

Abstract
A major obstacle in human immunodeficiency virus type 1 (HIV-1) eradication is the ability of the virus to remain latent in a subpopulation of the cells it infects. Latently infected cells can escape the viral immune response and persist for long periods of time, despite the presence of successful highly active antiretroviral therapy (HAART). Given the appropriate stimulus, latently infected cells can reactivate and start producing infectious virions. The susceptibility of these cell populations to HIV-1, their life span, their proliferative capacity, and their ability to periodically produce infectious virus subsequent to alterations in cellular physiology and/or immunologic controls are critical issues which determine the contribution of these cells to viral persistence.
Memory CD4+ T cells due to the long life span, which may be several years, and their ability to reactivate upon encounter with their cognate antigen or other stimulation, are considered a critical reservoir for maintenance of latent HIV-1 proviral DNA. Cells of the monocyte-macrophage lineage, which originate in the bone marrow (BM), are of particular importance in HIV-1 persistence due to their ability to cross the blood-brain barrier (BBB) and spread HIV-1 infection in the immunoprivileged central nervous system (CNS). Hematopoietic progenitor cells (HPCs) are also a potential HIV-1 reservoir, as several studies have shown that CD34+ HPCs carrying proviral DNA can be found in vivo in a subpopulation of HIV-1-infected patients. The ability of HPCs to proliferate and potentially generate clonal populations of infected cells of the monocyte-macrophage lineage may be crucial in HIV-1 dissemination. The contribution of these and other cell populations in HIV-1 persistence, as well as the possible strategies to eliminate latently infected cells are critically examined in this review.
Keywords: HIV-1, latency, eradication, persistence, reservoir, reactivation
Introduction
Despite the initial vigorous immune response, HIV-1 manages to establish a chronic infection, which ultimately leads to severe immunodeficiency in the majority of the infected patients despite treatment with HAART. HIV-1 utilizes a plethora of strategies to evade the host immune response, including the establishment of a latent infection within a subpopulation of susceptible cells. HIV-1 infection of activated CD4+ T cells leads to pronounced production of viral RNAs and proteins, prompt assembly of virions, and eventually cell death due to the cytopathic effects of the virus. On the contrary, within certain cell populations, viral replication is arrested and these cells can survive for prolonged periods of time, presenting a major obstacle to the eradication of HIV-1. Based on the lack of active viral replication in latently infected cell populations, HAART is no longer effective with respect to reducing HIV-1 proviral DNA burden and the immune system, even an intact one, cannot always recognize and clear silently infected cells.
Mechanisms of Latency
Following HIV-1 binding and entry, the viral genome has to be reverse transcribed into DNA, transported into the nucleus and integrated into the host genome for the virus to be able to replicate [1]. Due to blocks in reverse transcription and nuclear transport of the pre-integration complex, infection of resting CD4+ T cells often results in the transient state of pre-integration latency. From this state, the virus may either surpass the cellular blocks and move to a productive infection [2] or in the majority of cases, become inactivated and be cleared from the cell. Interestingly, selective transcription of Tat and Nef has been shown from pre-integrated provirus in resting CD4+ T cells; these viral proteins may induce activation, ultimately leading to integration of the viral genome within these cells [3]. Pre-integration latency in resting CD4+ T cells is quantitatively dominant over post-integration latency; at any given time, the fraction of infected cells in the state of pre-integration latency is larger than the fraction containing latently integrated HIV-1 DNA [4]. However, due to the comparatively short half-life or labile nature of pre-integration latency its contribution to viral persistence is limited.
In post-integration latency, the RNA viral genome has been reverse transcribed and the generated viral DNA has translocated to the nucleus, where it has stably integrated into the host genome. The level of the viral gene transcription is low, and little or no virus is produced. A cell can remain in this state of latency for its natural life span as the lack of viral proteins protects it from the cytopathic effects of the viral infection while it remains undetected by the host immune system. A number of factors, possibly acting simultaneously, may contribute to the repression of viral gene transcription resulting in the establishment of HIV-1 post-integration latency. Limited transcription factor availability in resting cells may impose a block in viral replication, which can be relieved following exposure of the cell to appropriate stimuli [5]. Generally, upon differentiation and/or activation of a given host cell population, the availability of numerous transcription factors in the cell nucleus is greatly enhanced, influencing the transcription of host and viral genes (Fig. 1). The availability of transcription factors may be particularly crucial in determining viral gene expression in the presence of Tat variants that have been shown to be weak transactivators of the LTR. Tat variants have been identified that are conditionally dysfunctional and contribute to viral latency under certain conditions but are able to trans-activate the LTR following appropriate stimulation [6, 7]. Numerous studies have shown an increase in HIV-1 genome transcriptional activity following activation of resting T cells, and macrophages [8-11]. Co-infections have also been shown to lead to reactivation from latency [12, 13].
Fig. (1). Activation of integrated HIV-1 LTR.
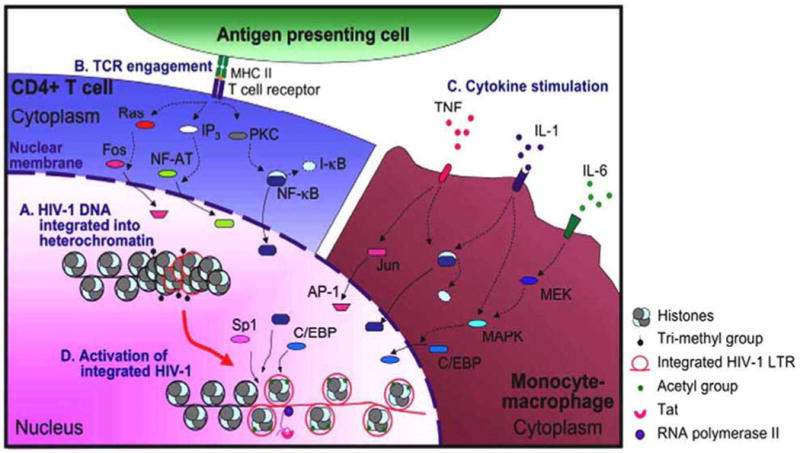
Integration of HIV-1 DNA into heterochromatin may suppress viral gene expression (A). Several stimuli, including TCR engagement in CD4+ T cells (B), and proinflammatory cytokine stimulation in monocyte-macrophages (C), induce signaling cascades that may ultimately lead to changes in the nuclear transcription factor milieu. Although available and induced transcription factors may differ between CD4+ T cells and cells of the monocyte-macrophage lineage, in both cases they facilitate histone methylation and acetylation that may promote conversion of heterochromatin to euchromatin. Cumulatively, these changes may allow viral gene expression and emergence from latency (D).
Integration of the viral genome into a repressive heterochromatin environment has been shown to favor viral latency [14]. However, conversion of euchromatin to heterochromatin is possible following chromatin modifications (Fig. 1). Trimethylation of histone H3 lysine 9 (H3 Lys9), which can be reversed, has been implicated in heterohromatin formation and transcriptional silencing of the integrated HIV-1 proviral genome [15-17]. Subsequent to HIV-1 integration, independent of the integration site, two nucleosomes (designated nuc-0 and nuc-1) are deposited at the viral promoter at precise locations with respect to cis-acting regulatory elements [18, 19]. Disruption or displacement of nuc-1 is a prerequisite for HIV-1 transcription [18]. Histones of nuc-0 and nuc-1 are deacetylated in HIV-1 latency, suggesting an important role for histone deacetylase (HDAC) in the HIV-1 latency. An important role in mediating HDAC1 recruitment to HIV-1 LTR has been shown for transcription factors Sp1, and c-Myc, leading in decreased LTR-driven viral gene expression [20]. Additionally, COUP-TF interacting protein 2 (CTIP2), C-promoter binding factor-1 (CBF-1), LSF, YY1, NF-κB p50-p50 homodimer, and thyroid hormone (T3) receptor have been shown to be involved in the recruitment of HDACs and facilitate the establishment of HIV-1 latency [16, 21-23]. Tat has been shown to interact and recruit histone acetyltransferases (HATs) p300/CBP, p300/CBP-associated factor (PCAF) and hGCN5, and is involved in reversing histone hypoacetylation. Tat-recruited HATs have been proposed to acetylate histones in LTR-proximal nucleosomes and as a result are able to induce LTR-driven transcription from an integrated chromatin microenvironment.
Interestingly, cellular microRNAs (miRNAs) have been proposed to inhibit histone acetylation and promote the establishment of HIV-1 latency. miRNAs are small single-stranded noncoding RNAs that participate in regulation of gene expression and innate host cell immunity against viruses, by binding to the target messenger RNAs (mRNA) and then degrading the mRNA or inhibiting mRNA translation [24]. It has been shown that cellular microRNAs (miRNAs) may inhibit HIV-1 gene expression by decreasing the expression of PCAF, and interfering with histone acetylation [25]. Additionally, miRNAs have been shown to participate in repressing HIV-1 gene expression by directly targeting the HIV-1 messenger RNA (mRNA). Particularly, five cellular miRNAs able to target the 3′ end of HIV-1 mRNAs have been shown to inhibit HIV-1 gene expression in resting CD4+ T cells. The five miRNAs have been shown to be upregulated in resting CD4+ T cells compared to activated CD4+ T cells [26, 27], further linking them to latency.
Cellular HIV-1 Reservoirs
Resting CD4+ T Cells
The pool of latently infected CD4+ T cells is established early in the course of the disease, during primary infection [28]. The fraction of resting CD4+ T cells harboring integrated HIV-1 genome during asymptomatic infection has been estimated to be in the order of <0.05%, which is two orders of magnitude less than the productively infected activated CD4+ T cells, with the total number of latently infected CD4+ T cells estimated to be around 105-107 cells per patient [4]. Despite the small size of this infected cell compartment, this population is considered to be a major obstacle to HIV-1 eradication and may allow viral persistence despite the presence of vigorous immune responses and HAART.
