASN.V13272. [DOI] [PubMed] [Google Scholar]
- 32.Amlal H, LeGoff C, Vernimmen C, Soleimani M, Paillard M, Bichara M. ANG II controls Na(+)-K+(NH4+)-2Cl- cotransport via 20-HETE and PKC in medullary thick ascending limb. Am J Physiol. 1998;274:C1047–C1056. doi: 10.1152/ajpcell.1998.274.4.C1047. [DOI] [PubMed] [Google Scholar]
- 33.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol. 1998;274:F148–F155. doi: 10.1152/ajprenal.1998.274.1.F148. [DOI] [PubMed] [Google Scholar]
- 34.Wang CT, Navar LG, Mitchell KD. Proximal tubular fluid angiotensin II levels in angiotensin II-induced hypertensive rats. J Hypertens. 2003;21:353–360. doi: 10.1097/00004872-200302000-00027. [DOI] [PubMed] [Google Scholar]
- 35.Seikaly MG, Arant BS, Jr, Seney FD., Jr Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest. 1990;86:1352–1357. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navar LG, Lewis L, Hymel A, Braam B, Mitchell KD. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J Am Soc Nephrol. 1994;5:1153–1158. doi: 10.1681/ASN.V541153. [DOI] [PubMed] [Google Scholar]
- 37.Herrera M, Silva GB, Garvin JL. A high-salt diet dissociates NO synthase-3 expression and NO production by the thick ascending limb. Hypertension. 2006;47:95–101. doi: 10.1161/01.HYP.0000196274.78603.85. [DOI] [PubMed] [Google Scholar]
- 38.Shin SJ, Lai FJ, Wen JD, Lin SR, Hsieh MC, Hsiao PJ, Tsai JH. Increased nitric oxide synthase mRNA expression in the renal medulla of water-deprived rats. Kidney Int. 1999;56:2191–2202. doi: 10.1046/j.1523-1755.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 39.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. II. Role of PI3-kinase and Hsp90. Am J Physiol Renal Physiol. 2004;287:F281–F288. doi: 10.1152/ajprenal.00383.2003. [DOI] [PubMed] [Google Scholar]
- 40.Ortiz PA, Hong NJ, Garvin JL. Luminal flow induces eNOS activation and translocation in the rat thick ascending limb. Am J Physiol Renal Physiol. 2004;287:F274–F280. doi: 10.1152/ajprenal.00382.2003. [DOI] [PubMed] [Google Scholar]
- 41.Plato CF, Garvin JL. Alpha(2)-adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol. 2001;281:F679–F686. doi: 10.1152/ajprenal.2001.281.4.F679. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Pearlman A, Luo Z, Wilcox CS. Hydrogen peroxide mediates a transient vasorelaxation with tempol during oxidative stress. Am J Physiol Heart Circ Physiol. 2007;293:H2085–H2092. doi: 10.1152/ajpheart.00968.2006. [DOI] [PubMed] [Google Scholar]
- 43.Huie RE, Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 44.Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- 45.Whiteman M, Siau JL, Halliwell B. Lack of tyrosine nitration by hypochlorous acid in the presence of physiological concentrations of nitrite. Implications for the role of nitryl chloride in tyrosine nitration in vivo. J Biol Chem. 2003;278:8380–8384. doi: 10.1074/jbc.M211086200. [DOI] [PubMed] [Google Scholar]
- 46.Kase H, Hashikabe Y, Uchida K, Nakanishi N, Hattori Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J Hypertens. 2005;23:1375–1382. doi: 10.1097/01.hjh.0000173520.13976.7d. [DOI] [PubMed] [Google Scholar]
- 47.Zhang CI. Peroxynitrite and the regulation of Na(+), K(+)-ATPase activity by angiotensin II in the rat proximal tubule. Nitric Oxide. 2002;7:30–35. doi: 10.1016/s1089-8603(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 48.Guo W, Adachi T, Matsui R, Xu S, Jiang B, Zou MH, Kirber M, Lieberthal W, Cohen RA. Quantitative assessment of tyrosine nitration of manganese superoxide dismutase in angiotensin II-infused rat kidney. Am J Physiol Heart Circ Physiol. 2003;285:H1396–H1403. doi: 10.1152/ajpheart.00096.2003. [DOI] [PubMed] [Google Scholar]
- 49.El-Ramessy Azza B, Tawfik Huda E, Matragoon Suraporn, Ali Tayyeba K, Caldwell Ruth B, Caldwell Robert W. Peroxynitrite mediates diabetes-induced endothelial dysfunction by reducing eNOS expression: Possible role of Rho Kinase (ROCK) activation. Circulation. 2006;114:II_330–330. Abstract. [Google Scholar]
- 50.Nagareddy PR, Xia Z, MacLeod KM, McNeill JH. N-acetylcysteine prevents nitrosative stress-associated depression of blood pressure and heart rate in streptozotocin diabetic rats. J Cardiovasc Pharmacol. 2006;47:513–520. doi: 10.1097/01.fjc.0000211744.93701.25. [DOI] [PubMed] [Google Scholar]
- 51.Mason RP, Kubant R, Heeba G, Jacob RF, Day CA, Medlin YS, Funovics P, Malinski T. Synergistic effect of amlodipine and atorvastatin in reversing LDL-induced endothelial dysfunction. Pharm Res. 2008;25:1798–1806. doi: 10.1007/s11095-007-9491-1. [DOI] [PubMed] [Google Scholar]
- 52.Heeba G, Hassan MK, Khalifa M, Malinski T. Adverse balance of nitric oxide/peroxynitrite in the dysfunctional endothelium can be reversed by statins. J Cardiovasc Pharmacol. 2007;50:391–398. doi: 10.1097/FJC.0b013e31811f3fd0. [DOI] [PubMed] [Google Scholar]
- 53.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topal G, Brunet A, Millanvoye E, Boucher JL, Rendu F, Devynck MA, David-Dufilho M. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic Biol Med. 2004;36:1532–1541. doi: 10.1016/j.freeradbiomed.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 56.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 57.Hall JE, Guyton AC, Trippodo NC, Lohmeier TE, McCaa RE, Cowley AW., Jr Intrarenal control of electrolyte excretion by angiotensin II. Am J Physiol. 1977;232:F538–F544. doi: 10.1152/ajprenal.1977.232.6.F538. [DOI] [PubMed] [Google Scholar]
- 58.Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. [Review] [33 refs] J Am Soc Nephrol. 1999;10 (Suppl 12):S258–S265. [PubMed] [Google Scholar]
- 59.Granger JP, Alexander BT. Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol Scand. 2000;168:161–168. doi: 10.1046/j.1365-201x.2000.00655.x. [DOI] [PubMed] [Google Scholar]
- 60.Cowley AW., Jr Role of the renal medulla in volume and arterial pressure regulation. Am J Physiol. 1997;273:R1–15. doi: 10.1152/ajpregu.1997.273.1.R1. [DOI] [PubMed] [Google Scholar]