

Abstract
Children exposed prenatally to alcohol can display a variety of neural deficits, including an altered development of the corpus callosum (CC), the largest interhemispheric axon pathway in the brain. Furthermore, these children show functional abnormalities that are related to brain regions with significant numbers of CC connections. Little is known about how alcohol imparts influence on CC development, but one possible mechanism is by affecting the corpus callosum projection neurons (CCpn) directly. The purpose of this study was to quantify the effects of prenatal alcohol exposure on the number, size and distribution of CCpn within the visual cortex. The visual cortex was selected specifically due to the many vision-related deficits noted in fetal alcohol exposed children and because the critical role of the CC in visual cortex development is well documented. Sprague-Dawley rat pups received one of four alcohol dosages during gestational days (G) 1–20, or reared as nutritional or untreated control animals. Each litter was categorized according to the peak blood alcohol concentration (BAC) experienced. Pups were removed from each litter on days equivalent to G29, G36, G43, and G50, for histology and measurement. Callosal axons were labelled retrogradely to their CCpn using DiI and the CCpn were then examined using confocal laser scanning microscopy. Differences between alcohol-exposed and control animals were observed in CCpn cell body size, number, and location with the cortex. This was particularly true of animals exposed to high doses of alcohol. In addition, some trends of CCpn development were found to be unchanged as a result of prenatal alcohol exposure. The results demonstrate clear differences in the development of CCpn in the visual cortex between alcohol-exposed and control animals and suggest that this development is particularly affected in those animals exposed to high doses of alcohol.
Keywords: corpus callosum, visual cortex, ethanol, development
Introduction
The corpus callosum (CC) is the largest axon pathway in the brain. In addition to providing interhemispheric communication, it also plays a critical role in the development of the visual system (Elberger 1993; 1994a; b). Children exposed to alcohol prenatally have been shown to have defects in the gross morphology of the CC, including a reduction in size (Clarren, 1986; Mattson et al., 1992; Mattson et al., 1994; Riley et al., 1995) or complete absence (Jones and Smith, 1975; Peiffer et al., 1979; Wisniewski et al., 1983; Jeret et al., 1987; Mattson et al., 1992; Riley et al., 1995). Fetal alcohol exposure has also been linked to a myriad of visual system deficits, including reduced visual acuity, nearsightedness, eye misalignment and optic nerve hypoplasia, (Pinazo-Duran et al., 1997; Stromland and Pinazo-Duran 2002; Stromland, 2004), suggesting a possible role for the CC in these defects.
Developmental reductions in the number of corpus callosum projection neurons (CCpn -those cells giving rise to the callosal axons) and CC terminal projections have been reported in the visual, auditory and somatosensory cortex of normal rats (Lund et al., 1984; Miller and Vogt, 1984; Olavarria and van Sluyters, 1985; Payne et al., 1988) suggesting the presence of transitory CC connections early in development. The presence of such transitory callosal axons was demonstrated in all regions of the visual cortex in both rat and cat models by Elberger (1993; 1994a; b). These transitory axons showed a gradual decline in numbers and were almost eliminated by the end of the first postnatal month, coinciding with the time period during which an intact CC has been demonstrated to be critical for normal visual development in the cat. This suggests that the presence of the transitory connections during a specific time period may be one critical factor for determining normal visual functioning (Elberger, 1993; 1994a; b).
Our hypothesis is that exposure to alcohol may reduce the number of these transitory connections, or delay their appearance in, or elimination from, the visual cortex, thus resulting in permanent visual defects. Although animal models of fetal alcohol exposure have demonstrated few gross morphological abnormalities in CC structure (Wainwright and Fritz, 1985; Zimmerberg and Scalzi, 1989; Zimmerberg and Mickus, 1990; Livy and Elberger, 2001), the cytoarchitecture of the CCpn has been found to be affected. For example, Miller (1997) found that the density and laminar distribution of CCpn was altered in the somatosensory cortex of ethanol-exposed rats. Qiang et al., (2002) found that the CCpn in the visual cortical areas 17 and 18a of rat pups exposed prenatally to alcohol displayed an altered radial and tangential distribution within the cortex, as well as an altered pattern of dendritic arborization.
In the present study, DiI was used as a retrograde tracer to label the CCpn of rat offspring exposed to alcohol throughout the length of their gestation in order to quantify the effects of alcohol on the number, size and distribution of CCpn within the cortex of the brain. Alcohol exposure consisted of a range of doses to determine possible threshold levels for causing effects. The timing of alcohol exposure coincided with the first two trimesters-equivalent to human gestation (Bayer et al., 1993; Dobbing, 1981; Dobbing and Sands, 1973; 1979), a period of time before and during the generation of neocortical neurons (Fukumitsu et al., 2006; Kennedy and Elliot, 1985; Miller, 1986; 1987; 1989). Morphological analyses were carried out in offspring during the time period shown previously to have transitory CC axons (Elberger 1994a,b). Differences in CCpn cell body size, number and location were found between control and ethanol-treated rats, particularly in those receiving high doses of ethanol.
Materials and Methods
Subjects
Virgin female Sprague-Dawley rats, obtained at about 200g body weight, and proven male breeders (Harlan Laboratories, Birmingham, AL) were used. All animals were housed at the AAALAC–accredited facility at the University of Tennessee Health Science Center on a 12 hr light/dark cycle (lights on at 6AM). All experimental protocols were approved by the University of Tennessee Health Science Center IACUC. Females were quarantined for two weeks and then handled for five days to acclimate them to the intragastric gavage procedure (see below). For breeding, one male was housed with one or two females. Vaginal smears were taken each morning and checked for the presence of sperm. Conception (sperm-positive) was designated as gestational day 1 (G1). Pregnant females were housed individually for the duration of the pregnancy and pup rearing.
Alcohol Protocol
Conception-positive females were assigned to one of three treatment groups: alcohol (EtOH), pair-fed (PF), and chow (Chow). EtOH rats received one daily administration of 1.5g, 2.25g, 4.0g, or 6.0g ethanol/Kg body weight using a 25% w/v solution of ethanol in double-distilled water on G1-20, the time approximating trimesters 1 and 2 in human gestation (Bayer et al., 1993; Dobbing, 1981; Dobbing and Sands, 1973; 1979). Pair-fed rats were weight-matched to an EtOH female. These females received a dose of maltose-dextrin (MD; Bio-Serv, Frenchtown, NJ) that was isocaloric and isovolumetric to the EtOH dose received by their weight-matched EtOH female. They were then given the same amount of food consumed by their weight-matched EtOH female. Food for all animals consisted of a standard laboratory rat chow. All animals had free access to water. Females in the Chow group received free access to both food and water.