Two populations of resting CD4+ T cells exist, the naïve and the memory CD4+ T cells. Infection of both of these populations is typically present in HIV-1-infected patients [29]. Naïve CD4+ T cells and the majority of memory CD4+ T cells are CCR5 negative, while, similarly to activated CD4+ T cells, they have a high level expression of CXCR4. Although X4-tropic viruses can readily infect resting CD4+ T cells, these strains of virus typically emerge only late in the course of the disease, and it has been shown that early in disease most HIV-1-infected resting CD4+ T cells harbor R5-tropic provirus [30]. Limited or no CCR5 expression is not the sole barrier that HIV-1 encounters when infecting resting CD4+ T cells, as following viral entry into resting CD4+ T cells, blocks in viral reverse transcription and nuclear import are normally present in quiescent T cells [31]. For these reasons, the presence of latently infected CD4+ T cells, which are consistently present in all HIV-1-infected patients, has been rather puzzling. It has been hypothesized that the memory CD4+ T cells, which carry HIV-1 provirus, had been infected while they were in an activated state and survived long enough to move into a resting memory state. This process, although possible, might not be efficient since activated CD4+ T cells, shortly after infection with HIV-1, typically succumb to the cytopathic effects of the virus, or to the cytolytic effects of the host immune response. A more likely explanation suggests that for an infected CD4+ T cell to become a memory cell, infection should occur when the cell is already in the process of reverting to the resting state; during that short time frame the cell might still be permissive for the early steps in the virus life cycle but unable to support fully productive viral replication [32].
Alternatively, some evidence indicates that a CD4+ T cell may be permissive to HIV-1 infection without being activated [33]. It has been proposed that naïve CCR5− CD4+ T cells may in fact have a very low level expression of CCR5 that can not be detected by standard methods of flow cytometry. Consequently, their infection by CCR5-utilizing viruses may be possible, as long as expression of CD4 is relatively high [34]. Additionally, it has been demonstrated that the microenvironment of human lymphoid tissues may be sufficient to support infection of naïve CD4+ T cells by HIV-1 [35] (Fig. 2). Although the mechanism of this induced susceptibility is unknown, it is likely that interaction of the CD4+ T cell with their autologous B cells or dendritic cells (DCs) might be a contributing factor [36, 37]. Interestingly, Nef-mediated signals from HIV-1-infected macrophages have been shown to promote HIV-1 infection of resting CD4+ T cells [38]. The activation status of CD4+ T cells appears to be a key factor in determining their susceptibility to infection [39]; however, subtle activating signals may be able to render a cell susceptible to HIV-1 infection, without necessarily inducing any detectable changes towards an activated phenotype. There is evidence that stimulation with cytokines can be sufficient for increased susceptibility of resting CD4+ T cells to HIV-1 infection; interleukin (IL)-2, IL-4, IL-7, IL-15 and to a lesser extent IL-6, all have been shown to have such an effect [40]. Similarly, chemokines CCL19 and CCL21 have been shown to promote latent infection of resting CD4+ T cells without inducing any detectable activation markers [41].
Fig. (2). Cellular HIV-1 reservoirs in the lymph nodes.
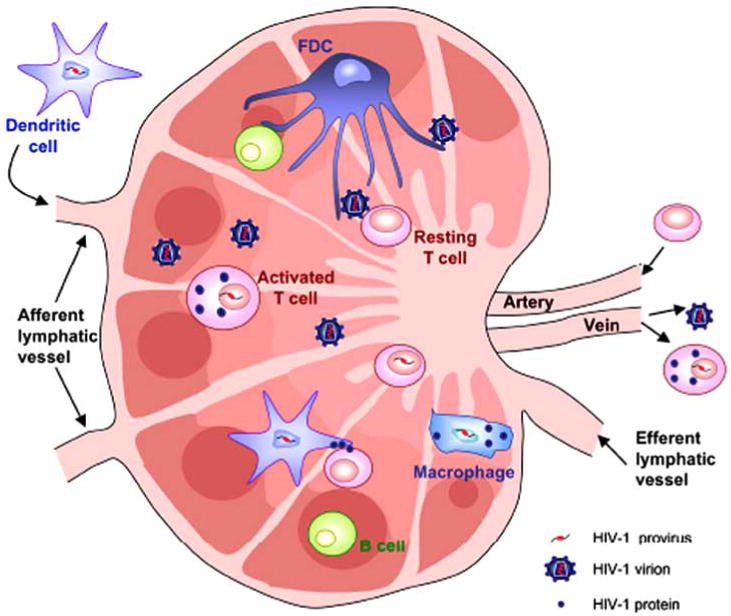
Lymph nodes are a site of pronounced viral production. The majority of virions are being produced by activated T cells but DCs and resting T cells may also be infected. Interaction of resting T cells with their autologous DCs or B cells, or with follicular dendritic cells may render them permissive to HIV-1 infection.
The presence of provirus in a resting CD4+ T cell is typically associated with latency; nevertheless, there are a number of reports suggesting that some of these cells may actively produce virus, especially in viremic patients [42]. In a study by Zhang et al. it was shown that HIV-1 and SIV could be propagated from both resting and activated CD4+ T cells. The activated T cells were responsible for the production of a bulk of the virus while the resting CD4+ T cells were producing less virus but were able to persist after retroviral therapy [43]. In vitro studies further support the continuous replication within the resting memory CD4+ T population, since it was shown that the CD4+ T cells, which were infected while being activated, continue to produce small amounts of virus persistently after reverting back to a resting state and never establish a truly silent proviral latency [44].
Memory T cells are the ideal reservoir for long-term maintenance of a viral genome due to their long life span. In addition, because they are quiescent cells not undergoing cell division or going through differentiation or activation processes and are only maintaining basic cellular processes for survival and cellular functions that will enable the cell to respond to appropriate immunologic stimuli their transcriptional machinery is set in a mode that greatly favors viral latency with minimal capability of supporting basal viral transcription. The limited viral production that may take place within these cells may be responsible for reseeding the pool of productively infected cells, while it may be insufficient to trigger the cytopathic effects typically observed in HIV-1-activated CD4+ T cells. However, under activating conditions these cells are capable of vigorous production of virus. The contribution of the naïve CD4+ T cell population in the dissemination of the virus and the establishment of latency is less clear. Their relative short life span, unless activated, is certainly not consistent with what one would expect for a cell functioning as a long-term reservoir of HIV-1 proviral DNA. However, given that the majority of T cells in a healthy individual are in the naïve state, the fact that they can be infected with HIV-1 is of crucial importance despite their short average life span.
Monocytes and Macrophages
The presence of latent proviral HIV-1 DNA in the memory CD4+ T cell population has been undoubtedly proven; and given the long life span of these cells, many investigators have supported the notion that this may be the only physiologically important, long-term, cellular reservoir for HIV-1 [45]. However, Chun et al. have shown that latent HIV-1 proviral DNA within the resting CD4+ T cell compartment is not the only and potentially not the major source of rebounding virus after cessation of HAART. This was revealed by comparing replication competent HIV-1 from resting CD4+ T cells to virus that re-emerged promptly after discontinuation of therapy, indicating the presence of viral reservoirs within cells other than the resting CD4+ T cells [46].
A number of features make the cells of the monocyte-macrophage lineage a potential HIV-1 reservoir. Monocytes circulate in the blood for up to 3 days and then migrate to various tissues where they differentiate into macrophages. Depending on the tissue, the life span of these cells can range from a few days up to several months. Contrary to what happens to activated CD4+ T cells, HIV-1 infection does not impact monocyte-macrophage viability to the same extent [47-49]. In contrast, viral replication is more persistent in nature, which may lead to a continuous low-level virus production mode lasting for the natural life-span of the cells. Limited expression of viral proteins may have a profound physiologic significance, enabling these cells to escape the immune response.
Initiation of HAART in patients is accompanied by a 99% decrease in plasma viremia within two weeks, attributed to the rapid elimination of cell free virus and clearance of the productively infected CD4+ T cells. A second phase of decay follows and has been attributed to longer-lived cells, presumably cells of the monocyte-macrophage lineage, with a half-life of 1-4 weeks [50]. Activation of latently infected lymphocytes and release of sequestered virions could also contribute to this phase of decay. The size of the infected monocyte-macrophage pool is estimated to be rather small compared to the infected T cell pool. It is estimated that HIV-1 proviral DNA is present in less than 1% of monocytes [51], while in a later study it was shown that only ∼50 in 106 lymph node macrophages were infected [4]. The limited extent of infection is attributed to the low CD4 surface expression which leads to reduced viral binding [48], but also to barriers in reverse transcription [52] and nuclear import leading to abortive infection [53] of this cell population. However, cells of the monocyte-macrophage lineage may, in part, carry a wide range of HIV-1 strains [54] and consist of up to 10% of all productively infected cells in early stages of disease, and may increase significantly at late stages of disease when CD4+ T cells are depleted [55].
Studies conducted when only AZT therapy was available indicated that monocytes harbor latent HIV-1 proviral DNA in all stages of the disease and in spite of the presence of antiviral therapy [51]. Later studies further confirmed this observation, by demonstrating that infectious HIV-1 could be detected in circulating monocytes from patients on HAART for prolonged periods of time [56, 57]. Importantly, these monocytes produced undetectable amounts of HIV-1 RNA under basal conditions, but the virus could reactivate following appropriate stimulation [56]. Reactivation in macrophages has been shown in vivo in response to opportunistic infections. Common pathogens such as Mycobacterium avium and Pneumocystis carinii have been shown to induce virus production from macrophages in lymph nodes, further highlighting the role of macrophages as an HIV-1 proviral DNA reservoir [12]. Subsequent studies revealed more cases of HIV-1 reactivation in macrophages following infection with other pathogens, such as human herpesvirus-8 (HHV-8) [58]. Recently, HIV-1 reactivation in promonocytic cell lines has been shown following cell to cell contact with CD4+ T cells [59].
Another key feature of macrophages that enables them to act as an HIV-1 reservoir has been their role in spreading the virus to CD4+ T cells. Monocyte-derived macrophages infected with HIV-1 have been shown to fuse with autologous and heterologous CD4+ T cells and thus transmit the virus [60, 61]. Furthermore, Nef-expressing macrophages have been demonstrated to facilitate resting T cell infection in a paracrine fashion, therefore expanding the HIV-1 reservoir by promoting latent infection of cells [38]. The ability of infected macrophages to recruit and activate lymphocytes, thus contributing to the dissemination of the virus, has also been shown [62], while the role of Nef in this process is crucial [63].
The role of cells of the monocyte-macrophage lineage as an HIV-1 reservoir has been further complicated by their resistance to protease inhibitors (PIs). PIs inactivate the virus-encoded aspartyl protease, resulting in inhibition of Gag and Pol processing which has been shown to occur during the late stages of HIV-1 replication [64]. As a result, PIs inhibit the formation of new virions from cells already infected with HIV-1. In this respect, they differ from the reverse transcriptase inhibitors, which act before the integration of the virus to the host genome and hinder new infections [65]. Reverse transcriptase inhibitors have no effect on latently infected cells, in which HIV-1 already has been reversed transcribed and integrated into the genome. In a comparative study, the activity of a number of PIs on chronically infected cells of the monocyte-macrophage lineage was found to be several fold lower than in chronically infected lymphocytes [66]. It has been suggested that the resistance of cells of the monocyte-macrophage lineage to PIs is due to their expression of P-glycoprotein transporters, which limits drug availability [67]. In this regard, the activity of PIs in chronically infected moncytic cells was found to be 7- to 26-fold less compared to acutely infected cells of the monocyte-macrophage lineage, indicating that the drug may be mostly ineffective on the longer-lived monocyte-macrophage reservoir [66].
Resident Macrophages of the CNS
Four types of macrophages are found in the CNS, the perivascular macrophages, the meningeal macrophages, the macrophages of the choroid-plexus, and the microglia [68]. These cells all belong to the monocyte-macrophage lineage and are the major phagocytic and antigen presenting cells of the CNS. Just as other tissue macrophages, they can be infected by HIV-1 and HIV-2, and by SIV in non-human primate models; their infection is CD4 dependent and typically involves CCR5, although CXCR4 and CCR3 have also been implicated [69, 70].
Perivascular macrophages and microglia are the major HIV-1 producing cells in the CNS (Fig. 3) and their infection and activation is heavily involved in the pathogenesis of HIV-1-associated dementia (HAD) [68, 71]. However, some controversy exists over their relative contribution to disease, as these cells cannot be easily distinguished from each other. A number of surface markers have been proposed for their identification; CD14 and CD45 have been reported as markers present on macrophages and not on microglia [72], however after activation microglia appear to upregulate these molecules [73]. Localization within the brain has been used as a surrogate marker for their identification. Perivascular macrophages are found in the parenchymal side of the blood-brain barrier surrounding the blood vessels while microglia (also referred to as parenchymal microglia) are found dispersed throughout the brain. Meningeal macrophages are located in the meninges, while choroids plexus macrophages are present at the interface between blood and the cerebrospinal fluid; considerably less information is available on the contribution of these cells to the viral load of the CNS and the pathogenesis of HAD [74-77].
Fig. (3). Cellular HIV-1 reservoirs in the CNS.
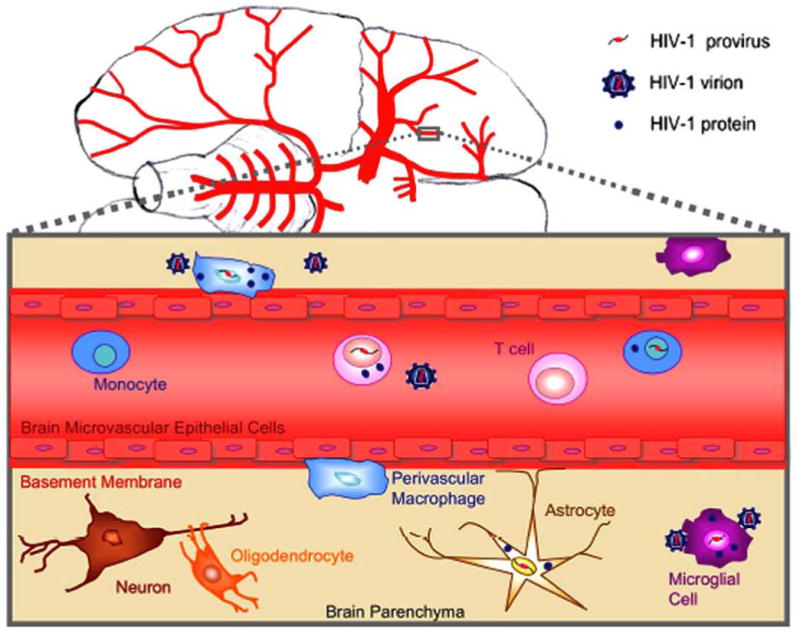
Perivascular macrophages and microglia are the major HIV-1 producing cells in the CNS. HIV-1 infection of astrocytes is considered to be non-productive.
The turnover rate of each of the macrophage and microglial cell populations of the CNS varies and their role in the spread and persistence of the virus depends upon it. Meningeal macrophages have a short turnover rate and are rapidly replaced by monocytes originating from the bone marrow. Perivascular macrophages and macrophages of the choroid-plexus are also replaced by cells of bone marrow origin, but at a slower rate [78, 79]. Repopulation of perivascular macrophages is thought to be the mechanism primarily responsible for the dissemination of HIV-1 from the peripheral blood to the CNS, a hypothesis often referred to as the “Trojan horse” mechanism. The opposite process is also assumed possible; perivascular macrophages are able to sample antigens and leave the CNS, potentially harboring HIV, and travel from the perivascular space to the cerebrospinal fluid, and subsequently to the subarachnoid space, to end up in the draining lymph nodes [80, 81]. Trafficking of perivascular macrophages back into the lymph nodes could be involved in reseeding of the periphery with viruses that have been residing in the CNS.
On the other hand, microglia persist for much longer periods of time and are able to proliferate in situ [82], therefore their role as viral reservoirs may be crucial. Limited repopulation of the microglial compartment by cells originating from the bone marrow has been shown in rodents and in humans, suggesting that, under physiologic conditions, microglial cells can survive throughout the normal life span of humans [78, 83]. It is believed that HIV-1 infection of the microglial cell compartment can take place during acute HIV-1 infection when the viral load is high, but replication of the virus is limited by the host immune response and a natural resistance of these cells to HIV-1 infection [84, 85]. However, as the immune response is progressively weakened and the presence of proinflammatory stimuli increases, the virus is able to reactivate from latency, and initiate new rounds of infection. At early stages of HIV-1 disease, only a few productively infected microglial cells can be found; however, during late stage disease their numbers significantly increase [73].
A crucial role in promoting viral latency in microglia has been described for the transcription factor CTIP2 through recruitment of HDAC1 or HDAC2 to the viral promoter and promoting local deacetylation of histone 3 (H3) [86, 87]. Furthermore, CTIP2 has been shown to be associated with histone methyltransferase Suv39H1, which increases local H3 methylation and creates a binding site for heterochromatin protein 1 (HP1). All these factors cooperatively promote the formation of the heterochromatin environment and repress HIV-1 gene expression in microglial cells [87].
Astrocytes
Astrocytes are the most abundant cell type of the CNS. Their role is to establish and maintain an optimal microenvironment for the nourishment and function of neurons. In doing so, astrocytes participate in the maintenance of the BBB, in neuronal repair, and in a bidirectional communication with neurons. Additionally, astrocytes participate in the immune defense, particularly in the presence of inflammation, during which they can be induced to secrete immune active molecules and upregulate receptors that facilitate interaction with cells of the immune system [88].
Viral entry appears to be a limiting step in HIV-1 infection of astrocytes; however, once the virus has entered an astrocyte it has been shown to be able to establish a persistent infection [89, 90]. The extent and nature of astrocyte infection by HIV-1 has been under investigation for a number of years. HIV-1 DNA and RNA are often found in astrocytes of postmortem brain tissues of acquired immunodeficiency syndrome (AIDS) patients, indicating that this cell type can be infected by HIV-1 [91, 92]. However, viral proteins, other than Nef, are rarely detected, suggesting that HIV-1 infection of astrocytes is non-productive or persistent in nature and likely does not lead to considerable virus production in vivo [93]. Astrocytes express several chemokine receptors, including CCR5 and CXCR4 [94], however CD4 expression has not been shown in astrocytes and CD4 blocking antibodies do not inhibit infection of astrocytes [95-97]. A number of surface molecules have been proposed as potential HIV-1 receptors, such as the mannose receptor [98], however, the in vivo significance has not been conclusively proven.
In vitro HIV-1 infection has been shown in a number of glioblastoma and astrocytoma cell lines as well as in primary fetal tissue-derived astrocytes. Typically, astrocyte cultures exposed to HIV-1 show initial transient virus production in a subpopulation of cells [99] and virus production peaks at around day 2-7 post infection [100]. After the initial acute infection period, HIV-1 production decreases to very low or undetectable levels and persistent infection of HIV-1 is established. Persistently infected astrocytes are capable of transmitting the virus to both monocytic cell lines and to T cell lines [96, 101-103]. Importantly, the virus that is transmitted from astrocytes can give rise to fully productive infection, further supporting the notion that astrocytes can be a source of persistence and dissemination of virus in the brain. Moreover, studies have shown reactivation of virus from latently infected astrocytes, following stimulation with tumor necrosis factor (TNF)-α and IL-1β [104, 105]. TNF-α has also been shown to stimulate reactivation from latency in brain-derived progenitor cells, suggesting that a pluripotent cell could also serve as a reservoir [106]. Pretreatment of astrocytes with interferon (IFN)-γ has similarly been reported to prime astrocytes for productive HIV-1 infection while granulocyte-macrophage colony stimulating factor (GM-CSF) and TNF-α did not [107]. IFN-γ was shown to exert its effects through alleviating the inhibitory effect of the transcription factor T cell factor-4 (TCF-4) [108, 109]. An additional role of astrocytes in the dissemination of HIV-1 has been recently suggested; HIV-1 uptake and release from astrocytes may not require replication of the virus; instead the virus could be engulfed into vesicle-like structures and subsequently be released and transmitted to CD4+ T cells [110], similarly to what has been suggested for dendritic cells (DCs) [111]. However, it is currently unknown if such a mode of transmission from astrocytes takes place in vivo and for how long HIV-1 can remain infectious when sequestered by astrocytes.