The alcohol administration protocol was strictly timed so that the animals received their alcohol dose at the same time each day. Food was removed from the cage at 8AM to allow food to clear from the stomach and allow alcohol absorption to be unimpeded. Food was weighed each day to determine amount consumed. Females were weighed and given their respective doses of EtOH or MD at 12PM, after which food was returned to the cage. Solutions were delivered by intragastric gavage using a curved 20G × 2½″ stainless steel feeding tube with a 3.0mm ball (Popper & Sons, Inc, New Hyde Park, NY) lubricated with a small drop of corn oil to facilitate its travel down the esophagus. Blood samples were collected from the tails of both the EtOH and PF dams every hour for four hours following alcohol delivery (i.e. at 1PM, 2PM, 3PM, and 4PM) on days 3, 10, 13 and 20 of the treatment regimen. For most of the dams, samples were collected using 100μL heparinized capillary tubes (Drummond Scientific Co., Broomall, CA) and transferred to 10mL gas chromatography vials containing 100μL of an internal standard solution. Vials were sealed using a septum-sealed lid and stored at room temperature until analysed by headspace gas chromatography at the federally licensed Shelby County Health Department Toxicology Laboratory to determine the blood alcohol concentration (BAC). For a smaller percentage of dams, samples were collected using 10μL heparinized capillary tubes (Analox, Lunenburg, MA). Tubes were spun in a hematocrit centrifuge, and then plasma was tested in an Analox GL5 Analyser after calibration to alcohol standards. Similar BAC levels were obtained using the two methods. Trough samples were taken approximately 24 hours later.
Pup Handling Protocol
Treatment administrations for EtOH and MD females were completed on G20. All females were then allowed free access to food (standard laboratory rat chow) and water. Pups were born at about G21-23 in PF and Chow females but at G22-24 in EtOH females. The number, sex, and body weight of the pups was recorded on the day of birth (postnatal day 0 (P0)). Litters were culled to 8–10 pups at this time. Pups were re-weighed on G29, G36, G43, and G50 (roughly equivalent to P7, P14, P21, and P28). On each assessment day, two pups (preferably one male and one female) were removed from the litter and processed for histology and measurement.
Pups were euthanized using sodium pentobarbital and perfused intracardially with 0.9% saline followed by a fixative consisting of 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.2). After removal of the head, the scalp and occipital bone were removed and the head was placed in fresh fixative overnight. The brain was then extracted from the skull, trimmed (removal of brain stem, optic nerves, olfactory bulbs, and membranes) and postfixed in fresh fixative for about a week. The brain was then weighed and stored in fixative until the histology was performed.
Histology and Measurement
Brains were bisected at the midsagittal plane. Membrane labelling of the callosal axons extending to the CCpn was achieved using crystals of DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate; Molecular Probes, OR) between 50 and 100μm in diameter. DiI crystals were placed along the entire length of the CC at the midsagittal plane to maximize the labelling of all callosal axons (see Fig. 1). Although the majority of connections through the CC are homotopic, heterotopic connections have also been documented (Ivy and Killackey, 1981; Segraves and Rosenquist, 1982a; b; Ding and Elberger, 2001). Therefore, labelling along the entire CC length would include any callosal axons with cells of origin in the visual cortex, even if they traversed the CC through areas other than the splenium. After insertion of the dye crystals, the hemispheres were placed in phosphate buffer (pH 7.2) and stored in the dark at 37°C to increase the rate of dye diffusion. Brains were stored for 6 - 12 weeks, with older animals receiving the longer diffusion times.
Figure 1.

CCpn labeled with DiI in the visual cortex demonstrating the cell labeling in both the infragranular and supragranular regions of the cortex. Scale bar = 50 μm.
Hemispheres were vibratome-sectioned at 100μm in the coronal plane. Sections were submerged in 65% glycerin in 0.1M phosphate-buffered saline (PBS; pH 7.2) at 4°C overnight, with a few drops of 0.5% m-phenylenediamine (fluorescent Nissl, Sigma) added to every sixth section. Sections were then mounted on glass slides using the 65% glycerin in PBS solution, coverslipped, and stored at 4°C until viewed.
Digital images were collected from each hemisphere using a Bio-Rad MRC1024 Confocal Laser Scanning Microscope equipped with a Krypton-Argon laser; the 568nm line was used to image the DiI and the 488nm line was used to image the fluorescent Nissl. The following procedure was used to standardize the CC region that was analyzed in pups of different ages. Five sections of the DiI labeled callosal axons were imaged from each hemisphere using the midline crossing of the splenial callosal fibres as a landmark. Due to the growth of the occipital cortex in relation to the position of the splenium, the collected images varied around the landmark section with pup age. In P0 and G29 pups, images were collected from the landmark section plus the two sections caudal and rostral to it. In G36 pups, the landmark section plus three sections caudal and one section rostral were collected, and in G43 and G50 pups, the landmark section plus the four sections caudal were collected. In the above section collection formulas, the fluorescent Nissl sections were exempted if they fell within the range of sections collected for the DiI images. Instead, the Nissl-stained sections either from within the range of DiI sections, or just rostral and caudal to the DiI section series were collected and assessed independently.
Confocal microscopy was used to optically section the 100μm thick sections at 5 μm intervals in the z-dimension using 10× magnification. The image space was a 768×512 pixel box with the long dimension oriented radially through the middle of area 17 in the visual cortex; this provided the greatest consistency, given that results might differ in other visual cortical areas. This allowed the inclusion of all labelled CCpn from layers I through VI. Collected images were measured using LaserSharp (BioRad, CA) image analysis software. Measurements included the number of cell bodies labelled by the DiI, the radial depth of each soma (from the middle of the soma to the pial surface), and the area of the soma profile. Area measurements were obtained from 20 cells sampled randomly across the image space from both the supragranular (II and III) and infragranular (V and VI) layers.