Dendritic Cells (DCs)
There are various types of DCs differing both in their maturation stage and their origin. DCs are primarily classified as myeloid (MDCs) and plasmacytoid DCs (PDCs), both of which are potent professional antigen presenting cells (APC) but vary in the expression of a number of surface molecules and in some functional aspects. Both MDCs and PDCs can be infected with HIV-1 although there are differences in their susceptibility to the various strains of HIV-1 [112, 113].
DCs are crucial in the dissemination of HIV-1 following primary infection as experimental results have suggested that they might be the first cell type to be infected after intravaginal inoculation [114]. Following capture of virions at the primary site of infection, DCs mature and migrate into the lymph nodes thus providing HIV-1 with access to an immense pool of CD4+ T cells. Importantly, MDCs and PDCs transfer HIV-1 preferentially to antigen-specific CD4+ T cells, undermining the development of an efficient immune response against HIV-1 [113].
DCs participate in the transmission of HIV-1 in two ways; by being infected and transmitting newly synthesized virus (cis-infection) or by harboring and transporting infectious virus without being infected themselves (trans-infection) [115]. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) has been shown to play a key role in trans-infection of DCs, by capturing and mediating internalization of virions in non-degradative compartments within DCs where they can retain infectivity for a few days [111]. Trans-infection may be crucial in the dissemination of HIV-1, however, internalized virus most likely represents a short-term reservoir of HIV-1 with limited contribution to the chronic persistence of the virus. Recent results have suggested that infection of DCs is required for transfer of the virus to CD4+ T cells and this capability is maintained for periods of time greater than 3 days [116, 117].
It remains an open question for how long infected DCs can survive and contribute to the pool of virus production. In vitro studies imply that MDCs can survive and retain infectivity for more than 45 days [118], however it is possible that their in vivo half-life is much shorter. Earlier studies have suggested that epidermal Langerhans cells have a half life of about 15 days [119], while DCs in mucosal tissues have a turnover rate of just 2-3 days [120]. Some additional results arguing against a role of DCs as a long-term HIV-1 reservoir came from studies involving individuals undergoing HAART; HIV-1 proviral DNA was undetectable in peripheral blood DCs from patients with undetectable levels of viral RNA [121], suggesting that these cells may not serve as a reservoir in individuals on virus-suppressive HAART.
Interestingly, activation does not seem to increase HIV-1 replication in infected DCs, as observed with monocytes and T cells. On the contrary, immature DCs appear to be permissive to HIV-1 while mature DCs are not and can only participate in trans-infection of T cells [122]. These results were confirmed by a more recent study which showed that LPS-induced maturation inhibits the ability of DCs to support HIV-1 replication, however, it was also shown that the presence of activated T cells enhanced the ability of DCs to support viral replication [123].
Follicular Dendritic Cells (FDCs)
FDCs are located in the B-cell follicles of secondary lymphoid organs and play a crucial role in the maintenance of long-term CD4+ T cell-mediated antibody responses [124]. FDCs are capable of capturing antigen and preserving it on their cell surface in its natural conformation for extended periods of time, similarly they are able to trap HIV-1 and become a reservoir for the virus, although they are not productively infected [125]. Virus found on FDCs is bound by antibodies [126], however it is still infectious; actually, it has been suggested that antibodies are required for the maintenance of HIV-1 infectivity on FDCs [127]. Importantly, it has been shown that FDCs can convert HIV-1 from a neutralized form into an infectious form even in the presence of excess of neutralizing antibody [128].
In the asymptomatic phase of the infection, the pool of virions trapped on the surface of FDCs is stable and exceeds the viral load in the infected cell by at least an order of magnitude, and the load in plasma by a few orders of magnitude [129]. Studies in mice where no productive HIV-1 infection can take place have shown that virus on FDCs can remain infectious for more than 9 months, and have a half-life of about 2 months [130]. On the other hand, free virions in the blood have a half-life of only about 6 hours [50], which emphasizes the significance of FDCs as a long-term reservoir, especially when one considers that the lymphoid organs are the primary site of HIV-1 replication [131] and replenishment of the pool of virions on the FDCs has been shown to be possible even under HAART. Six weeks of HAART treatment appeared to be capable of decreasing the viral load on FDCs by 3-4 orders of magnitude [132], however, it remains uncertain whether this reservoir can be completely cleared or whether a small pool of infectious virus remains trapped on FDCs and is being replenished even during periods of aviremia.
Hematopoietic Progenitor Cells (HPCs)
Although there is extensive literature regarding the infection of HPCs, there is still considerable controversy in this area of research. Several investigators have shown that sub-populations of CD34+ HPCs express CD4, CCR5, and/or CXCR4 [133-138] and the receptor and coreceptor expression correlates with the susceptibility of these cells to HIV-1 [139-143]. However, studies investigating infection of very primitive HPC populations have failed to conclusively show infection of these cells [144, 145]. Detection of HIV-1-infected CD34+ HPCs in vivo has been possible for only a subpopulation of patients, ranging from 14 to 36% [146-149]. Despite the limited infection of the HPC compartment by HIV-1, their role as a viral reservoir may be crucial due to their potential for proliferation and generation of infected cell lineages such as monocytes that may spread the virus to immunoprivileged sites. Relevant to this hypothesis, a recent study by Bailey et al. has suggested that most of the viral sequences in the plasma of HIV-1-infected patients comes from a cell population with proliferating capacity which is distinct from CD4+ T cells [150].
Other Potential Cellular Reservoirs
A small subpopulation of natural killer (NK) cells expresses CD4 on their surface and have been reported to harbor HIV-1 DNA for prolonged periods of time in patients undergoing HAART [151]. However, other studies have failed to detect infected NK cells in a similar population of patients [152] and it is questionable whether these cells contribute to the latent HIV-1 reservoir.
Mast cells express CCR5 and CXCR4 as well as low levels of CD4 and it has been shown that they can be infected when they circulate as immature cells in the blood [153, 154]. Most importantly, it has been shown that Toll-like receptor signaling can reactivate HIV-1 replication in latently infected mast cells [155], which may serve as an inducible HIV-1 reservoir (Fig. 4).
Fig. (4). Cellular HIV-1 reservoirs in the blood.
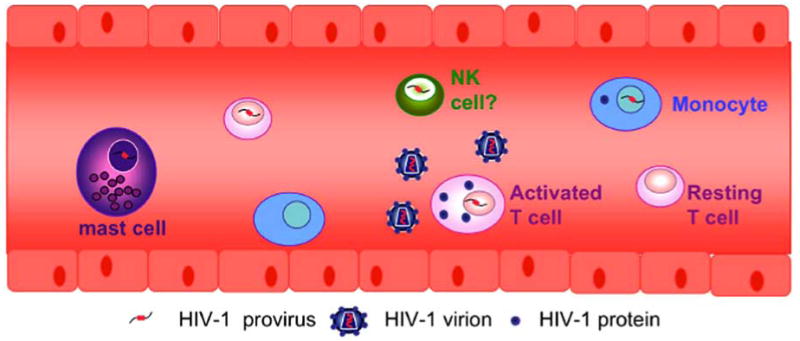
In addition to CD4+ T cells and monocytes, infected mast cells and potentially NK cells may also be present in the blood of HIV-1-infected patients.
Neurons and oligodendrocytes have been reported to be infected with HIV-1. However, there are also several reports that have failed to identify HIV-1 nucleic acids in both of these cell types of the CNS, and it generally believed that if HIV-1 is able to enter these cells it is not able to replicate; reviewed in [100].
Efforts Towards the Eradication of HIV-1 Reservoirs
Although the introduction of HAART has considerably prolonged the survival and improved the quality of life of HIV-1-infected patients, eradication of the virus has, so far, not been possible. Initial optimistic estimations suggesting that eradication may be achieved during 2-3 years of HAART [50] have been followed by the realization that 60 or more years of successful HAART may be required to completely eliminate HIV-1 from an infected individual [156], which obviously emphasizes the need for new approaches to achieve HIV-1 eradication. Several strategies have been proposed that may lead to this goal including structured treatment interruption (STI) and immune activation, which may be combined with intensification of HAART.
STI is based on the hypothesis that HAART interruption may boost HIV-specific immunity, through controlled exposure to autologous virus over limited time periods. It was assumed that following several interruptions that the immune response would be reinforced to the extent of being able to control viral replication in the absence of HAART [157]. Although a significant amount of knowledge has been gained, STI clinical trials have not been successful in decreasing the size of latently infected cell pool and have been associated with worse clinical outcomes than continuous therapy [158-162].
Immune activation therapy aims to drive resting CD4+ T cells out of latency, and may be combined with intensification of HAART to protect uninfected cells from the potential increase in viral load during immune activation. Numerous CD4+ T cell activating agents have been used in clinical trials including IL-2 [163, 164], OKT-3 [165, 166], and valproic acid [167, 168], while IL-7 has also been suggested [169]. Hydroxyurea usually in combination with didanosine [170, 171], and enfuvirtide [167, 168] have been used to intensify HAART. Although evidence exists for a decrease in the infected CD4+ T cell pool, following discontinuation of therapy, plasma virus rebounded in all cases. In addition, toxicity associated with these HAART intensification protocols is often a limiting factor in implementing these treatments. Since the observed decrease in the infected CD4+ T cell pool has been viewed as a partial success, efforts are currently directed towards improving immune activation and intensification protocols.