Data were analyzed using ANOVA with age and alcohol dosage as independent variables. Statistics were performed using SPSS v.12.0 software.
Results
General
A total of 159 pups from 23 pregnant females were assessed (see Tables 1 and 2). Figure 1 shows the DiI-labelled CCpn within the supragranular and infragranular regions. During DiI insertion into the midsagittal callosal area, no observations of callosal agenesis or apparent differences in midsagittal callosal area were noted.
Table 1.
Mean and Standard Error of Alcohol-Exposed Pregnant Female Data.
| Treatment Group | Number of Females | peak BAC (mg/dL) | Litter Size | Gestation Length (days) |
|---|---|---|---|---|
| control | 6 | 0 | 13.67 (0.84) | 22.67 (0.33) |
| 1.5g EtOH | 3 | 83.67 (11.32) | 13.33 (1.86) | 23.00 (0.00) |
| 2.25g EtOH | 3 | 124.00 (24.42) | 12.33 (1.67) | 23.00 (0.58) |
| 4.0g EtOH | 3 | 196.00 (14.01) | 14.67 (1.20) | 22.67 (0.33) |
| 6.0g EtOH | 8 | 379.25 (23.56) | 10.25 (1.01) | 23.50 (0.27) |
Table 2.
Pup Data - Number of Pups (top number), Mean and Standard Error of Body Weight (grams – middle number) and Brain Weight (grams – bottom number) for each Treatment Category
| Treatment Group | Pup Age |
||||
|---|---|---|---|---|---|
| P0 | G29 | G36 | G43 | G50 | |
| control | 7 | 12 | 9 | 10 | 10 |
| 6.20 (0.25) | 13.38 (0.61) | 29.00 (0.91) | 48.75 (1.94) | 78.93 (4.17) | |
| 0.256 (0.01) | 0.601 (0.02) | 1.116 (0.02) | 1.375 (0.02) | 1.485 (0.03) | |
|
| |||||
| 1.5g EtOH | 6 | 6 | 6 | 6 | 6 |
| 6.05 (0.30) | 13.85 (0.53) | 31.58 (0.69) | 53.27 (1.31) | 91.13 (5.86) | |
| 0.247 (0.01) | 0.610 (0.01) | 1.135 (0.03) | 1.364 (0.02) | 1.465 (0.02) | |
|
| |||||
| 2.25g EtOH | 4 | 6 | 6 | 6 | 5 |
| 6.58 (0.13) | 13.93 (0.44) | 31.15 (1.66) | 52.63 (1.43) | 91.36 (2.42) | |
| 0.262 (0.02) | 0.621 (0.01) | 1.141 (0.03) | 1.383 (0.01) | 1.529 (0.02) | |
|
| |||||
| 4.0g EtOH | 7 | 5 | 6 | 5 | 7 |
| n/a | 12.80 (0.78) | 32.23 (2.84) | 49.12 (4.84) | 81.26 (5.36) | |
| 0.241 (0.01) | 0.586 (0.03) | 1.164 (0.03) | 1.334 (0.06) | 1.501 (0.02) | |
|
| |||||
| 6.0g EtOH | 4 | 4 | 3 | 8 | 7 |
| n/a | 11.40 (1.11) | 23.43 (3.12) | 39.5 | 59.90 (9.40) | |
| 0.163 (0.01) | 0.542 (0.05) | 1.010 (0.05) | 1.289 (0.06) | 1.530 (0.05) | |
A comparison between the Chow and PF animals indicated no significant difference for any of the dependent measurements (p > 0.05), therefore these two groups were combined into one control group. Similarly, no significant main effects of sex were found (p > 0.05) and therefore data for both sexes were pooled at each age point.
The number of pregnant females in each group, mean BAC, litter sizes, and length of gestation are shown in Table 1. One-way ANOVA indicated no differences among groups for litter size or length of gestation.
The number of pups in which DiI diffusion was successful to the point of permitting measurement for each alcohol dosage is shown in Table 2. In a few cases, and specific to the older animals, DiI diffusion created non-specific background staining that prevented an accurate assessment of cell morphology. In these cases, the animal was not included in the study. Body weight measures indicated a significant interaction (F14,112 = 1.88; p = 0.036) between age and treatment, with those animals receiving alcohol at 6.0g/Kg showing a reduction in the rate of weight gain at the time of perfusion relative to the other treatment groups. Brain weight measures showed a significant main effect of age (F4,134 = 1261.69; p < 0.0001; increase in size with age) and of treatment (F4,134 = 3.41; p = 0.011; brain weight appeared to be reduced in those animals that received 6.0g EtOH relative to the other treatment groups).
Cell Area
Infragranular
Table 3 shows the mean cell areas for each of the treatment conditions. A two-way ANOVA between age and treatment indicated a significant interaction (F16,133 = 3.92; p < 0.0001). Cell areas in most of the alcohol-treated animals were smallest in the P0 animals and then rose to their largest sizes at G29, followed by a gradual decrease in size. In the 6.0g EtOH animals, cell area increased to a peak at G36, followed by a gradual size decrease. Cell area in the control animals increased between P0 and G29, but then remained relatively constant from G29 through G50. In almost all cases, the area of the infragranular cells exceeded that of the supragranular cells.
Table 3.
Mean (top number) and Standard Error (bottom number) of Infragranular Cell Areas.
| Treatment Group | Infragranular Cell Areas (μm2) |
||||
|---|---|---|---|---|---|
| P0 | G29 | G36 | G43 | G50 | |
| control | 184.5 | 227.33 | 245.71 | 227.25 | 238.41 |
| 4.52 | 11.68 | 6.35 | 6.43 | 11.35 | |
|
| |||||
| 1.5g EtOH | 174.71 | 254.5289 | 218.22 | 206.82 | 215.77 |
| 8.05 | 12.65 | 9.69 | 7.42 | 4.16 | |
|
| |||||
| 2.25g EtOH | 187.28 | 273.10 | 231.99 | 209.66 | 211.55 |
| 5.10 | 10.74 | 8.85 | 10.42 | 8.87 | |
|
| |||||
| 4.0g EtOH | 175.90 | 287.89 | 249.43 | 265.55 | 245.74 |
| 6.12 | 11.75 | 13.38 | 12.69 | 9.16 | |
|
| |||||
| 6.0g EtOH | 140.82 | 258.89 | 279.44 | 255.68 | 215.86 |
| 6.17 | 10.37 | 3.75 | 11.06 | 4.95 | |
Supragranular
Table 4 shows the mean cell areas for each of the treatment conditions in the supragranular cells. A two-way ANOVA between age and treatment indicated a significant interaction (F16,133 = 3.745; p < 0.0001). In general, the pattern of cell area differences was the same as that seen in the infragranular cells. Cell areas in most of the alcohol-treated animals were smallest at P0 and increased to a peak at G29, followed by a gradual decrease in area. Cell areas in the 6.0g EtOH group peaked at G36 followed by a gradual decline while those in the control animals again remained relatively constant from G29 through G50.
Table 4.
Mean (top number) and Standard Error (bottom number) of Supragranular Cell Areas.
| Treatment Group | Supragranular Cell Areas (μm2) |
||||
|---|---|---|---|---|---|
| P0 | G29 | G36 | G43 | G50 | |
| Control | 179.87 | 188.82 | 213.64 | 199.38 | 222.48 |
| 6.10 | 8.66 | 6.54 | 5.98 | 11.42 | |
|
| |||||
| 1.5g EtOH | 172.43 | 209.81 | 192.90 | 180.52 | 189.51 |
| 6.79 | 6.44 | 10.82 | 5.57 | 13.72 | |
|
| |||||
| 2.25g EtOH | 164.11 | 223.14 | 198.72 | 178.39 | 192.12 |
| 2.63 | 6.65 | 5.87 | 8.13 | 11.89 | |
|
| |||||
| 4.0g EtOH | 160.70 | 228.54 | 216.68 | 234.75 | 223.53 |
| 5.92 | 10.02 | 11.61 | 12.65 | 9.88 | |
|
| |||||
| 6.0g EtOH | 150.10 | 245.58 | 263.62 | 225.31 | 206.57 |
| 6.87 | 26.52 | 19.81 | 6.63 | 6.55 | |
Cell Depth Within Neocortex
Figure 2 shows the mean cell depths for each treatment group at each age. A two-way ANOVA between age and treatment indicated a significant interaction (F16,134 = 2.48; p = 0.0005). As shown, the distance of the CCpn from the pia membrane increased with age (p < 0.0001), but no significant main effect of treatment was apparent due to a paradoxical difference between the results found at P0 and those from later ages. At P0, the CCpn in the control and 1.5g EtOH groups were farther from the pia than those in the remaining alcohol groups, and increasing alcohol exposure appeared to decrease that distance from the pia. This relationship was reversed in the animals at G29; more of the CCpn were found deeper in the cortex of the alcohol-exposed animals. The treatment groups demonstrated relatively little variation in cell depth from G36 through G50.
Figure 2.

Mean and standard error of the depth of the CCpn from the pia membrane. There was a significant age × treatment interaction (p<0.0005) with the animals at birth showing a closer proximity to the pia with increasing BAC level and the older animals showing a reversed relationship.
This difference in cell positioning between control and the 6.0g EtOH animals is demonstrated in Figure 3. Fig. 3a shows the CCpn in a G29 Chow animal while Fig. 3b shows the CCpn in a G29 6.0g EtOH animal. The CCpn in the EtOH animal appear farther from the pia membrane than those in the control animal. It should be noted that the cell depths referred to are the mean cell depths, and that the CCpn were found to reside at all layers within the cortex in all treatment groups. However, in the older animals, very few cells were found to reside in the supragranular layers.
Figure 3.

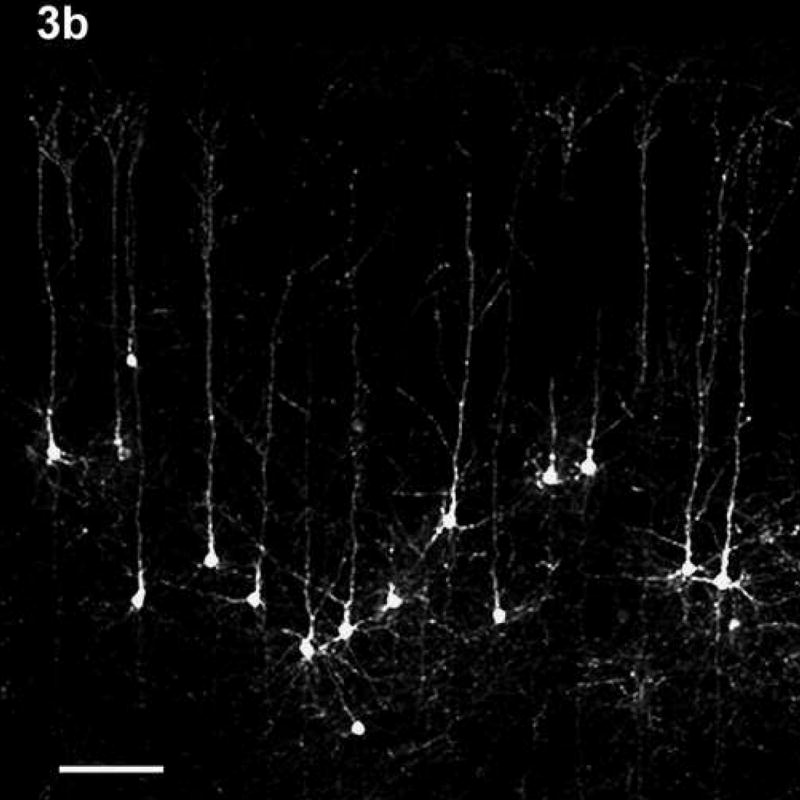
The position of the CCpn in a G29 CHOW animal (3a) and a G29 6.0g EtOH animal (3b). Note the difference in positioning of the CCpn in the two treatment groups – cells in the EtOH-treated animal appear farther away from the pia membrane than those in the control animal, despite the similar overall cortical depth. Scale bars = 100 μm.