A critical question in the field of HIV-1 latency is whether a low level replication continuously replenishes the viral reservoir or whether the longevity of the latently infected cells is responsible for the stability of the reservoir. Currently, neither possibility can be excluded, and while the increased decay rate of the viral reservoir observed in some HAART intensification studies suggests ongoing replication [172, 173], the absence of viral evolution in patients undergoing successful HAART therapy supports the notion that the reservoir is intrinsically stable [174, 175]. Approaches to generate new antiviral agents, including ones that would be more effective in the reservoirs of the CNS, which would be added to the current HAART regimens [176] could only be successful if the “ongoing replication” hypothesis is true. On the contrary, if the “intrinsic stability” hypothesis is true, inducing reactivation of the virus from the latently infected cells would be a more suitable approach. However, in order to eradicate HIV-1, reactivation from all of the potential cellular reservoirs would need to be addressed. Immune activation therapies have, so far, been directed primarily towards driving resting CD4+ T cells out of latency, while the monocyte-macrophage and other reservoirs have largely been neglected. Additional strategies to target silent HIV-1 proviral DNA within the context of cellular chromatin are under investigation.
The development of technology such as RNA interference [177] to irreversibly silence the viral promoter within the cells of any HIV-1 reservoir would circumvent the need for activation of viral gene expression and the toxicities associated with cellular activation and HAART intensification protocols in order to clear proviral DNA from any distinct cellular reservoir. Although these experimental directions are worth pursuit, their clinical application is complicated by issues pertaining to specificity and delivery. The use of padlock probes may also be of value in silencing the promoter within the context of the integrated provirus [178]. Tissue-specific drug delivery methods are also being explored, and are particularly interesting when addressing purging of HIV-1 from a restricted anatomical reservoir such as the CNS [179]. Additionally, gene therapy, potentially targeting HPCs [180], is a novel strategy that could, in the future, have an important role in treating HIV-1 infection, however, currently is not ready to be put in clinical practice.
Acknowledgments
Salary support was provided to the authors by the United States Public Health Service / National Institutes of Health through grants awarded to B. Wigdahl from the National Institute of Neurological Disorders and Stroke, NS32092 and NS46263, and the National Institute of Drug Abuse, DA19807.
References
- 1.Greene WC, Peterlin BM. Charting HIV's remarkable voyage through the cell: Basic science as a passport to future therapy. Nat Med. 2002;8:673–80. doi: 10.1038/nm0702-673. [DOI] [PubMed] [Google Scholar]
- 2.Zamborlini A, Lehmann-Che J, Clave E, et al. Centrosomal pre-integration latency of HIV-1 in quiescent cells. Retrovirology. 2007;4:63. doi: 10.1186/1742-4690-4-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu Y, Marsh JW. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science. 2001;293:1503–6. doi: 10.1126/science.1061548. [DOI] [PubMed] [Google Scholar]
- 4.Chun TW, Carruth L, Finzi D, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Rohr O, Marban C, Aunis D, Schaeffer E. Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J Leukoc Biol. 2003;74:736–49. doi: 10.1189/jlb.0403180. [DOI] [PubMed] [Google Scholar]
- 6.Emiliani S, Van Lint C, Fischle W, et al. A point mutation in the HIV-1 Tat responsive element is associated with postintegration latency. Proc Natl Acad Sci USA. 1996;93:6377–81. doi: 10.1073/pnas.93.13.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emiliani S, Fischle W, Ott M, et al. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the U1 cell line. J Virol. 1998;72:1666–70. doi: 10.1128/jvi.72.2.1666-1670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun TW, Engel D, Mizell SB, Ehler LA, Fauci AS. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J Exp Med. 1998;188:83–91. doi: 10.1084/jem.188.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc Natl Acad Sci USA. 2003;100:12955–60. doi: 10.1073/pnas.2233345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devadas K, Hardegen NJ, Wahl LM, et al. Mechanisms for macrophage-mediated HIV-1 induction. J Immunol. 2004;173:6735–44. doi: 10.4049/jimmunol.173.11.6735. [DOI] [PubMed] [Google Scholar]
- 11.Alexaki A, Quiterio SJ, Liu Y, et al. PMA-Induced Differentiation of a Bone Marrow Progenitor Cell Line Activates HIV-1 LTR-Driven Transcription. DNA Cell Biol. 2007;26:387–94. doi: 10.1089/dna.2006.0542. [DOI] [PubMed] [Google Scholar]
- 12.Orenstein JM, Fox C, Wahl SM. Macrophages as a source of HIV during opportunistic infections. Science. 1997;276:1857–61. doi: 10.1126/science.276.5320.1857. [DOI] [PubMed] [Google Scholar]
- 13.Wahl SM, Greenwell-Wild T, Peng G, et al. Mycobacterium avium complex augments macrophage HIV-1 production and increases CCR5 expression. Proc Natl Acad Sci USA. 1998;95:12574–9. doi: 10.1073/pnas.95.21.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–77. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 16.du Chene I, Basyuk E, Lin YL, et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–35. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 18.Verdin E, Paras P, Jr, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–59. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verdin E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J Virol. 1991;65:6790–9. doi: 10.1128/jvi.65.12.6790-6799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. Journal of virology. 2007;81:10914–23. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–95. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol. 2002;22:2965–73. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coull JJ, Romerio F, Sun JM, et al. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–9. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du T, Zamore PD. Beginning to understand microRNA function. Cell Res. 2007;17:661–3. doi: 10.1038/cr.2007.67. [DOI] [PubMed] [Google Scholar]
- 25.Triboulet R, Mari B, Lin YL, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315:1579–82. doi: 10.1126/science.1136319. [DOI] [PubMed] [Google Scholar]
- 26.Huang J, Wang F, Argyris E, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–7. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- 27.Han Y, Siliciano RF. Keeping quiet: microRNAs in HIV-1 latency. Nat Med. 2007;13:1138–40. doi: 10.1038/nm1007-1138. [DOI] [PubMed] [Google Scholar]
- 28.Chun TW, Engel D, Berrey MM, et al. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci USA. 1998;95:8869–73. doi: 10.1073/pnas.95.15.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ostrowski MA, Chun TW, Justement SJ, et al. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J Virol. 1999;73:6430–5. doi: 10.1128/jvi.73.8.6430-6435.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierson T, Hoffman TL, Blankson J, et al. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J Virol. 2000;74:7824–33. doi: 10.1128/jvi.74.17.7824-7833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Zhang H, Siliciano JD, Siliciano RF. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J Virol. 2005;79:2199–210. doi: 10.1128/JVI.79.4.2199-2210.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Persaud D, Zhou Y, Siliciano JM, Siliciano RF. Latency in human immunodeficiency virus type 1 infection: no easy answers. J Virol. 2003;77:1659–65. doi: 10.1128/JVI.77.3.1659-1665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swiggard WJ, Baytop C, Yu JJ, et al. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol. 2005;79:14179–88. doi: 10.1128/JVI.79.22.14179-14188.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chanel C, Staropoli I, Baleux F, et al. Low levels of co-receptor CCR5 are sufficient to permit HIV envelope-mediated fusion with resting CD4 T cells. AIDS. 2002;16:2337–40. doi: 10.1097/00002030-200211220-00016. [DOI] [PubMed] [Google Scholar]
- 35.Eckstein DA, Penn ML, Korin YD, et al. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity. 2001;15:671–82. doi: 10.1016/s1074-7613(01)00217-5. [DOI] [PubMed] [Google Scholar]
- 36.Dopper S, Wilflingseder D, Prodinger WM, et al. Mechanism(s) promoting HIV-1 infection of primary unstimulated T lymphocytes in autologous B cell/T cell co-cultures. Eur J Immunol. 2003;33:2098–107. doi: 10.1002/eji.200323932. [DOI] [PubMed] [Google Scholar]
- 37.Arrighi JF, Pion M, Garcia E, et al. DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J Exp Med. 2004;200:1279–88. doi: 10.1084/jem.20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swingler S, Brichacek B, Jacque JM, et al. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature. 2003;424:213–9. doi: 10.1038/nature01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oswald-Richter K, Grill SM, Leelawong M, Unutmaz D. HIV infection of primary human T cells is determined by tunable thresholds of T cell activation. Eur J Immunol. 2004;34:1705–14. doi: 10.1002/eji.200424892. [DOI] [PubMed] [Google Scholar]
- 40.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med. 1999;189:1735–46. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saleh S, Solomon A, Wightman F, et al. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood. 2007;110:4161–4. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 42.Chun TW, Justement JS, Lempicki RA, et al. Gene expression and viral prodution in latently infected, resting CD4+ T cells in viremic versus aviremic HIV-infected individuals. Proc Natl Acad Sci USA. 2003;100:1908–13. doi: 10.1073/pnas.0437640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Schuler T, Zupancic M, et al. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 1999;286:1353–7. doi: 10.1126/science.286.5443.1353. [DOI] [PubMed] [Google Scholar]