Cell Number
The mean number of cells for each treatment condition is shown in Fig. 4. A two-way ANOVA between age and treatment indicated a significant interaction (F16,134 = 2.487; p = 0.0024). In most of the treatment groups, the number of cells increased to a peak at G36, followed by a sharp decline through G43 and G50. The exception to this was those pups in the 6.0g EtOH group, which did not show a clear relationship between cell number and age. It was interesting to note the general trend to a decrease in cell numbers with increasing alcohol exposure in the G29 and G36 groups but the inverse relationship at age G50.
Figure 4.

Mean and standard error of the number of CCpn measured for each treatment group. There was a significant age × treatment interaction (p=0.0024). In most of the treatment conditions, the number of cells increased to a peak at G36, followed by a sharp decline through G43 and G50. The exception to this was those pups in the 6.0g group, which did not show a clear relationship between CCpn number and age.
In all but the 6.0g EtOH treatment group, the increase in cell number to the peak at G36 was gradual. This gradual increase is reflective of a natural increase in the number of CCpn whose axons gradually reach the midline so that they become labeled by DiI, and/or an increase in the number of cells migrating into the cortex to become CCpn. The apparent decline in CCpn number was rapid in the control, 1.5g and 2.25g EtOH groups but more gradual in the 4.0g EtOH group. Therefore, it appears that the decline in the numbers of labeled CCpn is related to the level of alcohol exposure during prenatal development, suggesting either an acceleration in the elimination of transitory CCpn axons that is related to amount of prenatal alcohol exposure, or an increased CCpn, and possibly cortical, cell loss related to the amount of prenatal alcohol exposure.
Discussion
Pregnant females exposed to alcohol during the course of their gestation showed an altered pattern of CC development. In the control animals, the number of CCpn in the visual cortex increased through G36, followed by a sharp decline in the older ages. This confirms the accepted view of callosal development in the visual cortex, as follows. The CCpn are generated and migrate out to reside in all layers of the cortex. Such cells extend axons through the corpus callosum (hence the ability of DiI to label these axons retrogradely to the CCpn) to make connections in the contralateral hemisphere. However, these connections are transitory as indicated by the sharp reduction in the number of labeled CCpn only 7 days later. This confirms our results of transitory CC connections using DiI in normal rat and normal cat (Elberger, 1993; 1994a; b) as well as those of others.
In most of the alcohol-exposed pups, the number of cells also increases through G36 and then decreases; however the rate of increase to peak and the subsequent decline varies according to alcohol dosage. Cell numbers from animals in the 1.5g and 2.25g EtOH groups follow a developmental pattern that is similar to the control animals, suggesting that these alcohol levels do not interfere with the normal development and migration of CCpn. Animals in the 4.0g EtOH group show an increase in CCpn number up to a relatively small peak at G36 but thereafter very little change, suggesting that these higher levels of alcohol may delay the development and migration of the CCpn sufficiently to overlap with the elimination of the transitory axons that are present. Animals in the 6.0g EtOH group showed no relationship between CCpn number and age, suggesting that high-dose prenatal alcohol exposure limits the migration of cells into the cortex to become CCpn, and/or limits the ability of cortical cells destined to become CCpn to extend axonal processes to the midline and hence become labeled with the DiI. The latter explanation would account for reduced CC size in cases of heavy prenatal alcohol consumption that has been observed in humans (Clarren, 1986; Mattson et al., 1992; Mattson et al., 1994; Riley et al., 1995). Previous behavioural and physiological studies have shown that the CC connections must be there during critical time periods for visual development in order to establish normal visual functions (Elberger, 1979; 1980; 1981; 1982; 1984a;b; 1988; 1989; 1990; Elberger and Smith, 1985; Timney et al., 1985). For example, if CC axons are eliminated during postnatal week 2 in the cat, the visual cortex will be significantly reorganized. This would suggest that if axons were delayed in their arrival to the contralateral cortex until the second week, the same result would ensue.
Animals in the 6.0g EtOH group showed a considerable decrease in cell number between P0 and G29. One possible explanation for this would be a precipitous loss of CCpn during this time period. A second possible explanation would be that there is a precipitous premature elimination of transitory CCpn axons during this time period. A third possible explanation would be that there is a less homogeneous, more clustered, distribution of CCpn in the 6.0g EtOH group than in the other alcohol treatment groups or controls, so that numbers measured at each of the 5 age points represent the statistical likelihood of having a cluster within the microscope field being measured. Although we have observed some CCpn to demonstrate this type of irregular distribution, no such clustering was noted uniquely in the 6.0g EtOH animals.
The altered pattern of cell body area seen in the CCpn of the alcohol-exposed animals may be related to differences in dendritic patterning. The relative reduction in the number of CCpn in alcohol-treated animals as compared to controls at G29 suggests that fewer cells must receive contralateral projection fibre input, requiring an increase in the complexity of the dendrites of the CCpn. Indeed, Qiang et al. (2002) reported that animals exposed to high-dose (4.0g/Kg and 6.0g/Kg) prenatal alcohol showed an increase in the number and length of dendrites at G29, an effect that was attenuated as the animals became older. The generation and maintenance of more numerous and longer dendritic branches would require larger cells to support the increased cellular metabolism, hence the increase in cell size. By G36, the generation of additional cells in the alcohol-exposed animals would reduce the need for such complex cell structures, resulting in cell sizes similar to the control.
The distance of the CCpn from the pia increases with age in the control animals, suggesting that more of the cells residing in the supragranular layers are eliminated. Alcohol-exposed animals follow a similar pattern of development except for those in the 6.0g EtOH group; in this group, the CCpn appear to maintain their relative positioning within the cortex, suggesting an even distribution of CCpn elimination throughout the cortical levels. It is possible that these animals retain a greater proportion of their callosal connections due to the relative reduction in the number of CCpn. Alternatively, those animals exposed to higher alcohol levels demonstrated a delayed development for CCpn and therefore it may also be possible that more of the CCpn would have been eliminated at an older age than was examined in the present study.
The defects observed in the number, size and positioning of CCpn in those animals exposed to high levels of alcohol suggest that the corpus callosum is an effective model for the study of alcohol effects on the developing brain. It should be noted that the midsagittal area of the rat corpus callosum does not appear to be affected by similar parameters of alcohol exposure (Livy and Elberger, 2001). Therefore, although the results presented here demonstrate an effect of alcohol on the CCpn, these effects are not profound enough to affect severely the overall production of CCpn or their commissural fibres. This is in stark contrast to the sometimes severe callosal defects reported in the human literature. It is possible that the developing callosal cells and fibres in the human are more susceptible to prenatal alcohol exposure. However, it may also be that alcohol exposure patterns in humans are more complex and variable, and often associated with other environmental stressors (poor nutrition, other drugs of abuse) that may have a compounding and confounding effect on axon pathway development. It also remains unclear whether the CC is the only axon pathway affected by alcohol exposure. Livy et al. (1997) demonstrated an increase in the number of axons in the anterior commissure (AC) of mice displaying callosal agenesis. This increase was accomplished without a corresponding increase in the midsagittal area of the AC. If similar changes were evident in the axon pathways of humans with altered CC development, they would go unnoticed in MRI scans.
The CCpn changes described pertain to rats exposed to alcohol throughout their gestation, however it is possible that the effects of the alcohol exposure are specific to certain periods of development. For example, the critical time period for callosal effects to occur may be confined to either the first or second trimester, and may not necessarily require exposure for the duration of both trimesters. Alternatively, callosal connections may be differentially affected by alcohol exposure during different time periods. Evidence for this alternative hypothesis is found in preliminary results we obtained comparing the different effects on CCpn size and distribution within the cortex when alcohol exposure was limited to G1-10, G11-20 or G1-20. Prenatal alcohol exposure in rats from G1-10 produced similar effects on CCpn development, including the abnormal number and length of CCpn dendrites. These effects of gestational timing may be particularly important when interpreting the present results for human development.
Acknowledgments
This work was supported by NIH grant AA11325 awarded to AJE.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bayer SA, Altman J, Russo R, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determine patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Blood alcohol concentration and microencephaly: a dose response study in the neonatal rat. Teratology. 1988;37:223–231. doi: 10.1002/tera.1420370307. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Goodlett CR, West JR. Blood alcohol concentration and severity of microencephaly in neonatal rats depend on the pattern of alcohol administration. Alcohol. 1988;5:209–214. doi: 10.1016/0741-8329(88)90054-7. [DOI] [PubMed] [Google Scholar]
- Braisted JE, Tuttle R, O’leary DD. Thalamocortical axons are influenced by chemorepellent and chemoattractant activities localized to decision points along their path. Dev Biol. 1999;208:430–40. doi: 10.1006/dbio.1999.9216. [DOI] [PubMed] [Google Scholar]
- Clarren SK. Neuropathology in fetal alcohol syndrome. In: West JR, editor. Alcohol and Brain Development. New York: Oxford University Press; 1986. pp. 158–166. [Google Scholar]
- Coles CD, Brown RT, Smith IE, Platzman KA, Erickson S, Falek A. Effects of prenatal alcohol exposure at school age. I Physical and cognitive development Neurotoxicol Teratol. 1991;13:1–11. doi: 10.1016/0892-0362(91)90084-a. [DOI] [PubMed] [Google Scholar]
- Dehay C, Kennedy H, Bullier J. Characterization of transient cortical projections from auditory, somatosensory, and motor cortices to visual areas 17, 18 and 19 in the kitten. J Comp Neurol. 1988;272:68–89. doi: 10.1002/cne.902720106. [DOI] [PubMed] [Google Scholar]
- Ding SL, Elberger AJ. Postnatal development of biotinylated dextran amine-labeled corpus callosum axons projecting from the visual and auditory cortices to the visual cortex of the rat. Exp Brain Res. 2001;136:179–193. doi: 10.1007/s002210000576. [DOI] [PubMed] [Google Scholar]
- Dobbing J. The later development of the brain and its vulnerability. In: Davis JA, Dobbing J, editors. Scientific Foundations of Pediatrics. 2. London: William Heineman Medical Books; 1981. pp. 841–847. [Google Scholar]
- Dobbing J, Sands J. Quantitative growth and development of the human brain. Arch Dis Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The role of the corpus callosum in the development of interocular eye alignment and the organization of the visual field in the cat. Exp Brain Res. 1979;36:71–85. doi: 10.1007/BF00238468. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The effect of neonatal section of the corpus callosum on the development of depth perception in young cats. Vision Res. 1980;20:177–187. doi: 10.1016/0042-6989(80)90160-1. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. Ocular dominance in striate cortex is altered by neonatal section of the posterior corpus callosum in the cat. Exp Brain Res. 1981;41:280–291. doi: 10.1007/BF00238885. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The corpus callosum is a critical factor for developing maximum visual acuity. Dev Brain Res. 1982;5:350–353. doi: 10.1016/0165-3806(82)90134-1. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The existence of a separate, brief critical period for the corpus callosum to affect visual development. Behav Brain Res. 1984a;11:223–231. doi: 10.1016/0166-4328(84)90214-6. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The minimum extent of corpus callosum connections required for normal visual development in the cat. Human Neurobiol. 1984b;3:115–120. [PubMed] [Google Scholar]