- 44.Gondois-Rey F, Biancotto A, Pion M, et al. Production of HIV-1 by resting memory T lymphocytes. AIDS. 2001;15:1931–40. doi: 10.1097/00002030-200110190-00004. [DOI] [PubMed] [Google Scholar]
- 45.Pierson T, McArthur J, Siliciano RF. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol. 2000;18:665–708. doi: 10.1146/annurev.immunol.18.1.665. [DOI] [PubMed] [Google Scholar]
- 46.Chun TW, Davey RT, Jr, Ostrowski M, et al. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med. 2000;6:757–61. doi: 10.1038/77481. [DOI] [PubMed] [Google Scholar]
- 47.Ho DD, Rota TR, Hirsch MS. Infection of monocyte/macrophages by human T lymphotropic virus type III. J Clin Invest. 1986;77:1712–5. doi: 10.1172/JCI112491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicholson JK, Cross GD, Callaway CS, McDougal JS. In vitro infection of human monocytes with human T lymphotropic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) J Immunol. 1986;137:323–9. [PubMed] [Google Scholar]
- 49.Swingler S, Mann AM, Zhou J, Swingler C, Stevenson M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007;3:1281–90. doi: 10.1371/journal.ppat.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–91. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 51.McElrath MJ, Steinman RM, Cohn ZA. Latent HIV-1 infection in enriched populations of blood monocytes and T cells from seropositive patients. J Clin Invest. 1991;87:27–30. doi: 10.1172/JCI114981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Brien WA. HIV-1 entry and reverse transcription in macrophages. J Leukoc Biol. 1994;56:273–7. doi: 10.1002/jlb.56.3.273. [DOI] [PubMed] [Google Scholar]
- 53.Diamond TL, Roshal M, Jamburuthugoda VK, et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279:51545–53. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Y, Zhu H, Wilcox CK, et al. Blood monocytes harbor HIV type 1 strains with diversified phenotypes including macrophage-specific CCR5 virus. J Infect Dis. 2008;197:309–18. doi: 10.1086/524847. [DOI] [PubMed] [Google Scholar]
- 55.Smith PD, Meng G, Salazar-Gonzalez JF, Shaw GM. Macrophage HIV-1 infection and the gastrointestinal tract reservoir. J Leukoc Biol. 2003;74:642–9. doi: 10.1189/jlb.0503219. [DOI] [PubMed] [Google Scholar]
- 56.Lambotte O, Taoufik Y, de Goer MG, et al. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2000;23:114–9. doi: 10.1097/00126334-200002010-00002. [DOI] [PubMed] [Google Scholar]
- 57.Zhu T, Muthui D, Holte S, et al. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J Virol. 2002;76:707–16. doi: 10.1128/JVI.76.2.707-716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caselli E, Galvan M, Cassai E, et al. Human herpesvirus 8 enhances human immunodeficiency virus replication in acutely infected cells and induces reactivation in latently infected cells. Blood. 2005;106:2790–7. doi: 10.1182/blood-2005-04-1390. [DOI] [PubMed] [Google Scholar]
- 59.Qi X, Koya Y, Saitoh T, et al. Efficient induction of HIV-1 replication in latently infected cells through contact with CD4+ T cells: involvement of NF-kappaB activation. Virology. 2007;361:325–34. doi: 10.1016/j.virol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 60.Crowe SM, Mills J, Elbeik T, et al. Human immunodeficiency virus-infected monocyte-derived macrophages express surface gp120 and fuse with CD4 lymphoid cells in vitro: a possible mechanism of T lymphocyte depletion in vivo. Clin Immunol Immunopathol. 1992;65:143–51. doi: 10.1016/0090-1229(92)90217-c. [DOI] [PubMed] [Google Scholar]
- 61.Crowe SM, Mills J, Kirihara J, et al. Full-length recombinant CD4 and recombinant gp120 inhibit fusion between HIV infected macrophages and uninfected CD4-expressing T-lymphoblastoid cells. AIDS Res Hum Retroviruses. 1990;6:1031–7. doi: 10.1089/aid.1990.6.1031. [DOI] [PubMed] [Google Scholar]
- 62.Fantuzzi L, Belardelli F, Gessani S. Monocyte/macrophage-derived CC chemokines and their modulation by HIV-1 and cytokines: a complex network of interactions influencing viral replication and AIDS pathogenesis. J Leukoc Biol. 2003;74:719–25. doi: 10.1189/jlb.0403175. [DOI] [PubMed] [Google Scholar]
- 63.Swingler S, Mann A, Jacque J, et al. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med. 1999;5:997–103. doi: 10.1038/12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–85. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 65.Tarrago-Litvak L, Andreola ML, Nevinsky GA, Sarih-Cottin L, Litvak S. The reverse transcriptase of HIV-1: from enzymology to therapeutic intervention. FASEB J. 1994;8:497–503. doi: 10.1096/fasebj.8.8.7514143. [DOI] [PubMed] [Google Scholar]
- 66.Perno CF, Newcomb FM, Davis DA, et al. Relative potency of protease inhibitors in monocytes/macrophages acutely and chronically infected with human immunodeficiency virus. J Infect Dis. 1998;178:413–22. doi: 10.1086/515642. [DOI] [PubMed] [Google Scholar]
- 67.Kim RB, Fromm MF, Wandel C, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–94. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci. 2002;25:537–62. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- 69.He J, Chen Y, Farzan M, et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–9. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 70.Lavi E, Strizki JM, Ulrich AM, et al. CXCR-4 (Fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons. Am J Pathol. 1997;151:1035–42. [PMC free article] [PubMed] [Google Scholar]
- 71.Garden GA. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia. 2002;40:240–51. doi: 10.1002/glia.10155. [DOI] [PubMed] [Google Scholar]
- 72.Williams KC, Corey S, Westmoreland SV, et al. Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001;193:905–15. doi: 10.1084/jem.193.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002;12:442–55. doi: 10.1111/j.1750-3639.2002.tb00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petito CK. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J Neurovirol. 2004;10(Suppl 1):21–4. doi: 10.1080/753312748. [DOI] [PubMed] [Google Scholar]
- 75.Petito CK, Chen H, Mastri AR, et al. HIV infection of choroid plexus in AIDS and asymptomatic HIV-infected patients suggests that the choroid plexus may be a reservoir of productive infection. J Neurovirol. 1999;5:670–7. doi: 10.3109/13550289909021295. [DOI] [PubMed] [Google Scholar]
- 76.Bagasra O, Lavi E, Bobroski L, et al. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS. 1996;10:573–85. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 77.Harouse JM, Wroblewska Z, Laughlin MA, et al. Human choroid plexus cells can be latently infected with human immunodeficiency virus. Ann Neurol. 1989;25:406–11. doi: 10.1002/ana.410250414. [DOI] [PubMed] [Google Scholar]
- 78.Williams K, Alvarez X, Lackner AA. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia. 2001;36:156–64. doi: 10.1002/glia.1105. [DOI] [PubMed] [Google Scholar]
- 79.Hickey WF, Vass K, Lassmann H. Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultra-structural survey of rat chimeras. J Neuropathol Exp Neurol. 1992;51:246–56. doi: 10.1097/00005072-199205000-00002. [DOI] [PubMed] [Google Scholar]
- 80.Kida S, Steart PV, Zhang ET, Weller RO. Perivascular cells act as scavengers in the cerebral perivascular spaces and remain distinct from pericytes, microglia and macrophages. Acta Neuropathol (Berl) 1993;85:646–52. doi: 10.1007/BF00334675. [DOI] [PubMed] [Google Scholar]
- 81.Harling-Berg C, Knopf PM, Merriam J, Cserr HF. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol. 1989;25:185–93. doi: 10.1016/0165-5728(89)90136-7. [DOI] [PubMed] [Google Scholar]
- 82.Suh HS, Kim MO, Lee SC. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J Immunol. 2005;174:2712–9. doi: 10.4049/jimmunol.174.5.2712. [DOI] [PubMed] [Google Scholar]
- 83.Krall WJ, Challita PM, Perlmutter LS, Skelton DC, Kohn DB. Cells expressing human glucocerebrosidase from a retroviral vector repopulate macrophages and central nervous system microglia after murine bone marrow transplantation. Blood. 1994;83:2737–48. [PubMed] [Google Scholar]
- 84.Chakrabarti L, Hurtrel M, Maire MA, et al. Early viral replication in the brain of SIV-infected rhesus monkeys. Am J Pathol. 1991;139:1273–80. [PMC free article] [PubMed] [Google Scholar]
- 85.Davis LE, Hjelle BL, Miller VE, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–9. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- 86.Rohr O, Lecestre D, Chasserot-Golaz S, et al. Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol. 2003;77:5415–27. doi: 10.1128/JVI.77.9.5415-5427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marban C, Redel L, Suzanne S, et al. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 2005;33:2318–31. doi: 10.1093/nar/gki529. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 88.Speth C, Dierich MP, Sopper S. HIV-infection of the central nervous system: the tightrope walk of innate immunity. Mol Immunol. 2005;42:213–28. doi: 10.1016/j.molimm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 89.Canki M, Thai JN, Chao W, et al. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J Virol. 2001;75:7925–33. doi: 10.1128/JVI.75.17.7925-7933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bencheikh M, Bentsman G, Sarkissian N, Canki M, Volsky DJ. Replication of different clones of human immunodeficiency virus type 1 in primary fetal human astrocytes: enhancement of viral gene expression by Nef. J Neurovirol. 1999;5:115–24. doi: 10.3109/13550289909021993. [DOI] [PubMed] [Google Scholar]
- 91.Takahashi K, Wesselingh SL, Griffin DE, et al. Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol. 1996;39:705–11. doi: 10.1002/ana.410390606. [DOI] [PubMed] [Google Scholar]