- Elberger AJ. Developmental interactions between the corpus callosum and the visual system in cats. Behav Brain Res. 1988;30:119–134. doi: 10.1016/0166-4328(88)90142-8. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. Binocularity and single cell acuity are related in striate cortex of corpus callosum sectioned and normal cats. Exp Brain Res. 1989;77:213–216. doi: 10.1007/BF00250583. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. Spatial frequency thresholds of single striate cortical cells in neonatal corpus callosum sectioned cats. Exp Brain Res. 1990;82:617–627. doi: 10.1007/BF00228803. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. Distribution of transitory corpus callosum axons projecting to developing cat visual cortex revealed by DiI. J Comp Neurol. 1993;333:326–342. doi: 10.1002/cne.903330303. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. Transitory corpus callosum axons projecting through developing rat visual cortex revealed by DiI. Cereb Cort. 1994a;4:279–299. doi: 10.1093/cercor/4.3.279. [DOI] [PubMed] [Google Scholar]
- Elberger AJ. The corpus callosum provides a massive transitory input to the visual cortex of cat and rat during early postnatal development. Behav Brain Res. 1994b;64:15–24. doi: 10.1016/0166-4328(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Elberger AJ, Smith EL., III The critical period for corpus callosum section to affect cortical binocularity. Exp Brain Res. 1985;57:213–223. doi: 10.1007/BF00236526. [DOI] [PubMed] [Google Scholar]
- Fukumitsu H, Ohtsuka M, Murai R, Nakamura H, Itoh K, Furukawa S. Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J Neurosci. 2006;26:13218–13230. doi: 10.1523/JNEUROSCI.4251-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, Marcussen BL, West JR. A single day of alcohol exposure during the brain growth spurt induces brain weight restriction and cerebellar Purkinje cell loss. Alcohol. 1990;7:107–114. doi: 10.1016/0741-8329(90)90070-s. [DOI] [PubMed] [Google Scholar]
- Hamre KM, West JR. The effects of the timing of ethanol exposure during the brain growth spurt on the number of cerebellar Purkinje and granule cell nuclear profiles. Alcohol Clin Exp Res. 1993;17:610–622. doi: 10.1111/j.1530-0277.1993.tb00808.x. [DOI] [PubMed] [Google Scholar]
- Honig MG, Hume RI. DiI and DiO: versatile fluorescent dyes for neuronal labelling and pathway tracing. Trends Neurosci. 1989;12:337–341. [PubMed] [Google Scholar]
- Houzel JC, Carvalho ML, Lent R. Interhemispheric connections between primary visual areas: beyond the midline rule. Braz J Med Biol Res. 2002;35:1441–1453. doi: 10.1590/s0100-879x2002001200005. [DOI] [PubMed] [Google Scholar]
- Innocenti GM. Growth and reshaping of axons in the establishment of visual callosal connections. Science. 1981;212:824–827. doi: 10.1126/science.7221566. [DOI] [PubMed] [Google Scholar]
- Innocenti GM, Caminiti R. Postnatal shaping of callosal connections from sensory areas. Exp Brain Res. 1980;38:381–394. doi: 10.1007/BF00237518. [DOI] [PubMed] [Google Scholar]
- Innocenti GM, Clarke S. The organization of immature callosal connections. J Comp Neurol. 1984;230:287–309. doi: 10.1002/cne.902300212. [DOI] [PubMed] [Google Scholar]
- Innocenti GM, Clarke S, Kraftsik R. Interchange of callosal and association projections in the developing visual cortex. J Neurosci. 1986;6:1384–409. doi: 10.1523/JNEUROSCI.06-05-01384.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy GO, Killackey HP. The ontogeny of the distribution of callosal projection neurons in the rat parietal cortex. J Comp Neurol. 1981;195:367–389. doi: 10.1002/cne.901950302. [DOI] [PubMed] [Google Scholar]
- Jacobson S, Trojanowski JQ. The cells of origin of the corpus callosum in rat, cat and rhesus monkey. Brain Res. 1974;74:149–155. doi: 10.1016/0006-8993(74)90118-8. [DOI] [PubMed] [Google Scholar]
- Jeret JS, Serur D, Wisniewski KE, Lubin RA. Clinicopathological findings associated with agenesis of the corpus callosum. Brain Dev. 1987;9:225–264. doi: 10.1016/s0387-7604(87)80042-6. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. The fetal alcohol syndrome. Teratology. 1975;12:1–10. doi: 10.1002/tera.1420120102. [DOI] [PubMed] [Google Scholar]
- Katz LC, Burkhalter A, Dreyer WJ. Fluorescent latex microspheres as a retrograde neuronal marker for in vivo and in vitro studies of visual cortex. Nature. 1984;310:498–500. doi: 10.1038/310498a0. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Elliott M. Cell proliferation in the embryonic mouse neocortex following acute maternal alcohol consumption. Int J Dev Neurosci. 1985;3:311–315. doi: 10.1016/0736-5748(85)90063-2. [DOI] [PubMed] [Google Scholar]
- Koester SE, O’Leary DDM. Connectional distinction between callosal and subcortically projecting cortical neurons is determined prior to axon extension. Dev Biol. 1993;160:1–14. doi: 10.1006/dbio.1993.1281. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Elberger AJ. Effect of prenatal alcohol exposure on midsagittal commissure size in rats. Teratology. 2001;63:15–22. doi: 10.1002/1096-9926(200101)63:1<15::AID-TERA1003>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Wahslten D. Retarded formation of the hippocampal commissure in embryos from mouse strains lacking a corpus callosum. Hippocampus. 1997;7:2–14. doi: 10.1002/(SICI)1098-1063(1997)7:1<2::AID-HIPO2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Schalomon PM, Roy M, Zacharias MC, Pimenta J, Lent R, Wahlsten D. Increased axon number in the anterior commissure of mice lacking a corpus callosum. Exp Neurol. 1997;146:491–501. doi: 10.1006/exnr.1997.6564. [DOI] [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurtoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Lund JS, Lund RD. The termination of callosal fibers in the paravisual cortex of the rat. Brain Res. 1986;17:35–45. doi: 10.1016/0006-8993(70)90306-9. [DOI] [PubMed] [Google Scholar]
- Lund RD, Chang FLF, Land PW. The development of callosal projections in normal and one-eyed rats. Dev Brain Res. 1984;14:139–142. doi: 10.1016/0165-3806(84)90018-x. [DOI] [PubMed] [Google Scholar]
- Maier SE, Chen WJA, Miller JA, West JR. Fetal alcohol exposure and temporal vulnerability: Regional differences in alcohol-induced microencephaly as a function of the timing of binge-like alcohol exposure during rat brain development. Alcohol Clin Exp Res. 1997;21:1418–1428. doi: 10.1111/j.1530-0277.1997.tb04471.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Riley EP, Jernigan TL. Fetal alcohol syndrome: A case report of neuropsychological, MRI, and EEG assessment of two children. Alcohol Clin Exp Res. 1992;16:1001–1003. doi: 10.1111/j.1530-0277.1992.tb01909.x. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Jernigan TL, Riley EP. MRI and prenatal alcohol exposure. Alcohol Hlth Res World. 1994;18:49–52. [PMC free article] [PubMed] [Google Scholar]
- Miller MW. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science. 1986;233:1308–1311. doi: 10.1126/science.3749878. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effect of prenatal exposure to alcohol on the distribution and time of origin of corticospinal neurons in the rat. J Comp Neurol. 1987;257:372–382. doi: 10.1002/cne.902570306. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effects of prenatal exposure to ethanol on neocortical development. II Cell proliferation in the ventricular and subventricular zones of the rat J Comp Neurol. 1989;287:326–338. doi: 10.1002/cne.902870305. [DOI] [PubMed] [Google Scholar]
- Miller MW. Effects of prenatal exposure to ethanol on callosal projection neurons in rat somatosensory cortex. Brain Res. 1997;766:121–128. doi: 10.1016/s0006-8993(97)00533-7. [DOI] [PubMed] [Google Scholar]
- Miller MW, Vogt BA. The postnatal growth of the callosal connections of primary and secondary visual cortex in the rat. Dev Brain Res. 1984;14:304–309. doi: 10.1016/0165-3806(84)90319-5. [DOI] [PubMed] [Google Scholar]
- Olavarria J, Van Sluyters RC. Organization and postnatal development of callosal connections in the visual cortex of the rat. J Comp Neurol. 1985;239:1–26. doi: 10.1002/cne.902390102. [DOI] [PubMed] [Google Scholar]
- Ozaki HS, Wahlsten D. Prenatal formation of the normal mouse corpus callosum: A quantitative study with carbocyanine dyes. J Comp Neurol. 1992;323:81–90. doi: 10.1002/cne.903230107. [DOI] [PubMed] [Google Scholar]
- Payne B, Pearson H, Cornwell P. Development of visual and auditory cortical connections in the cat. In: Peters A, Jones EG, editors. Cerebral Cortex. Vol. 7. New York: Plenum Press; 1988. pp. 309–389. [Google Scholar]
- Peiffer J, Majewski F, Fischbach H, Bierich JR, Volk B. Alcohol embryo- and fetopathy. J Neurol Sci. 1979;41:125–137. doi: 10.1016/0022-510x(79)90033-9. [DOI] [PubMed] [Google Scholar]
- Pinazo-Duran MD, Renau-Piqueras J, Guerri C, Stromland K. Optic nerve hypoplasia in fetal alcohol syndrome: an update. Eur J Ophthalmol. 1997;7:262–270. doi: 10.1177/112067219700700311. [DOI] [PubMed] [Google Scholar]
- Qiang M, Wang M, Elberger AJ. Second trimester prenatal alcohol exposure alters development of rat corpus callosum. Neurotoxicol Teratol. 2002;24:719–732. doi: 10.1016/s0892-0362(02)00267-2. [DOI] [PubMed] [Google Scholar]
- Riley EP, Mattson SN, Sowell ER, Jernigan TL, Sobel DF, Jones KL. Abnormalities of the corpus callosum in children prenatally exposed to alcohol. Alcohol Clin Exp Res. 1995;19:1198–1202. doi: 10.1111/j.1530-0277.1995.tb01600.x. [DOI] [PubMed] [Google Scholar]
- Samson HH, Grant KA. Ethanol-induced microcephaly in neonatal rats: Relation to dose. Alcohol Clin Exp Res. 1984;8:201–203. doi: 10.1111/j.1530-0277.1984.tb05839.x. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, O’Leary DD. Early development of the somatotopic map and barrel patterning in rat somatosensory cortex. J Comp Neurol. 1994;346:80–96. doi: 10.1002/cne.903460106. [DOI] [PubMed] [Google Scholar]
- Segraves MA, Rosenquist AC. The distribution of the cells of origin of callosal projections in cat visual cortex. J Neurosci. 1982a;2:1079–1089. doi: 10.1523/JNEUROSCI.02-08-01079.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segraves MA, Rosenquist AC. The afferent and efferent callosal connections of retinotopically defined areas in cat cortex. J Neurosci. 1982b;2:1090–1107. doi: 10.1523/JNEUROSCI.02-08-01090.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IE, Coles CD, Lancaster J, Fernhoff PM, Falek A. The effect of volume and duration of prenatal ethanol exposure on neonatal physical and behavioral development. Neurobehav Toxicol Teratol. 1986;8:375–381. [PubMed] [Google Scholar]
- Stromland K. Visual impairment and ocular abnormalities in children with fetal alcohol syndrome. Addict Biol. 2004;9:153–157. doi: 10.1080/13556210410001717024. [DOI] [PubMed] [Google Scholar]
- Stromland K, Pinazo-Duran MD. Ophthalmic involvement in the fetal alcohol syndrome: clinical and animal model studies. Alcohol Alcohol. 2002;37:2–8. doi: 10.1093/alcalc/37.1.2. [DOI] [PubMed] [Google Scholar]
- Timney B, Elberger AJ, Vandewater ML. Binocular depth perception in the cat following early corpus callosum section. Exp Brain Res. 1985;60:19–26. doi: 10.1007/BF00237014. [DOI] [PubMed] [Google Scholar]
- Vercelli A, Innocenti GM. Morphology of visual callosal neurons with different locations, contralateral targets or patterns of development. Exp Brain Res. 1993;94:393–404. doi: 10.1007/BF00230198. [DOI] [PubMed] [Google Scholar]
- Wainwright P, Fritz G. Effect of moderate prenatal alcohol exposure on postnatal brain and behavioral development in BALB/c mice. Exp Neurol. 1985;89:237–249. doi: 10.1016/0014-4886(85)90279-1. [DOI] [PubMed] [Google Scholar]
- Wainwright P, Gagnon M. Moderate prenatal alcohol exposure interacts with strain in affecting brain development in BALB/c and C57BL/6 mice. Exp Neurol. 1985;88:84–94. doi: 10.1016/0014-4886(85)90115-3. [DOI] [PubMed] [Google Scholar]
- Wisniewski K, Dambska M, Sher JH, Qazi Q. A clinical neuropathological study of the fetal alcohol syndrome. Neuropediatrics. 1983;14:197–201. doi: 10.1055/s-2008-1059578. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, Mickus LA. Sex differences in corpus callosum: influence of prenatal alcohol exposure and maternal undernutrition. Brain Res. 1990;537:115–122. doi: 10.1016/0006-8993(90)90347-e. [DOI] [PubMed] [Google Scholar]
- Zimmerberg B, Scalzi LV. Commissural size in neonatal rats: effects of sex and prenatal alcohol exposure. Int J Dev Neurosci. 1989;7:81–86. doi: 10.1016/0736-5748(89)90046-4. [DOI] [PubMed] [Google Scholar]