- 92.Trillo-Pazos G, Diamanturos A, Rislove L, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol. 2003;13:144–54. doi: 10.1111/j.1750-3639.2003.tb00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- 94.Dorf ME, Berman MA, Tanabe S, Heesen M, Luo Y. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111:109–21. doi: 10.1016/s0165-5728(00)00371-4. [DOI] [PubMed] [Google Scholar]
- 95.Ma M, Geiger JD, Nath A. Characterization of a novel binding site for the human immunodeficiency virus type 1 envelope protein gp120 on human fetal astrocytes. J Virol. 1994;68:6824–28. doi: 10.1128/jvi.68.10.6824-6828.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Clapham PR, Weber JN, Whitby D, et al. Soluble CD4 blocks the infectivity of diverse strains of HIV and SIV for T cells and monocytes but not for brain and muscle cells. Nature. 1989;337:368–70. doi: 10.1038/337368a0. [DOI] [PubMed] [Google Scholar]
- 97.Lavi E, Kolson DL, Ulrich AM, Fu L, Gonzalez-Scarano F. Chemokine receptors in the human brain and their relationship to HIV infection. J Neurovirol. 1998;4:301–11. doi: 10.3109/13550289809114531. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y, Liu H, Kim BO, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78:4120–33. doi: 10.1128/JVI.78.8.4120-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brack-Werner R, Kleinschmidt A, Ludvigsen A, et al. Infection of human brain cells by HIV-1: restricted virus production in chronically infected human glial cell lines. AIDS. 1992;6:273–85. [PubMed] [Google Scholar]
- 100.Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 101.Neumann M, Felber BK, Kleinschmidt A, et al. Restriction of human immunodeficiency virus type 1 production in a human astrocytoma cell line is associated with a cellular block in Rev function. J Virol. 1995;69:2159–67. doi: 10.1128/jvi.69.4.2159-2167.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiodi F, Fuerstenberg S, Gidlund M, Asjo B, Fenyo EM. Infection of brain-derived cells with the human immunodeficiency virus. J Virol. 1987;61:1244–7. doi: 10.1128/jvi.61.4.1244-1247.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sabri F, Tresoldi E, Di Stefano M, et al. Nonproductive human immunodeficiency virus type 1 infection of human fetal astrocytes: independence from CD4 and major chemokine receptors. Virology. 1999;264:370–84. doi: 10.1006/viro.1999.9998. [DOI] [PubMed] [Google Scholar]
- 104.Tornatore C, Meyers K, Atwood W, Conant K, Major E. Temporal patterns of human immunodeficiency virus type 1 transcripts in human fetal astrocytes. J Virol. 1994;68:93–102. doi: 10.1128/jvi.68.1.93-102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tornatore C, Nath A, Amemiya K, Major EO. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J Virol. 1991;65:6094–100. doi: 10.1128/jvi.65.11.6094-6100.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lawrence DM, Durham LC, Schwartz L, et al. Human immunodeficiency virus type 1 infection of human brain-derived progenitor cells. J Virol. 2004;78:7319–28. doi: 10.1128/JVI.78.14.7319-7328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carroll-Anzinger D, Al-Harthi L. Gamma interferon primes productive human immunodeficiency virus infection in astrocytes. J Virol. 2006;80:541–4. doi: 10.1128/JVI.80.1.541-544.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carroll-Anzinger D, Kumar A, Adarichev V, Kashanchi F, Al-Harthi L. Human immunodeficiency virus-restricted replication in astrocytes and the ability of gamma interferon to modulate this restriction are regulated by a downstream effector of the Wnt signaling pathway. J Virol. 2007;81:5864–71. doi: 10.1128/JVI.02234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rossi A, Mukerjee R, Ferrante P, et al. Human immunodeficiency virus type 1 Tat prevents dephosphorylation of Sp1 by TCF-4 in astrocytes. J Gen Virol. 2006;87:1613–23. doi: 10.1099/vir.0.81691-0. [DOI] [PubMed] [Google Scholar]
- 110.Clarke JN, Lake JA, Burrell CJ, et al. Novel pathway of human immunodeficiency virus type 1 uptake and release in astrocytes. Virology. 2006;348:141–55. doi: 10.1016/j.virol.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 111.Geijtenbeek TB, Kwon DS, Torensma R, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–97. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 112.Lore K, Smed-Sorensen A, Vasudevan J, Mascola JR, Koup RA. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J Exp Med. 2005;201:2023–33. doi: 10.1084/jem.20042413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smed-Sorensen A, Lore K, Vasudevan J, et al. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol. 2005;79:8861–9. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spira AI, Marx PA, Patterson BK, et al. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J Exp Med. 1996;183:215–25. doi: 10.1084/jem.183.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dong C, Janas AM, Wang JH, Olson WJ, Wu L. Characterization of human immunodeficiency virus type 1 replication in immature and mature dendritic cells reveals dissociable cis- and trans-infection. J Virol. 2007;81:11352–62. doi: 10.1128/JVI.01081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Turville SG, Santos JJ, Frank I, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. 2004;103:2170–9. doi: 10.1182/blood-2003-09-3129. [DOI] [PubMed] [Google Scholar]
- 117.Burleigh L, Lozach PY, Schiffer C, et al. Infection of dendritic cells (DCs), not DC-SIGN-mediated internalization of human immunodeficiency virus, is required for long-term transfer of virus to T cells. J Virol. 2006;80:2949–57. doi: 10.1128/JVI.80.6.2949-2957.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Popov S, Chenine AL, Gruber A, Li PL, Ruprecht RM. Long-term productive human immunodeficiency virus infection of CD1a-sorted myeloid dendritic cells. J Virol. 2005;79:602–8. doi: 10.1128/JVI.79.1.602-608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kalter DC, Greenhouse JJ, Orenstein JM, et al. Epidermal Langerhans cells are not principal reservoirs of virus in HIV disease. J Immunol. 1991;146:3396–404. [PubMed] [Google Scholar]
- 120.Pugh CW, MacPherson GG, Steer HW. Characterization of non-lymphoid cells derived from rat peripheral lymph. J Exp Med. 1983;157:1758–79. doi: 10.1084/jem.157.6.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Otero M, Nunnari G, Leto D, et al. Peripheral blood Dendritic cells are not a major reservoir for HIV type 1 in infected individuals on virally suppressive HAART. AIDS Res Hum Retroviruses. 2003;19:1097–103. doi: 10.1089/088922203771881194. [DOI] [PubMed] [Google Scholar]
- 122.Granelli-Piperno A, Delgado E, Finkel V, Paxton W, Steinman RM. Immature dendritic cells selectively replicate macrophagetropic (M-tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M- and T-tropic virus to T cells. J Virol. 1998;72:2733–7. doi: 10.1128/jvi.72.4.2733-2737.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.MacDougall TH, Shattock RJ, Madsen C, Chain BM, Katz DR. Regulation of primary HIV-1 isolate replication in dendritic cells. Clin Exp Immunol. 2002;127:66–71. doi: 10.1046/j.1365-2249.2002.01715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Szakal AK, Tew JG. Follicular dendritic cells: B-cell proliferation and maturation. Cancer Res. 1992;52:5554s–6s. [PubMed] [Google Scholar]
- 125.Schmitz J, van Lunzen J, Tenner-Racz K, et al. Follicular dendritic cells retain HIV-1 particles on their plasma membrane, but are not productively infected in asymptomatic patients with follicular hyperplasia. J Immunol. 1994;153:1352–9. [PubMed] [Google Scholar]
- 126.Keele BF, Tazi L, Gartner S, et al. Characterization of the follicular dendritic cell reservoir of human immunodeficiency virus type 1. J Virol. 2008;82:5548–61. doi: 10.1128/JVI.00124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith-Franklin BA, Keele BF, Tew JG, et al. Follicular dendritic cells and the persistence of HIV infectivity: the role of antibodies and Fcgamma receptors. J Immunol. 2002;168:2408–14. doi: 10.4049/jimmunol.168.5.2408. [DOI] [PubMed] [Google Scholar]
- 128.Heath SL, Tew JG, Tew JG, Szakal AK, Burton GF. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature. 1995;377:740–4. doi: 10.1038/377740a0. [DOI] [PubMed] [Google Scholar]
- 129.Haase AT, Henry K, Zupancic M, et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science. 1996;274:985–9. doi: 10.1126/science.274.5289.985. [DOI] [PubMed] [Google Scholar]
- 130.Smith BA, Gartner S, Liu Y, et al. Persistence of infectious HIV on follicular dendritic cells. J Immunol. 2001;166:690–6. doi: 10.4049/jimmunol.166.1.690. [DOI] [PubMed] [Google Scholar]
- 131.Pantaleo G, Graziosi C, Demarest JF, et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–8. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 132.Cavert W, Notermans DW, Staskus K, et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science. 1997;276:960–4. doi: 10.1126/science.276.5314.960. [DOI] [PubMed] [Google Scholar]
- 133.Carr JM, Ramshaw HS, Li P, Burrell CJ. CD34+ cells and their derivatives contain mRNA for CD4 and human immunodeficiency virus (HIV) co-receptors and are susceptible to infection with M- and T-tropic HIV. J Gen Virol. 1998;79(Pt 1):71–5. doi: 10.1099/0022-1317-79-1-71. [DOI] [PubMed] [Google Scholar]
- 134.Shen H, Cheng T, Preffer FI, et al. Intrinsic human immunodeficiency virus type 1 resistance of hematopoietic stem cells despite coreceptor expression. J Virol. 1999;73:728–37. doi: 10.1128/jvi.73.1.728-737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deichmann M, Kronenwett R, Haas R. Expression of the human immunodeficiency virus type-1 coreceptors CXCR-4 (fusin, LESTR) and CKR-5 in CD34+ hematopoietic progenitor cells. Blood. 1997;89:3522–8. [PubMed] [Google Scholar]
- 136.Nicholls SE, Lucas G, Graham GJ, et al. Macrophage-inflammatory protein-1alpha receptor expression on normal and chronic myeloid leukemia CD34+ cells. J Immunol. 1999;162:6191–9. [PubMed] [Google Scholar]
- 137.Banda NK, Simon GR, Sipple JD, et al. Depletion of CD34+ CD4+ cells in bone marrow from HIV-1-infected individuals. Biol Blood Marrow Transplant. 1999;5:162–72. doi: 10.1053/bbmt.1999.v5.pm10392962. [DOI] [PubMed] [Google Scholar]
- 138.Aiuti A, Turchetto L, Cota M, et al. Human CD34(+) cells express CXCR4 and its ligand stromal cell-derived factor-1. Implications for infection by T-cell tropic human immunodeficiency virus Blood. 1999;94:62–73. [PubMed] [Google Scholar]
- 139.Folks TM, Kessler SW, Orenstein JM, et al. Infection and replication of HIV-1 in purified progenitor cells of normal human bone marrow. Science. 1988;242:919–22. doi: 10.1126/science.2460922. [DOI] [PubMed] [Google Scholar]
- 140.Steinberg HN, Crumpacker CS, Chatis PA. In vitro suppression of normal human bone marrow progenitor cells by human immunodeficiency virus. J Virol. 1991;65:1765–9. doi: 10.1128/jvi.65.4.1765-1769.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kitano K, Abboud CN, Ryan DH, et al. Macrophage-active colony-stimulating factors enhance human immunodeficiency virus type 1 infection in bone marrow stem cells. Blood. 1991;77:1699–705. [PubMed] [Google Scholar]
- 142.Ruiz ME, Cicala C, Arthos J, et al. Peripheral blood-derived CD34+ progenitor cells: CXC chemokine receptor 4 and CC chemokine receptor 5 expression and infection by HIV. J Immunol. 1998;161:4169–76. [PubMed] [Google Scholar]
- 143.Chelucci C, Hassan HJ, Locardi C, et al. In vitro human immunodeficiency virus-1 infection of purified hematopoietic progenitors in single-cell culture. Blood. 1995;85:1181–7. [PubMed] [Google Scholar]
- 144.Hariharan D, Li Y, Campbell DE, et al. Human immunodeficiency virus infection of human placental cord blood CD34+AC133+ stem cells and their progeny. AIDS Res Hum Retroviruses. 1999;15:1545–52. doi: 10.1089/088922299309838. [DOI] [PubMed] [Google Scholar]
- 145.Weichold FF, Zella D, Barabitskaja O, et al. Neither human immunodeficiency virus-1 (HIV-1) nor HIV-2 infects most-primitive human hematopoietic stem cells as assessed in long-term bone marrow cultures. Blood. 1998;91:907–15. [PubMed] [Google Scholar]
- 146.Neal TF, Holland HK, Baum CM, et al. CD34+ progenitor cells from asymptomatic patients are not a major reservoir for human immunodeficiency virus-1. Blood. 1995;86:1749–1756. [PubMed] [Google Scholar]
- 147.Zauli G, Re MC, Davis B, et al. Impaired in vitro growth of purified (CD34+) hematopoietic progenitors in human immunodeficiency virus-1 seropositive thrombocytopenic individuals. Blood. 1992;79:2680–7. [PubMed] [Google Scholar]
- 148.Slobod KS, Bennett TA, Freiden PJ, et al. Mobilization of CD34+ progenitor cells by granulocyte colony-stimulating factor in human immunodeficiency virus type 1-infected adults. Blood. 1996;88:3329–35. [PubMed] [Google Scholar]
- 149.Stanley SK, Kessler SW, Justement JS, et al. CD34+ bone marrow cells are infected with HIV in a subset of seropositive individuals. J Immunol. 1992;149:689–97. [PubMed] [Google Scholar]
- 150.Bailey JR, Sedaghat AR, Kieffer T, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80:6441–57. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Valentin A, Rosati M, Patenaude DJ, et al. Persistent HIV-1 infection of natural killer cells in patients receiving highly active antiretroviral therapy. Proc Natl Acad Sci USA. 2002;99:7015–20. doi: 10.1073/pnas.102672999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Delobel P, Sandres-Saune K, Cazabat M, et al. Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. AIDS. 2005;19:1739–50. doi: 10.1097/01.aids.0000183125.93958.26. [DOI] [PubMed] [Google Scholar]
- 153.Bannert N, Farzan M, Friend DS, et al. Human Mast cell progenitors can be infected by macrophagetropic human immunodeficiency virus type 1 and retain virus with maturation in vitro. J Virol. 2001;75:10808–14. doi: 10.1128/JVI.75.22.10808-10814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li Y, Li L, Wadley R, et al. Mast cells/basophils in the peripheral blood of allergic individuals who are HIV-1 susceptible due to their surface expression of CD4 and the chemokine receptors CCR3, CCR5, and CXCR4. Blood. 2001;97:3484–90. doi: 10.1182/blood.v97.11.3484. [DOI] [PubMed] [Google Scholar]
- 155.Sundstrom JB, Little DM, Villinger F, Ellis JE, Ansari AA. Signaling through Toll-like receptors triggers HIV-1 replication in latently infected mast cells. J Immunol. 2004;172:4391–401. doi: 10.4049/jimmunol.172.7.4391. [DOI] [PubMed] [Google Scholar]
- 156.Finzi D, Blankson J, Siliciano JD, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 157.Lisziewicz J, Lori F. Structured treatment interruptions in HIV/AIDS therapy. Microbes Infect. 2002;4:207–14. doi: 10.1016/s1286-4579(01)01529-5. [DOI] [PubMed] [Google Scholar]
- 158.Ortiz GM, Wellons M, Brancato J, et al. Structured antiretroviral treatment interruptions in chronically HIV-1-infected subjects. Proc Natl Acad Sci USA. 2001;98:13288–93. doi: 10.1073/pnas.221452198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Garcia F, Plana M, Ortiz GM, et al. The virological and immunological consequences of structured treatment interruptions in chronic HIV-1 infection. AIDS. 2001;15:F29–40. doi: 10.1097/00002030-200106150-00002. [DOI] [PubMed] [Google Scholar]
- 160.Deeks SG, Grant RM, Wrin T, et al. Persistence of drug-resistant HIV-1 after a structured treatment interruption and its impact on treatment response. AIDS. 2003;17:361–70. doi: 10.1097/00002030-200302140-00010. [DOI] [PubMed] [Google Scholar]
- 161.Danel C, Moh R, Minga A, et al. CD4-guided structured antiretroviral treatment interruption strategy in HIV-infected adults in west Africa (Trivacan ANRS 1269 trial): a randomised trial. Lancet. 2006;367:1981–9. doi: 10.1016/S0140-6736(06)68887-9. [DOI] [PubMed] [Google Scholar]
- 162.Ghosn J, Wirden M, Ktorza N, et al. No benefit of a structured treatment interruption based on genotypic resistance in heavily pre-treated HIV-infected patients. AIDS. 2005;19:1643–7. doi: 10.1097/01.aids.0000181322.17679.b2. [DOI] [PubMed] [Google Scholar]
- 163.Chun TW, Engel D, Mizell SB, et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat Med. 1999;5:651–5. doi: 10.1038/9498. [DOI] [PubMed] [Google Scholar]
- 164.Stellbrink HJ, van Lunzen J, Westby M, et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial) AIDS. 2002;16:1479–87. doi: 10.1097/00002030-200207260-00004. [DOI] [PubMed] [Google Scholar]
- 165.van Praag RM, Prins JM, Roos MT, et al. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J Clin Immunol. 2001;21:218–26. doi: 10.1023/a:1011091300321. [DOI] [PubMed] [Google Scholar]
- 166.Fraser C, Ferguson NM, Ghani AC, et al. Reduction of the HIV-1-infected T-cell reservoir by immune activation treatment is dose-dependent and restricted by the potency of antiretroviral drugs. AIDS. 2000;14:659–69. doi: 10.1097/00002030-200004140-00005. [DOI] [PubMed] [Google Scholar]
- 167.Lehrman G, Hogue IB, Palmer S, et al. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–55. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Siliciano JD, Lai J, Callender M, et al. Stability of the latent reservoir for HIV-1 in patients receiving valproic acid. J Infect Dis. 2007;195:833–6. doi: 10.1086/511823. [DOI] [PubMed] [Google Scholar]
- 169.Wang FX, Xu Y, Sullivan J, et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J Clin Invest. 2005;115:128–37. doi: 10.1172/JCI22574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nunnari G, Leto D, Sullivan J, et al. Seminal reservoirs during an HIV type 1 eradication trial. AIDS Res Hum Retroviruses. 2005;21:768–75. doi: 10.1089/aid.2005.21.768. [DOI] [PubMed] [Google Scholar]
- 171.Kulkosky J, Nunnari G, Otero M, et al. Intensification and stimulation therapy for human immunodeficiency virus type 1 reservoirs in infected persons receiving virally suppressive highly active antiretroviral therapy. J Infect Dis. 2002;186:1403–11. doi: 10.1086/344357. [DOI] [PubMed] [Google Scholar]
- 172.Ramratnam B, Ribeiro R, He T, et al. Intensification of antiretroviral therapy accelerates the decay of the HIV-1 latent reservoir and decreases, but does not eliminate, ongoing virus replication. J Acquir Immune Defic Syndr. 2004;35:33–7. doi: 10.1097/00126334-200401010-00004. [DOI] [PubMed] [Google Scholar]
- 173.Chun TW, Nickle DC, Justement JS, et al. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J Clin Invest. 2005;115:3250–5. doi: 10.1172/JCI26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sedaghat AR, Siliciano JD, Brennan TP, Wilke CO, Siliciano RF. Limits on replenishment of the resting CD4+ T cell reservoir for HIV in patients on HAART. PLoS Pathog. 2007;3:e122. doi: 10.1371/journal.ppat.0030122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sedaghat AR, Siliciano RF, Wilke CO. Low-level HIV-1 replication and the dynamics of the resting CD4+ T cell reservoir for HIV-1 in the setting of HAART. BMC Infect Dis. 2008;8:2. doi: 10.1186/1471-2334-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Geeraert L, Kraus G, Pomerantz RJ. Hide-and-seek: the challenge of viral persistence in HIV-1 infection. Annu Rev Med. 2008;59:487–501. doi: 10.1146/annurev.med.59.062806.123001. [DOI] [PubMed] [Google Scholar]
- 177.Lee NS, Rossi JJ. Control of HIV-1 replication by RNA interference. Virus Res. 2004;102:53–8. doi: 10.1016/j.virusres.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 178.Shi C, Parker AR, Hua L, et al. Anti-gene padlocks eliminate Escherichia coli based on their genotype. J Antimicrob Chemother. 2008;61:262–72. doi: 10.1093/jac/dkm482. [DOI] [PubMed] [Google Scholar]
- 179.Gorantla S, Dou H, Boska M, et al. Quantitative magnetic resonance and SPECT imaging for macrophage tissue migration and nanoformulated drug delivery. J Leukoc Biol. 2006;80:1165–74. doi: 10.1189/jlb.0206110. [DOI] [PubMed] [Google Scholar]
- 180.Strayer DS, Akkina R, Bunnell BA, et al. Current status of gene therapy strategies to treat HIV/AIDS. Mol Ther. 2005;11:823–42. doi: 10.1016/j.ymthe.2005.01.020. [DOI] [PubMed] [Google Scholar]